

Abstract
β-Lactamase enzymes (EC 3.5.2.6) are a significant threat to the continued use of β-lactam antibiotics to treat infections. A novel non-β-lactam β-lactamase inhibitor with activity against many class A and C and some class D β-lactamase variants, avibactam, is now available in the clinic in partnership with ceftazidime. Here, we explored the activity of avibactam against a variety of characterized isogenic laboratory constructs of β-lactamase inhibitor-resistant variants of the class A enzyme SHV (M69I/L/V, S130G, K234R, R244S, and N276D). We discovered that the S130G variant of SHV-1 shows the most significant resistance to inhibition by avibactam, based on both microbiological and biochemical characterizations. Using a constant concentration of 4 mg/liter of avibactam as a β-lactamase inhibitor in combination with ampicillin, the MIC increased from 1 mg/liter for blaSHV-1 to 256 mg/liter for blaSHV S130G expressed in Escherichia coli DH10B. At steady state, the k2/K value of the S130G variant when inactivated by avibactam was 1.3 M−1 s−1, versus 60,300 M−1 s−1 for the SHV-1 β-lactamase. Under timed inactivation conditions, we found that an approximately 1,700-fold-higher avibactam concentration was required to inhibit SHV S130G than the concentration that inhibited SHV-1. Molecular modeling suggested that the positioning of amino acids in the active site of SHV may result in an alternative pathway of inactivation when complexed with avibactam, compared to the structure of CTX-M-15–avibactam, and that S130 plays a role in the acylation of avibactam as a general acid/base. In addition, S130 may play a role in recyclization. As a result, we advance that the lack of a hydroxyl group at position 130 in the S130G variant of SHV-1 substantially slows carbamylation of the β-lactamase by avibactam by (i) removing an important proton acceptor and donator in catalysis and (ii) decreasing the number of H bonds. In addition, recyclization is most likely also slow due to the lack of a general base to initiate the process. Considering other inhibitor-resistant mechanisms among class A β-lactamases, S130 may be the most important amino acid for the inhibition of class A β-lactamases, perhaps even for the novel diazabicyclooctane class of β-lactamase inhibitors.
INTRODUCTION
The β-Lactam antibiotics (penicillins, cephalosporins, and carbapenems) are a cornerstone of therapy for many Gram-negative bacterial infections. These antibiotics act by binding to the penicillin-binding proteins of the bacterial cell membrane, preventing peptidoglycan cross-linking and eventually leading to cell lysis and death. Recent studies also showed that β-lactams create a “futile cycle” that involves a lytic transglycosylase (Slt) and leads to bacterial cell death (1). Unfortunately, Gram-negative bacteria contain β-lactamase enzymes in their periplasmic space that are able to hydrolyze the amide bond of β-lactams, preventing these compounds from reaching their sites of action. In order to combat the action of these enzymes, the synthesis of additional classes of β-lactams (i.e., future generations of cephalosporins, carbapenems, and monobactams) and the synthesis of β-lactamase inhibitors (BLIs) (i.e., sulbactam, tazobactam, and clavulanic acid) were undertaken. Despite the clinical success of β-lactams and BLIs, β-lactamase enzymes evolved to exhibit resistance to both of these strategies. A survey of these newly described β-lactamases reveals that point mutations leading to extended-spectrum β-lactamases (ESBLs; enzymes resistant to later-generation cephalosporins) and inhibitor-resistant (IR) β-lactamases (enzymes which exhibit resistance to inactivation, particularly by clavulanic acid) are the major mechanism by which resistance is manifested.
The SHV-1 β-lactamase is a class A enzyme that is chromosomally encoded in Klebsiella pneumoniae. SHV-1 typically acquires ESBL activity with a substitution at Ambler position G238 or E240 (2–4). In contrast, IR variants usually possess a substitution at Ambler position M69, S130, K234, or N276 (3, 4). Many of these IR variants have appeared clinically (Table 1), indicating that substitutions at these amino acids pose a significant threat to our current commonly prescribed β-lactam/BLI combinations (amoxicillin-clavulanate [Augmentin], piperacillin-tazobactam [Zosyn], and ampicillin-sulbactam [Unasyn]) (4).
TABLE 1.
IR SHV enzymes identified in clinical isolatesa
| Amino acid | β-Lactamase(s) |
|---|---|
| Met69Ile | SHV-49, SHV-52, SHV-92 |
| Ser130Gly | SHV-10 |
| Lys234Arg | SHV-56, SHV-72, SHV-73, SHV-84 |
| Asn276Asp | SHV-129 |
Adapted from reference 3.
The inactivation of a β-lactamase enzyme by a BLI (such as tazobactam, sulbactam, or clavulanic acid) involves an attack by the active site serine (in the case of SHV, S70) on the amide bond, formation of the acyl enzyme, and then subsequent rearrangement steps that lead to an imine or enamine intermediate that results in transient or long-lived inhibition of the enzyme (Fig. 1A) (4). In order to understand this mechanism of inhibition, mutagenesis studies were performed to create amino acid substitutions in the SHV-1 β-lactamase that led to resistance to inactivation by clavulanic acid (5–14). Notably, many of these IR β-lactamase variants were hypothesized to share a mechanism involving S130 that prevents or slows permanent inhibitor inactivation of the enzyme (5–14).
FIG 1.

(A) Proposed acylation mechanism of SHV-1 by clavulanic acid (4–7). (B) Proposed acylation mechanism of CTX-M-15 by avibactam (17).
Avibactam is a novel non-β-lactam BLI that was recently approved by the Food and Drug Administration, in combination with ceftazidime, for the treatment of complicated intraabdominal infections and complicated urinary tract infections (15). Avibactam is a diazabicyclooctane (DBO) inhibitor that does not have a β-lactam ring at the core of its structure. DBOs form a reversible inhibitory reaction that proceeds through rapid formation of an acyl (carbamyl) enzyme and can undergo recyclization of the ring structure to reform the active compound, albeit at a much slower rate (16). The inactivation of class A β-lactamase enzymes by avibactam is proposed to involve similar amino acid residues, compared to the β-lactam-based inhibitors (Fig. 1B) (17).
We sought to understand the efficacy of avibactam against a variety of SHV enzymes with known IR amino acid substitutions to determine if avibactam is able to inactivate β-lactamases that are resistant to inhibition by clavulanic acid. Our studies also begin to explore the mechanism of carbamylation and recyclization of avibactam against the class A β-lactamase SHV-1. In addition, we identify the role of important amino acid residues involved in avibactam inactivation of this class A β-lactamase and anticipate resistance patterns that may be observed with existing variants of SHV-1 as this drug enters the clinic. This information is essential as future DBO BLIs continue to be developed.
MATERIALS AND METHODS
Mutagenesis.
The blaSHV-1 gene was directionally subcloned into the pBC SK(−) phagemid vector. Mutagenesis studies in this construct were previously published (7, 11–14).
MIC measurements.
MIC measurements were performed by the agar dilution method according to the Clinical Laboratory and Standards Institute (CLSI) protocol (18). Briefly, bacterial cultures were grown overnight at 37°C in Mueller-Hinton (M-H) broth. The cultures were diluted, and a Steers replicator was used to stamp 10 μl of each dilution onto the Mueller-Hinton agar plates. Plates were incubated overnight at 37°C, and the MIC was read as the β-lactam concentration at which bacterial growth was no longer observed. We tested ampicillin (AMP; Sigma-Aldrich), AMP-avibactam (AVI; AstraZeneca), AMP-sulbactam (SUL; Astatech), AMP-clavulanic acid (CLAV; USP), piperacillin (PIP; Sigma-Aldrich), and PIP-tazobactam (TAZO; Chem-Impex). AVI, SUL, and CLAV were added to plates at a constant concentration of 4 mg/liter with increasing concentrations of AMP. PIP-TAZO was maintained at a constant 8:1 ratio.
Protein purification.
SHV-1 and SHV S130G β-lactamases were purified from E. coli DH10B cells containing blaSHV-1 or blaSHV S130G carried on a pBC SK(−) phagemid as previously described (19). Briefly, cells were grown overnight in super optimal broth (SOB) at 37°C containing chloramphenicol for plasmid maintenance. After harvesting, cells were pelleted by centrifugation and frozen at −20°C. After 48 h, the pellets were thawed and suspended in 50 mM Tris-HCl, pH 7.4. Lysozyme (40 μg/ml), benzonase nuclease, MgSO4, and 1 mM EDTA were added as previously described (19). The cellular debris was then removed by centrifugation, and the lysate was separated overnight using preparative isoelectric focusing (pIEF). Regions of the pIEF gel that demonstrated the ability to hydrolyze nitrocefin (NCF; Becton, Dickinson and Company) were then further purified by gel filtration (GF) column chromatography on an ÄKTA fast protein liquid chromatography (FPLC) system to >95% purity as assessed by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). The protein concentrations were calculated using an absorbance of 280 nm and the extinction coefficient of each protein (ε), which was determined using the ProtParam tool at Expasy (http://us.expasy.org/tools).
β-Lactamase kinetics.
The kinetic constants of SHV-1 and SHV S130G β-lactamases for the hydrolysis of NCF, Km and kcat, were determined according to previously published methods (7). An Agilent 8453 diode spectrophotometer (Santa Clara, CA) was used to measure the absorbance of the hydrolysis of NCF at 482 nm (ε, 17,400 M−1 cm−1) at 25°C in 10 mM phosphate-buffered saline (PBS, pH 7.4). The β-lactam concentration was held constant at 6 nM for SHV-1 and 390 nM for SHV S130G, while the concentration of NCF was varied. A higher concentration of SHV S130G β-lactamase was required for all experiments, due to the lower catalytic efficiency for NCF (7). The data were plotted, and the Km for NCF was calculated using the Michaelis-Menten equation (equation 1) and Enzfitter (Biosoft Corporation, Ferguson, MO).
| (1) |
Inhibitory enzyme kinetics analyses were also performed with the purified SHV-1 and SHV S130G proteins, and these experiments are described below.
The proposed interaction scheme of a class A β-lactamase and avibactam is shown in equation 2 (16).
| (2) |
The inhibition constant Ki app was measured in a direct competition assay between NCF and avibactam at 25°C in PBS. NCF was held constant at 100 μM, and the concentration of avibactam was varied (20). In this assay, the SHV S130G β-lactamase was used at a concentration of 390 nM, and SHV-1 was tested at a concentration of 6 nM. The Excel program was used to plot the inverse initial steady-state velocity (1/vi) versus avibactam concentration ([I]), and the value of the y-intercept divided by the slope of the line was defined as Ki app obs. This value was corrected for the use of NCF according to equation 3 to obtain the Ki app.
| (3) |
The k2/K value was determined using timed inactivation curves for SHV-1 and the S130G variant with NCF as the reporter substrate at 25°C in PBS. SHV S130G was used at a concentration of 98 nM, and SHV-1 was used at a concentration of 6 nM. A kobs value was determined for each avibactam concentration by using Origin 3.5.1 according to equation 4.
| (4) |
The kobs values were plotted against the avibactam concentrations ([I]) to obtain a straight line. Equation 5 was then used to determine the observed value of k2/K, the second-order rate constant for enzyme acylation.
| (5) |
The observed k2/K value was corrected for the use of NCF according to equation 6 to obtain the k2/K value.
| (6) |
The partition ratio or turnover number (kcat/kinact; tn) was determined for each enzyme by incubating the purified enzyme with inhibitor at various inhibitor:enzyme ratios during a 24-h time period at 25°C in PBS. Then, an aliquot was removed and the hydrolysis of NCF was measured. The avibactam:β-lactamase ratio required to inhibit NCF hydrolysis by >90% versus an uninhibited control was defined as the kcat/kinact, as previously described (20).
Electrospray Ionization (ESI)-mass spectrometry (MS).
A Waters Synapt G2-Si high-resolution quadrupole time of flight mass spectrometer (Waltham, MA) equipped with a LockSpray dual electrospray ion source was used to acquire MS data for purified protein and protein-avibactam complexes in order to determine the nature and timeline of avibactam inactivation of SHV-1 and SHV S130G. Glu-1–fibrinopeptide B was used as the lock mass, and the Synapt G2-Si apparatus was calibrated with sodium iodide, using a mass range of 50 to 2,000 m/z. This calibration resulted in an error of ±1 atomic mass units. After protein purification, with or without incubation with avibactam, 0.2% formic acid was added to quench any reactions. SHV-1 and SHV S130G proteins were incubated (at 30 μM) with a 1:1 ratio of avibactam:protein for various time points: 1 min, 5 min, 9 min, 15 min, and 24 h, at room temperature in PBS. C18 Zip-Tips were used to remove residual salt and to concentrate the samples according to the manufacturer's protocol (Millipore, Billerica, MA). Then, direct infusion at a rate of 50 μl/min was used to perform MS analysis on the eluted protein sample diluted into 50% acetonitrile and 0.2% formic acid. Data were collected for 1 min. Lock mass spectra were collected prior to each sample in a similar manner. The tune settings for each data run were as follows: capillary voltage, 3.2kV; sampling cone, 30; source offset, 30; source temperature, 100°C; desolvation temperature, 450°C; cone gas, at 50 liters/h; desolvation gas, at 600 liters/h; nebulizer bar, 6.0. Spectra were analyzed using MassLynx v4.1. Spectra were modified for lock mass deviations by applying a gain factor and then deconvoluted using the MaxEnt1 program.
Molecular modeling.
The crystal structures of SHV-1 (PDB ID 1SHV) and SHV S130G (PDB ID 1TDL) β-lactamases were used to construct and validate Michaelis-Menten and acyl enzyme complexes of SHV-1 with avibactam using Discovery Studio (DS) 3.1 molecular modeling software (Accelrys Inc., San Diego, CA) as previously described (11, 20). Avibactam was first constructed using the Fragment Builder tools and minimized using a Standard Dynamics Cascade protocol of DS 3.1. Avibactam was then automatically docked into the active sites of SHV-1 and SHV S130G using the CDOCKER module of DS 3.1. This protocol uses a CHARMm-based molecular dynamics scheme to dock ligands into a receptor-binding site. The best conformations were automatically aligned into polar and apolar active site hot spots, and the best-scoring poses were reported; hydrogen atoms were not maintained.
To further optimize the docked poses (by adding hydrogen atoms and preventing steric clashes between the receptor and ligand), a CHARMm minimization step was used. In this step, the Smart Minimization algorithm was utilized (1,000 steps of steepest descent with a root mean squared (RMS) gradient tolerance of 3 Å, followed by conjugate gradient minimization with an RMS deviation (RMSD) minimization gradient of 0.001 Å. For the last minimization of the avibactam conformations into the active sites of SHV-1 and SHV S130G β-lactamases, an RMSD cutoff of 1 Å was chosen.
The resulting conformations of the SHV-1–avibactam and SHV S130G-avibactam complexes were analyzed, and the most favorable positioning of avibactam was chosen. Then, the complexes between the enzymes and inhibitor were created (11, 20). To check the stability of the complexes, an 8-ps Molecular Dynamics simulation (MDS) was conducted for the SHV-1–avibactam and S130G-avibactam Michaelis-Menten and acyl enzyme complexes as previously described and validated (21). A temperature of 300 K and a constant pressure were maintained during the heating/cooling, equilibration, and production stages of MDS. The long-range electrostatics were treated with particle mesh Ewald and explicit solvation with the periodic boundary condition. These were run without any constraints.
The X-ray crystallography coordinates of CTX-M-15-avibactam were obtained (PDB ID 4HBT). DS 3.1 was used to compare the X-ray crystallography structure with our acyl enzyme model of SHV-1–avibactam.
RESULTS AND DISCUSSION
Microbiological activity of avibactam in combination with ampicillin against IR SHV variants expressed in E. coli DH10B.
The variants at Ambler positions M69, S130, K234, R244, and N276 are known to confer resistance to clavulanic acid inactivation by different mechanisms (4). The limitations of clavulanic acid in inhibiting these enzymes were previously highlighted (7, 11, 12, 14, 22). Table 2 summarizes the MICs that were determined for ampicillin and ampicillin-avibactam for this collection of IR SHV variants. Ceftazidime-avibactam was not reported in this table, as all variants were susceptible to ceftazidime (MIC, ≤8 mg/liter).
TABLE 2.
MICs for E. coli strains containing single IR SHV β-lactamases in a pBC SK(−) plasmida
| E. coli DH10B | MIC (mg/liter) |
|||||
|---|---|---|---|---|---|---|
| AMP | AMP-AVI | AMP-SUL | AMP-CLAV | PIP | PIP-TAZO | |
| Emptyb | 0.5 | 0.25 | 0.5 | 0.5 | 2 | 1 |
| SHV-1 | >16,384 | 1 | >16,384 | 16 | >1,024 | 256 |
| M69I | 8,192 | 4 | 8,192 | 128 | 1,024 | 128 |
| M69L | 16,384 | 8 | 16,384 | 64 | 1,024 | 256 |
| M69V | 16,384 | 4 | 16,384 | 128 | 1,024 | 256 |
| S130A | 2 | 1 | 1 | 2 | 32 | 32 |
| S130G | 512 | 256 | 256 | 256 | 1,024 | 128 |
| S130T | 64 | 8 | 64 | 0.5 | 256 | 128 |
| K234A | 16 | 8 | 8 | 8 | 8 | 8 |
| K234R | 16,384 | 256 | 4,096 | 512 | >1,024 | 16 |
| R244A | 256 | 8 | 128 | 32 | 64 | 32 |
| R244K | 4,096 | 2 | 2,048 | 32 | 256 | 64 |
| R244S | 1,024 | 32 | 256 | 32 | 128 | 32 |
| N276A | 512 | 0.5 | 256 | 1 | 128 | 8 |
| N276D | 16,384 | 2 | 16,384 | 32 | 1,024 | 128 |
| N276E | 4,096 | 1 | 2,048 | 4 | 512 | 32 |
E. coli DH10B was purchased from Agilent, SHV-1 was obtained from Helfand et al. (7), amino acid position 69 variants came from Totir et al. (5), position 130 variants came from Helfand et al. (10), position 234 variants were from Winkler et al. (11), position 244 variants also came from Thomson et al. (14), and position 276 variants were obtained from Drawz et al. (12). AVI, SUL, and CLAV concentrations were held constant at 4 mg/liter, and TAZO was kept at an 8:1 ratio of PIP:TAZO. Values shown in bold are for the isolates that showed the highest AMP-AVI MIC.
Empty strain contains pBC SK(−) plasmid with no bla gene.
As shown in Table 2, avibactam was more effective at restoring the potency of ampicillin against the IR SHV variants than other β-lactamase inhibitors (sulbactam, tazobactam, and clavulanate). With ampicillin alone, all isogenic strains except E. coli producing the S130A and K234A variants were resistant to ampicillin (MIC range, 64 to 16,384 mg/liter). When avibactam was combined with ampicillin, 3 of 14 variants possessed ampicillin-avibactam MICs of ≥32 mg/liter. On the other hand, 12 of 14 and 9 of 14 variants were resistant to ampicillin-sulbactam and ampicillin-clavulanic acid, respectively. For piperacillin, 11 of 14 variants demonstrated a MIC of ≥128 mg/liter for piperacillin; the addition of tazobactam reduced that number to 6 of 14 variant strains. We also observed that SHV-1 in E. coli DH10B manifested a higher-than-expected MIC for the sulfone inhibitors, which may be a result of the increased β-lactamase expression level due to the high copy number of the pBC SK(−) phagemid and its promoter (22).
Interestingly, three IR SHV variants in E. coli DH10B displayed elevated MICs to both ampicillin-avibactam and other BLI–β-lactam combinations, S130G, K234R, and R244S. For the S130G variant, the ampicillin MIC was only lowered by one dilution after the addition of avibactam (from 512 mg/liter to 256 mg/liter) (Table 2). E. coli DH10B expressing the SHV K234R variant also showed an elevated ampicillin-avibactam MIC (MIC of 256 mg/liter for ampicillin-avibactam, versus MIC of 16,384 mg/liter for ampicillin alone). The R244S variant expressed in E. coli DH10B had a MIC of 1,024 mg/liter for ampicillin alone and 32 mg/liter for ampicillin-avibactam. As E. coli DH10B containing SHV S130G demonstrated the least reduction in the ampicillin MIC after the addition of avibactam, we decided to further explore this IR SHV to begin to understand the mechanistic basis for why it resists inhibition by avibactam in comparison to the wild-type SHV-1.
Avibactam inhibition of SHV-1 and S130G β-lactamases.
In order to better understand the differences in inactivation of each of these enzymes, the inhibition of purified SHV-1 and SHV S130G by avibactam was studied. Our analysis showed that the k2/K value of the S130G variant of SHV-1 by avibactam was less than that of wild-type SHV-1 (1.3 M−1 s−1 versus 60,300 M−1 s−1, respectively) (Table 3). Additionally, we found that an approximately 1,700-fold-higher avibactam concentration was required to achieve full SHV S130G inhibition compared to SHV-1 inhibition (Fig. 2). We interpreted this to mean that the loss of the hydroxyl group at position 130 as a result of the Gly substitution impaired either the initial productive contact between the β-lactamase and avibactam or the formation of the acyl enzyme bond between S70 and avibactam. Despite these differences in inactivation rates, the partition ratio (kcat/kinact) for both enzymes was identical at 1 in a 24-hour time period (Table 3), indicating that avibactam inactivates both β-lactamases at a stoichiometric ratio during a long time period and is not hydrolyzed.
TABLE 3.
Inhibitory steady-state kinetic parameters for SHV-1 and the SHV-1 S130G variant with AVI
| Parameter | SHV-1 | SHV-1 S130G |
|---|---|---|
| NCF Km (μM) | 12.5 ± 2.5 | 3,107 ± 2393 |
| NCF kcat/Km (μM−1 s−1) | 15.3 | 0.02 |
| AVI k2/K (M−1 s−1) | 60,300 | 1.3 |
| AVI Ki app (μM) | 0.022 ± 0.002 | 10,274 ± 1,000 |
| AVI 24-h partition ratio | 1 | 1 |
FIG 2.
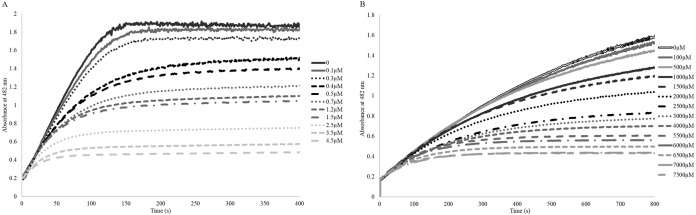
(A) Timed inactivation of SHV-1 by increasing concentrations of avibactam. (B) Timed inactivation of SHV S130G by increasing concentrations of avibactam. The SHV S130G variant takes longer to plateau because of the lower NCF catalytic efficiency of this enzyme. An approximately 1,700-fold-higher concentration of avibactam was required to inhibit the SHV S130G variant compared to SHV-1.
ESI-MS of SHV-1 and S130G β-lactamases.
The behaviors of SHV-1 and S130G β-lactamases incubated with a 1:1 ratio of avibactam:β-lactamase during a time period between 1 min and 24 h were followed using MS (Fig. 3). As anticipated from our kinetic analyses, avibactam readily carbamylates the SHV-1 β-lactamase. In contrast, we observed that the formation of the acyl enzyme species between SHV S130G and avibactam is much slower. As we examined the differences from 1 min to 15 min of incubation at a 1:1 ratio of inhibitor to enzyme, SHV S130G proceeded from a minimally carbamylated enzyme to the full acyl enzyme intermediate. In contrast, the SHV-1 β-lactamase was fully carbamylated by avibactam during the entire observation period. Importantly, we did not find evidence that either of these β-lactamases hydrolyzed avibactam under these conditions, as the carbamyl-enzyme complex between both β-lactamases and avibactam continued to be observed at the 24-h time point. Additionally, on-enzyme fragmentation of avibactam was not observed for either β-lactamase during this time course, as peaks of different masses were not identified. We cautiously interpret this to mean that avibactam does not undergo significant chemical rearrangements during the reaction and that a highly stable adduct is formed between SHV-1 and SHV S130G and avibactam.
FIG 3.
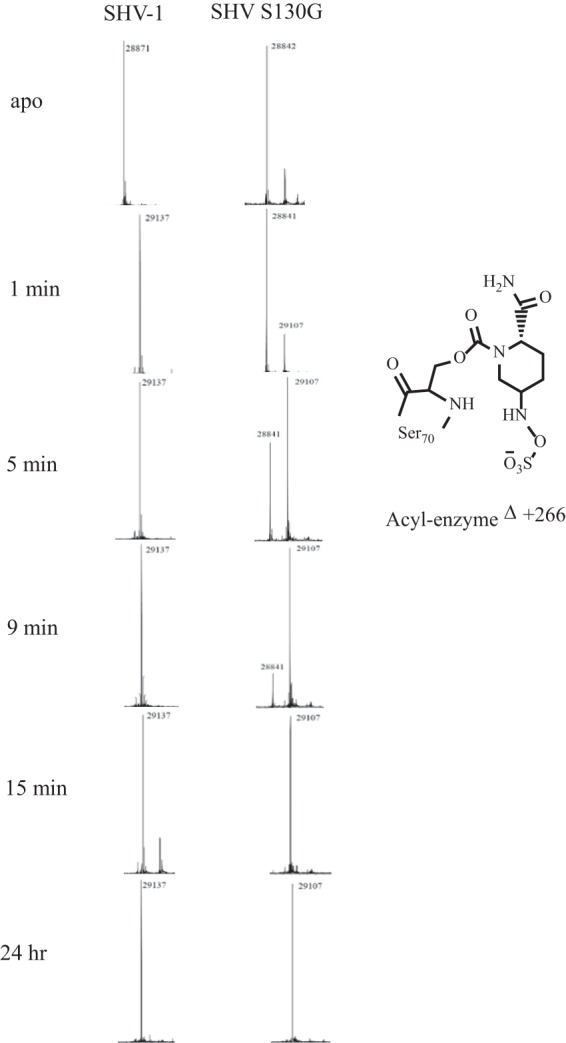
Mass spectrometry of SHV-1 and SHV S130G at various time points, ranging from 1 min to after a 24-h incubation of avibactam-enzyme at a 1:1 ratio.
The proposed avibactam acylation mechanism in CTX-M-15 and implications for avibactam acylation in the SHV-1 β-lactamase.
To begin to correlate the biochemical differences in binding and acylation between SHV-1 and the S130G variant to a catalytic mechanism, we considered the catalytic mechanism of class A β-lactamases and the proposed acylation mechanism of CTX-M-15 for avibactam.
Acylation of class A β-lactamases by β-lactams and BLIs is believed to follow two different pathways. Either E166 and a catalytic water molecule or K73 are believed to initiate catalysis (4, 17, 23–25). Evidence exists in the form of X-ray crystallography structures, thermodynamic calculations, nuclear magnetic spectroscopy, mutagenesis and kinetic measurements, and pH/pKa titration studies that supports both E166 and K73 acting as the initial general base for the acylation of class A β-lactamases (17, 25–31). Quantum mechanical calculations indicate that E166 is generally the most thermodynamically favored amino acid for initiation of acylation with the “K73 pathway” as an alternative (4, 24, 32). In addition to these important amino acids, studies also support the role of S130 in the acylation mechanism as a general acid/proton donor to the nitrogen amide in order to collapse the tetrahedral transition state (31). Despite significant effort, a clear consensus of which pathway predominates has not been reached. Moreover, the protonation states of K73 and E166 may change in the transition from apo-enzyme, to Michaelis-Menten complex, to acyl enzyme, as well as with different substrates or inhibitors in the active site of the enzyme (17, 25, 27–29). These changing protonation states may affect which acylation mechanism predominates under a given set of conditions.
Lahiri et al. hypothesized based on the X-ray crystallographic structure of the acyl enzyme of CTX-M-15–avibactam (PDB ID 4HBT) that a proton shuttle involving E166, a strategically placed water molecule, S70, avibactam, S130, K73, and back to E166 occurs (Fig. 1B and 4) (17). This mechanism was advanced based upon a high-resolution structure (<1 Å) showing unprotonated E166 in the apo-CTX-M-15 structure and protonation of this residue after binding of avibactam (17).
FIG 4.
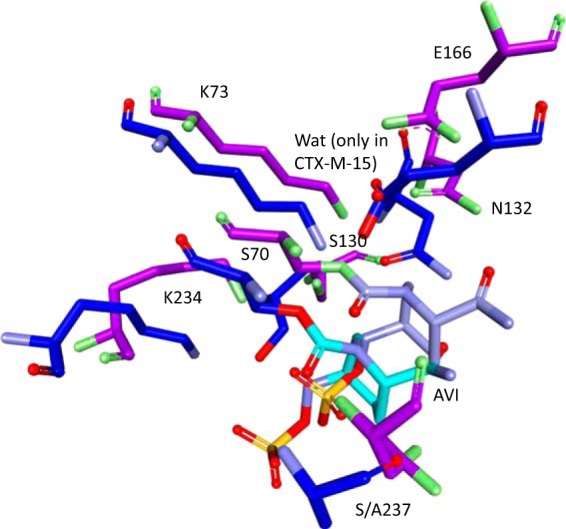
Overlay of the CTX-M-15–avibactam crystal structure (PDB ID 4HBT; purple) with the acyl enzyme model of SHV-1–avibactam (blue), showing significant movement of many of the important active site residues and an absence of water (Wat) molecules in the SHV-1–avibactam model.
We performed molecular modeling to examine the Michaelis-Menten and acyl enzyme complexes of both SHV-1 and SHV S130G with avibactam in order to gain deeper insight into the mechanism of avibactam inhibition and resistance.
In the SHV-1:avibactam Michaelis-Menten complex, avibactam is positioned within hydrogen-bonding distance of S70, K73, S130, E166, A237, and R244 and is poised within the oxyanion hole in a favorable position for carbamylation. In this model, the catalytic water molecule is absent; we advance that the water molecule may be displaced by the carboxamide of avibactam. Additionally, K73 is positioned more than 5 Å from S70 in all conformations of the simulation (Fig. 5A). In contrast, S130 is within hydrogen-bonding distance of S70. As the K73 and E166 acylation processes are not favored in this model (Fig. 6A), we postulate that S130 may serve an unexpected role as the general base for deprotonation of S70 during acylation of the SHV β-lactamase by avibactam (Fig. 6B). The observations of Lahiri et al. support this hypothesis, as they noted that due to the position of the carbamate bond of avibactam in the CTX-M-15–avibactam structure, E166, K73, or S130 may serve as the conjugate general base (17). Based on our model of SHV-1:avibactam, we advance that a proton shuttle occurs starting with K234, through S130, and then S70 (Fig. 6B). S130 would also further play a role in donating a proton to the nitrogen amide for collapse of the tetrahedral transition state in either potential acylation mechanism. Our other IR variants also support a different mechanism of inhibition of SHV-1 by avibactam compared to sulbactam, tazobactam, and clavulanic acid, as these variants (M69I/L/V, N276D) did not show elevated MICs to ampicillin-avibactam but the K234R and R244S variants did.
FIG 5.

(A) Michaelis-Menten complex model of SHV-1–avibactam. (B) SHV-1–avibactam acyl enzyme model. (C) Michaelis-Menten complex model of SHV S130G-avibactam. (D) SHV S130G-avibactam acyl enzyme model.
FIG 6.

Proposed acylation mechanism of avibactam for the SHV β-lactamase (A and B) and the S130G variant enzyme (C and D).
However, we propose that the flexibility of the enzyme active site may allow water to enter, which would permit acylation to proceed through a traditional pathway involving E166 (Fig. 6A). The movement of bulk water transiently into the enzyme active site was previously postulated to occur in the IR SHV R244S variant (14). Additionally, movement of K73 may also occur, allowing it to serve as the general base for acylation.
We must also state that that there are limitations to molecular modeling. Here, the time frame for MDS was 8 ps, and only one conformation (the most favorable) of avibactam was chosen for MDS. It is possible that with a longer MDS time frame or with a different initial avibactam conformation, the active site water molecules would be observed. This would allow E166 to serve as the general base or, alternatively, that K73 could move to be within hydrogen-binding distance of S70 to serve as the general base. From our previous molecular modeling and compound docking, we have observed active site water molecules in enzyme-inhibitor complexes after similar modeling parameters were used; avibactam appears to be the exception (12, 33). It is possible that competing acylation mechanisms may also exist.
Our MDS analysis of the SHV-1–avibactam acyl enzyme complex further revealed that the catalytic water molecule that is critical for initiating acylation is again absent. Finally, we observed that upon acylation in SHV-1, avibactam was shifted such that S130 was within 3.5 Å of the sulfate amide proton (Fig. 5B). Therefore, S130 may also play a role in the recyclization of avibactam from SHV-1, as proposed for CTX-M-15 (17). Thus, S130 may serve as the hydrogen acceptor to initiate recyclization. Furthermore, given that the K234R variant also possesses an elevated ampicillin-avibactam MIC, K234 may play a role in proton shuttling to S130, allowing recyclization to proceed instead of through K73, as proposed by Lahiri et al. (17). Further experimentation will be required to differentiate these mechanistic details.
Why is acylation impaired in the S130G variant of SHV?
The model of the SHV S130G:avibactam Michaelis-Menten complex disclosed that avibactam was uniquely positioned in the active site compared to the SHV-1:avibactam Michaelis-Menten complex, due to the loss of the S130 hydroxyl side chain (Fig. 5C). As a result, there were fewer potential hydrogen-bonding interactions between avibactam and the S130G variant enzyme in this model. However, we did observe that hydrogen-bonding interactions were possible between K234 and avibactam and between A237 and avibactam. As a Gly at position 130 does not have the hydroxyl group of Ser, this residue is unable to be involved in the acylation of the enzyme by avibactam as described above for SHV-1.
Previous X-ray crystallography of SHV S130G identified a water molecule in the active site of the enzyme that compensated for loss of this hydroxyl group (8). In our model of SHV S130G-avibactam, we did not observe this water molecule during our MDS of the S130G-avibactam representations (Fig. 5C). Two water molecules must enter the active site to allow acylation to proceed via a mechanism involving E166, as shown in Fig. 6C. Alternatively, K73 and S70 are within hydrogen-binding distance in the S130G-avibactam molecular model (2.6 Å), which may permit K73 to initiate acylation with only one water molecule required to enter the active site to compensate for the missing hydroxyl group of S130 (Fig. 6D). Again, we are limited by a lack of high-resolution crystallographic structures of SHV S130G revealing protonation states of K73 and E166. The same experiments required to identify the acylation (or competing acylation) mechanisms for SHV-1–avibactam are also needed to determine the major mechanism for SHV S130G. Overall, binding and acylation are less favorable in the S130G variant of SHV, as the number of potential hydrogen-bonding interactions between avibactam and the enzyme are decreased, and the hydroxyl group of S130 is important in any potential acylation mechanisms of SHV by avibactam.
The second catalytic water molecule was also absent in the SHV S130G acyl enzyme complex, again due to steric interference from the carboxamide of avibactam (Fig. 5D). An interaction with the sulfate amide proton of avibactam by the hydroxyl group of S130 is also no longer possible, as the Gly is now at position 130; thus, recyclization is less favored.
Conclusions.
The S130G substitution in the SHV class A β-lactamase, an amino acid change that is found in an IR SHV variant, leads to resistance to inhibition by avibactam, a novel non-β-lactam BLI. The mechanism of resistance seems to be slow onset of acylation of the S130G enzyme by avibactam. There are several possible amino acids that may initiate acylation of SHV and SHV S130G by avibactam according to our molecular modeling (K73, E166, and S130). As the protonation states of these residues are subject to debate, high-resolution X-ray crystallographic structures and more sophisticated molecular models and MDS are required to determine which acylation mechanism or mechanisms are favored.
Since ceftazidime is effective against bacteria containing non-ESBL SHV enzymes, the emergence of clinically significant avibactam-resistant isolates containing known IR substitutions alone in the clinic is highly unlikely. Nevertheless, we maintain that β-lactamases with a combination of ESBL and IR substitutions of the SHV type may evolve that exhibit resistance to ceftazidime-avibactam combination therapy, as was observed in some CTX-M-9 class A β-lactamase variants. In these variant enzymes, an ESBL substitution restored hydrolysis of cephalosporins to a sufficient quantity to allow an enzyme with the S130G substitution to display resistance to both molecules (34). Additionally, as the S130G substitution seems to provide resistance to many different classes of inhibitors and is present in SHV-10, which has been found clinically (Table 1), it is important to consider this substitution in addition to the K234R change, which seems to be intimately linked to the function of S130 and also a common clinical SHV variant, when developing future BLIs.
ACKNOWLEDGMENTS
We thank AstraZeneca Pharmaceuticals for providing the avibactam powder.
This work was funded by a grant from AstraZeneca Pharmaceuticals to K.P.W. and R.A.B. M.L.W. has been supported by Medical Scientist Training Program Training Grant, Case Western Reserve University T32 GM07250. K.P.W. is supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs and a Veterans Affairs Career Development Award. R.A.B. is supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Department of Veterans Affairs Merit Review Program (1I01BX001974), the Veterans Integrated Service Network 10 Geriatric Research, Education, and Clinical Center (VISN 10 GRECC), and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award Numbers R01 AI100560 and R01 AI063517.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Cho H, Uehara T, Bernhardt TG. 2014. β-Lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambler RP, Coulson AF, Frere JM, Ghuysen JM, Joris B, Forsman M, Levesque RC, Tiraby G, Waley SG. 1991. A standard numbering scheme for the class A β-lactamases. Biochem J 276:269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bush K, Palzkill T, Jacoby G. 2015. β-Lactamase classification and amino acid sequences for TEM, SHV and OXA extended-spectrum and inhibitor resistant enzymes. Lahey Clinic, Burlington, MA: http://www.lahey.org/Studies/ Accessed 1 July 2014. [Google Scholar]
- 4.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Totir MA, Padayatti PS, Helfand MS, Carey MP, Bonomo RA, Carey PR, van den Akker F. 2006. Effect of the inhibitor-resistant M69V substitution on the structures and populations of trans-enamine β-lactamase intermediates. Biochemistry 45:11895–11904. doi: 10.1021/bi060990m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sulton D, Pagan-Rodriguez D, Zhou X, Liu Y, Hujer AM, Bethel CR, Helfand MS, Thomson JM, Anderson VE, Buynak JD, Ng LM, Bonomo RA. 2005. Clavulanic acid inactivation of SHV-1 and the inhibitor-resistant S130G SHV-1 β-lactamase. Insights into the mechanism of inhibition. J Biol Chem 280:35528–35536. doi: 10.1074/jbc.M501251200. [DOI] [PubMed] [Google Scholar]
- 7.Helfand MS, Bethel CR, Hujer AM, Hujer KM, Anderson VE, Bonomo RA. 2003. Understanding resistance to β-lactams and β-lactamase inhibitors in the SHV β-lactamase: lessons from the mutagenesis of SER-130. J Biol Chem 278:52724–52729. doi: 10.1074/jbc.M306059200. [DOI] [PubMed] [Google Scholar]
- 8.Sun T, Bethel CR, Bonomo RA, Knox JR. 2004. Inhibitor-resistant class A β-lactamases: consequences of the Ser130-to-Gly mutation seen in Apo and tazobactam structures of the SHV-1 variant. Biochemistry 43:14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 9.Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, Shoichet BK. 2005. Structural consequences of the inhibitor-resistant Ser130Gly substitution in TEM β-lactamase. Biochemistry 44:9330–9338. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helfand MS, Taracila MA, Totir MA, Bonomo RA, Buynak JD, van den Akker F, Carey PR. 2007. Raman crystallographic studies of the intermediates formed by Ser130Gly SHV, a β-lactamase that confers resistance to clinical inhibitors. Biochemistry 46:8689–8699. doi: 10.1021/bi700581q. [DOI] [PubMed] [Google Scholar]
- 11.Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C, Caselli E, Prati F, van den Akker F, Bonomo RA. 2013. Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J Med Chem 56:1084–1097. doi: 10.1021/jm301490d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drawz SM, Bethel CR, Hujer KM, Hurless KN, Distler AM, Caselli E, Prati F, Bonomo RA. 2009. The role of a second-shell residue in modifying substrate and inhibitor interactions in the SHV β-lactamase: a study of Ambler position Asn276. Biochemistry 48:4557–4566. doi: 10.1021/bi9003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helfand MS, Hujer AM, Sonnichsen FD, Bonomo RA. 2002. Unexpected advanced generation cephalosporinase activity of the M69F variant of SHV β-lactamase. J Biol Chem 277:47719–47723. doi: 10.1074/jbc.M207271200. [DOI] [PubMed] [Google Scholar]
- 14.Thomson JM, Distler AM, Prati F, Bonomo RA. 2006. Probing active site chemistry in SHV β-lactamase variants at Ambler position 244. Understanding unique properties of inhibitor resistance. J Biol Chem 281:26734–26744. doi: 10.1074/jbc.M603222200. [DOI] [PubMed] [Google Scholar]
- 15.U.S. Food and Drug Administration. 2015. FDA approves new antibacterial drug Avycaz. U.S. FDA, Silver Spring, MD. [Google Scholar]
- 16.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier JD. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob Agents Chemother 57:2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing; twenty-fourth informational supplement. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 19.Lin S, Thomas M, Shlaes DM, Rudin SD, Knox JR, Anderson V, Bonomo RA. 1998. Kinetic analysis of an inhibitor-resistant variant of the OHIO-1 β-lactamase, an SHV-family class A enzyme. Biochem J 333:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papp-Wallace KM, Winkler ML, Gatta JA, Taracila MA, Chilakala S, Xu Y, Johnson JK, Bonomo RA. 2014. Reclaiming the efficacy of β-lactam–β-lactamase inhibitor combinations: avibactam restores the susceptibility of CMY-2-producing Escherichia coli to ceftazidime. Antimicrob Agents Chemother 58:4290–4297. doi: 10.1128/AAC.02625-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, Bonomo RA. 2012. Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: exploring the roles of Arg220 and Glu276. Antimicrob Agents Chemother 56:4428–4438. doi: 10.1128/AAC.05769-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomson JM, Distler AM, Bonomo RA. 2007. Overcoming resistance to β-lactamase inhibitors: comparing sulbactam to novel inhibitors against clavulanate resistant SHV enzymes with substitutions at Ambler position 244. Biochemistry 46:11361–11368. doi: 10.1021/bi700792a. [DOI] [PubMed] [Google Scholar]
- 23.Damblon C, Raquet X, Lian LY, Lamotte-Brasseur J, Fonze E, Charlier P, Roberts GC, Frere JM. 1996. The catalytic mechanism of β-lactamases: NMR titration of an active-site lysine residue of the TEM-1 enzyme. Proc Natl Acad Sci U S A 93:1747–1752. doi: 10.1073/pnas.93.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. 2005. Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc 127:15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 25.Golemi-Kotra D, Meroueh SO, Kim C, Vakulenko SB, Bulychev A, Stemmler AJ, Stemmler TL, Mobashery S. 2004. The importance of a critical protonation state and the fate of the catalytic steps in class A β-lactamases and penicillin-binding proteins. J Biol Chem 279:34665–34673. doi: 10.1074/jbc.M313143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tremblay LW, Xu H, Blanchard JS. 2010. Structures of the Michaelis complex (1.2 Å) and the covalent acyl intermediate (2.0 Å) of cefamandole bound in the active sites of the Mycobacterium tuberculosis β-lactamase K73A and E166A mutants. Biochemistry 49:9685–9687. doi: 10.1021/bi1015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. 1992. Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 Å resolution. Nature 359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 28.Rodkey EA, Drawz SM, Sampson JM, Bethel CR, Bonomo RA, van den Akker F. 2012. Crystal structure of a preacylation complex of the β-lactamase inhibitor sulbactam bound to a sulfenamide bond-containing thiol-β-lactamase. J Am Chem Soc 134:16798–16804. doi: 10.1021/ja3073676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minasov G, Wang X, Shoichet BK. 2002. An ultrahigh resolution structure of TEM-1 β-lactamase suggests a role for Glu166 as the general base in acylation. J Am Chem Soc 124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 30.Padayatti PS, Helfand MS, Totir MA, Carey MP, Carey PR, Bonomo RA, van den Akker F. 2005. High resolution crystal structures of the trans-enamine intermediates formed by sulbactam and clavulanic acid and E166A SHV-1 β-lactamase. J Biol Chem 280:34900–34907. doi: 10.1074/jbc.M505333200. [DOI] [PubMed] [Google Scholar]
- 31.Diaz N, Sordo TL, Merz KM Jr, Suarez D. 2003. Insights into the acylation mechanism of class A β-lactamases from molecular dynamics simulations of the TEM-1 enzyme complexed with benzylpenicillin. J Am Chem Soc 125:672–684. doi: 10.1021/ja027704o. [DOI] [PubMed] [Google Scholar]
- 32.Sgrignani J, Grazioso G, De Amici M, Colombo G. 2014. Inactivation of TEM-1 by avibactam (NXL-104): insights from quantum mechanics/molecular mechanics metadynamics simulations. Biochemistry 53:5174–5185. doi: 10.1021/bi500589x. [DOI] [PubMed] [Google Scholar]
- 33.Drawz SM, Taracila M, Caselli E, Prati F, Bonomo RA. 2011. Exploring sequence requirements for C3/C4 carboxylate recognition in the Pseudomonas aeruginosa cephalosporinase: insights into plasticity of the AmpC β-lactamase. Protein Sci 20:941–958. doi: 10.1002/pro.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ripoll A, Baquero F, Novais A, Rodriguez-Dominguez MJ, Turrientes MC, Canton R, Galan JC. 2011. In vitro selection of variants resistant to β-lactams plus β-lactamase inhibitors in CTX-M β-lactamases: predicting the in vivo scenario? Antimicrob Agents Chemother 55:4530–4536. doi: 10.1128/AAC.00178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]