

The amyloid hypothesis of Alzheimer's disease (AD) pathogenesis maintains that the key event is the production of specific C-terminal amyloid-β (Aβ) peptides following the abnormal proteolytic cleavage of the amyloid precursor protein (Vassar and Citron, 2000). Aβ peptides self-aggregate and are found in multiple forms from small soluble monomers and oligomers to much larger fibrils and plaques. The soluble Aβ oligomers that can diffuse throughout the brain are regarded as potent neurotoxins (Lambert et al., 1998). They demonstrate disease-specific accumulation in human brain and cerebrospinal fluid (CSF) (Georganopoulou et al., 2005). The close correlation between the concentrations of soluble Aβ oligomers and cognitive decline (Naslund et al., 2000; Hung et al., 2008) is evidence that the soluble Aβ oligomers are driving the pathogenesis of AD. The progressive dementia associated with AD is closely correlated with the loss of synapses, as measured by the loss of synaptic proteins including synaptophysin, synapsin-1 and synaptobrevin (DeKosky and Scheff, 1990; Terry et al., 1991; Heinonen et al., 1995; Sze et al., 1997). The addition of nanomolar concentrations of soluble Aβ oligomers (similar concentrations to those found in the CSF of AD patients (Lue et al., 1999; McLean et al., 1999; Bibl et al., 2006; Mc Donald et al., 2010)) leads to synapse degeneration in cultured neurons (Bate et al., 2010) and affects memory formation in vivo (Walsh et al., 2002).
Neurodegeneration in AD is preceded by the intraneuronal accumulation of Aβ (Takahashi et al., 2002). The chronic nature of AD suggests that it is the slow, progressive accumulation of Aβ that triggers neurodegeneration and hence the clinical symptoms. As neural regeneration cannot occur in the presence of toxic concentrations of Aβ, the first step in regeneration must be the removal of toxic Aβ. The concentration of Aβ in neurons is dependent upon both the rate of Aβ production and also the rate of Aβ degradation. While there are numerous studies examining Aβ production, the capacity of neurons to degrade Aβ once it has been formed has received little attention. A recent publication from my laboratory explored the hypothesis that the slow clearance of Aβ from neurons is a significant factor in the accumulation of Aβ within the brain that leads to synapse damage and dementia in AD (Simmons et al., 2014).
Soluble Aβ oligomers are found predominantly within detergent-resistant, cholesterol-dense membrane micro-domains which are referred to as lipid rafts (Lee et al., 1998; Kawarabayashi et al., 2004; Williamson et al., 2008). The targeting of Aβ to rafts may have important biological consequences, as many raft-associated proteins traffic through cells via “recycling pathways” which avoid the lysosomes (Nichols et al., 2001) (organelles responsible for protein degradation). In cultured neurons, Aβ oligomers behaved like a classic “raft protein”; they were not found within lysosomes and consequently were cleared slowly from neurons, with a half-life of greater than 5 days. Thus the targeting of Aβ oligomers to rafts may contribute to their gradual accumulation within neurons and subsequently their toxic effects. We postulated that the rate of Aβ degradation within neurons could be increased following pharmacological manipulation.
Cholesterol has multiple effects upon cell membranes and is instrumental in regulating membrane fluidity, protein trafficking, endocytosis and exocytosis (Nichols et al., 2001). Critically, the concentrations of cholesterol within cell membranes affect the formation and function of lipid rafts (Pike, 2004; Rajendran and Simons, 2005; Hancock, 2006). Cholesterol synthesis inhibitors that caused a mild reduction in cholesterol concentrations altered the fate of Aβ oligomers in cultured neurons (Simmons et al., 2014). Although cholesterol depletion did not affect the binding of Aβ oligomers to neurons, significantly less Aβ was found within lipid rafts. Greater amounts of Aβ were found within lysosomes and in cholesterol-depleted neurons the half-life of Aβ was reduced to less than 24 hours. Such results indicate that the pharmacological manipulation of neurons can significantly increase the clearance of Aβ.
As cholesterol is required for normal cell function cholesterol synthesis inhibitors can have multiple unwanted effects. Treatment with inhibitors of cytosolic phospholipases A2 (cPLA2), an enzyme which affects intracellular trafficking (de Figueiredo et al., 1998; Grimmer et al., 2005), or platelet-activating factor (PAF) antagonists had the same effects on Aβ metabolism in neurons as cholesterol depletion. These drugs reduced the activation of cholesterol ester hydrolases (CEH), enzymes that constitute a part of the cholesterol cycle which regulates the concentrations of cholesterol in cell membranes (Chang et al., 2006). CEHs release free cholesterol from the stores of biologically inert cholesterol esters. Aβ oligomers activate cPLA2 (Bate and Williams, 2011; Desbene et al., 2012) resulting in activation of CEH and increased cholesterol concentrations within specific membranes, without affecting total cellular cholesterol concentrations (Simmons et al., 2014). Critically, inhibitors of CEH had a similar effect to cholesterol synthesis inhibitors on the trafficking of Aβ in neurons. They reduced concentrations of Aβ in lipid rafts, increased Aβ in lysosomes, and reduced the half-life of Aβ to less than 24 hours.
Physiologically relevant (low nanomolar) concentrations of Aβ oligomers, similar to those found in the cerebrospinal fluid of AD patients (Lue et al., 1999; McLean et al., 1999; Bibl et al., 2006; Mc Donald et al., 2010), were incubated with cultured mouse cortical neurons. This concentration of Aβ oligomers did not affect neuronal survival but did cause significant synapse degeneration, as measured by the loss of synaptic proteins including synaptophysin and cysteine string protein. It is worth noting that in neuronal cultures synapse damage occurs at concentrations approximately 100-fold less than the concentrations required to cause neuronal death. Thus, these conditions mimic the early stages of AD in which synapse damage leads to clinical symptoms in the absence of any gross loss of neurons. The concentration of Aβ in these experiments was critical for two reasons. Firstly, the trafficking of Aβ may be concentration dependent, as supra-physiological concentrations of Aβ may induce abnormal trafficking pathways. Secondly, it is important to understand the trafficking of Aβ in normal rather than dying neurons.
In control neuronal cultures, synaptic density was reduced after a few hours and remained suppressed throughout the 5 days of the experiment. In contrast, when neuronal cultures treated with Aβ oligomers for 24 hours (resulting in the loss of synapses) were subsequently treated with 200 nM squalestatin, a cholesterol synthesis inhibitor, synapse density was significantly higher after 5 days (Figure 1). These results demonstrate a close correlation between the removal of Aβ from neurons and synapse regeneration.
Figure 1.
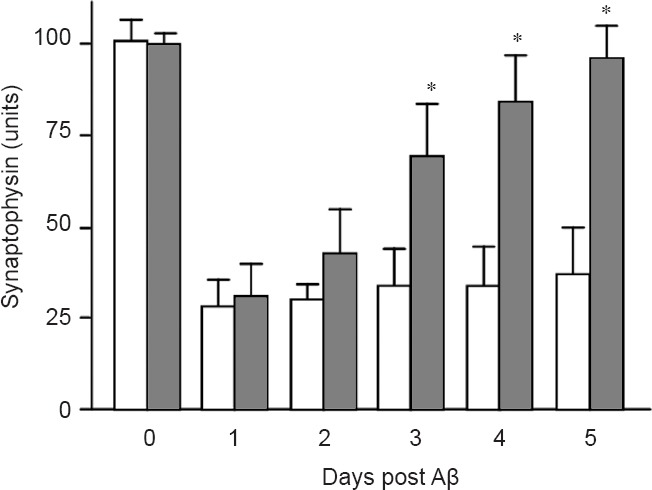
Synapse density is restored in treated neurons.
The amounts of synaptophysin (indicative of synapse density) in mouse cortical neurons treated with control medium (□) or with 200 nM squalestatin, a cholesterol synthesis inhibitor (■) 24 hours after the addition of 10 nM Aβ. Values are the mean ± SD from triplicate experiments (n = 6). *Synaptophysin significantly higher than in control neurons.
The gradual accumulation of Aβ within the brain leads to synapse degeneration and the progressive dementia associated with AD. The observation that Aβ oligomers are stable in neuronal lipid rafts and traffic via a recycling pathway that avoids the lysosomes may be an important factor in AD pathogenesis as it suggests that Aβ is cleared slowly from neurons. The demonstration that pharmacological manipulation of neurons can dramatically increase the rate of Aβ degradation and subsequently facilitate the regeneration of synapses may have important therapeutic implications. Although many experimental approaches block the development of pathology, in the real world therapeutic intervention will only be effective if it can reverse existing pathological changes. Reducing intraneuronal concentrations of Aβ may provide a mechanism that allows synapse regeneration.
References
- Bate C, Williams A. Amyloid-β-induced synapse damage is mediated via cross-linkage of the cellular prion protein. J Biol Chem. 2011;286:37955–37963. doi: 10.1074/jbc.M111.248724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Tayebi M, Williams A. Phospholipase A2 inhibitors protect against prion and Aβ mediated synapse degeneration. Mol Neurodegener. 2010;5:13. doi: 10.1186/1750-1326-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibl M, Mollenhauer B, Esselmann H, Lewczuk P, Klafki HW, Sparbier K, Smirnov A, Cepek L, Trenkwalder C, Ruther E, Kornhuber J, Otto M, Wiltfang J. CSF amyloid-β-peptides in Alzheimer's disease, dementia with Lewy bodies and Parkinson's disease dementia. Brain. 2006;129:1177–1187. doi: 10.1093/brain/awl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing trafficking and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- de Figueiredo P, Drecktrah D, Katzenellenbogen JA, Strang M, Brown WJ. Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc Natl Acad Sci U S A. 1998;95:8642–8647. doi: 10.1073/pnas.95.15.8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Desbene C, Malaplate-Armand C, Youssef I, Garcia P, Stenger C, Sauvee M, Fischer N, Rimet D, Koziel V, Escanye MC, Oster T, Kriem B, Yen FT, Pillot T, Olivier JL. Critical role of cPLA(2) in Abeta oligomer-induced neurodegeneration and memory deficit. Neurobiol Aging. 2012;33:1123 e17–29. doi: 10.1016/j.neurobiolaging.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:2273–2276. doi: 10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer S, Ying M, Walchli S, van Deurs B, Sandvig K. Golgi vesiculation induced by cholesterol occurs by a dynamin- and cPLA2-dependent mechanism. Traffic. 2005;6:144–156. doi: 10.1111/j.1600-0854.2005.00258.x. [DOI] [PubMed] [Google Scholar]
- Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7:456–462. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljarvi L, Koivisto E, Riekkinen PJ., Sr Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer's disease. Neuroscience. 1995;64:375–384. doi: 10.1016/0306-4522(94)00422-2. [DOI] [PubMed] [Google Scholar]
- Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Aβ dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy AK, Polishchuk RS, Lodge R, Roberts TH, Hirschberg K, Phair RD, Lippincott-Schwartz J. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol. 2001;153:529–541. doi: 10.1083/jcb.153.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Simons K. Lipid rafts and membrane dynamics. J Cell Sci. 2005;118:1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- Simmons C, Ingham V, Williams A, Bate C. Platelet-activating factor antagonists enhance intracellular degradation of amyloid-beta42 in neurons via regulation of cholesterol ester hydrolases. Alzheimers Res Ther. 2014;6:15. doi: 10.1186/alzrt245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropath Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Path. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Vassar R, Citron M. Aβ-generating enzymes: recent advances in b and g-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Williamson R, Usardi A, Hanger DP, Anderton BH. Membrane-bound β-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J. 2008;22:1552–1559. doi: 10.1096/fj.07-9766com. [DOI] [PubMed] [Google Scholar]