

Parkinson's disease (PD) is a slowly progressing neurodegenerative disorder clinically defined by key symptoms like bradykinesia, rigidity, resting tremor, and postural instability. Reduction of dopaminergic terminals in the caudate nucleus and putamen, i.e., the striatum, accompanied by decreased striatal levels of dopamine, and most likely consecutive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) are tightly linked to the onset of the motor symptoms. With the etiology of PD largely not yet well defined, there is accumulating evidence for increased immune activation in PD. The majority of studies favor a detrimental effect of microglia, the resident innate immune cells of the brain, on PD pathogenesis. In the presence of alpha-synuclein, inflammatory responses elicited by microglia may be initiated by pattern recognition receptors, e.g., toll-like receptors (TLR) leading to the distinct activation of signal transduction pathways. For instance, TLRs couple to signal adaptor proteins like myeloid differentiation primary response gene 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF), resulting ultimately in the activation of multiple, signal-dependent transcription factors including members of the nuclear factor κB (NF-κB) or AP-1 families. This leads to an enhanced production of pro-inflammatory cytokines like tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) or IL-6 (Figure 1). Indeed, increased levels of pro-inflammatory mediators have been detected within the SNpc and striatum supporting the existence of an underlying neuroinflammatory signature in PD (see review by Hirsch and Hunot, 2009). As a consequence, these inflammatory mediators may directly or via astrocyte activation perturb neuronal homeostasis (Saijo et al., 2009). However, the precise temporal and spatial sequence of this immunogenic response during the course of PD is not defined yet.
Figure 1.
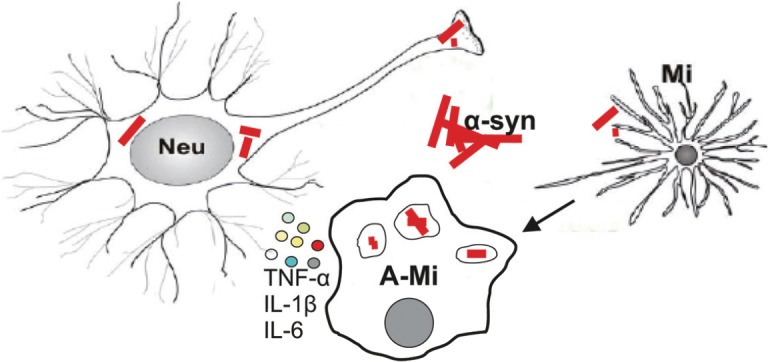
Dysfunctional and degenerating neurons (Neu) may activate microglia (A-Mi) via alpha-synuclein (α-syn), leading to the secretion of cytokines like TNF-α, IL-1β, and IL-6.
TNF-α: Tumor necrosis factor-α; IL: interleukin.
So far, numerous research efforts have focused on strategies to halt or at least to slow down neurodegenerative processes in PD. Despite the development of a plethora of “neuroprotective” agents, none of them have successfully entered the clinic. Besides dopaminergic replacement strategies, additional targets are an urgent need in particular due to the increasing incidence of PD in an aging society.
In recent years, the interest in the brain's endogenous regenerative potential has increased, but these events are very difficult to detect and poorly understood in humans. Neuroplasticity, the structural and functional reorganization within certain areas of the brain, includes physiological mechanisms like axonal sprouting, synaptogenesis, and neurogenesis. Distinct regions of the brain, i.e., the hippocampal dentate gyrus and the subventricular zone (SVZ)/olfactory bulb, bear the ability to generate new neurons throughout life. Neural precursor cells (NPCs) reside in the SVZ located adjacent to the striatum within the lateral ventricle wall. Newly generated cells within the SVZ physiologically migrate along the rostral aspect of the striatum as neuroblasts into the olfactory bulb, where they differentiate into dopaminergic or γ-aminobutyric acid (GABA) ergic interneurons. Recently, by employing a carbon-14 dating approach, it was shown in humans that new neurons integrate into the striatum throughout life, too (Ernst et al., 2014). It is tempting to speculate that the brain's capacity to generate new neurons is able to integrate these neurons into the target field of dopaminergic projections to locally replenish striatal dopamine levels.
Our main research focus is to study cell non-autonomous and autonomous factors modulating adult neurogenesis in PD (Marxreiter et al., 2013). Previously, we showed that striatal deafferentation induced by injection of the neurotoxin 6-hydroxydopamine (6-OHDA) results in reduced striatal dopaminergic input into the SVZ and attenuates NPC proliferation. Intriguingly, survival of newly generated neurons was increased in the glomerular layer of the olfactory bulb (Winner et al., 2006). We demonstrated that dopaminergic depletion is able to induce neuroplastic changes within the lesioned brain. Next, we pharmacologically enhanced adult SVZ neurogenesis with the aim to promote the generation of new mature neurons. Oral administration of the D2/D3 receptor agonist pramipexole promoted more newly generated neurons into a dopaminergic phenotype in the olfactory bulb (Winner et al., 2009). We also asked whether modulation of adult neurogenesis in the SVZ may be utilized to direct new neurons into the deafferented striatum. Indeed, combined intraventricular infusion of epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF-2) resulted in enhanced cell proliferation in the SVZ and facilitated migration of immature neuroblasts into the adjacent striatum. However, mature dopaminergic phenotype of these cells was not achieved (Winner et al., 2008). Taken together, adult neurogenesis within the SVZ is modified by a classical oral available dopamine agonist, but the maturation and integration of these newborn neurons was perturbed in the striatum after acute 6-OHDA lesioning. Since PD is associated with increased immune activation, we hypothesized that increased microglial activation and thereby the secretion of pro-inflammatory factors are detrimental for these newly born neurons within the striatum.
This prompted us to explore axonal and glial changes within the striatum using the identical model (Schlachetzki et al., 2014). To this aim, we exploited the acute, unilateral 6-OHDA model of PD by injection of the neurotoxin into the medial forebrain bundle (MFB) connecting the SNpc with the striatum. This lesion rapidly leads to striatal synaptic and axonal loss associated with striatal dopaminergic depletion. Given that regenerative responses may not occur immediately, we performed a comprehensive time-course study over 6 weeks. Indeed, we observed an almost complete reduction of striatal tyrosine hydroxylase (TH) and dopamine transporter (DAT) positive fibers within the lesioned striatum without axonal recovery. These findings support the limited capacity of the striatonigral system after chemically induced axotomy to restore neuronal circuitries and regain functionality. Next, we analyzed the microglial and astrocytic cell population on the lesioned side quantifying ionized calcium-binding adapter molecule 1 (Iba1) and glial fibrillar acid protein (GFAP) expressing cells, respectively. The number of Iba1 cells increased by 58% 1-week post-lesioning, and even increased further to 74% after 6 weeks. However, the detailed analysis of microglia did not reveal polarization into an activated phenotype as characterized by enlarged cell soma as well as thickening and retraction of branches. Moreover, no alterations were observed in gene expression levels of the pro-inflammatory cytokines TNF-α, IL-1β or IL-6 within the lesioned striatum. In addition, the number of GFAP positive astrocytes transiently almost doubled 2 weeks post-lesioning and was still increased by 40% after 6 weeks. To summarize, acute and profound striatal synaptic as well as axonal loss is long lasting without signs of resprouting, and more importantly, accompanied by a significant astro- and microgliosis, albeit exhibiting no signs of a pro-inflammatory activation state.
Next, we asked whether compensatory mechanisms are present within the contralateral, non-lesioned striatum. Unexpectedly, the intensity of TH positive fibers was significantly increased 6 weeks post-lesioning accompanied by a profound microgliosis without an astrocytic component. The key finding within the contralateral, non-lesioned striatum is the substantial compensatory effect within the axonal compartment accompanied solely by microglia. Our results highlight not only endogenous compensatory mechanisms within the striatum but also the dichotomic nature of microglia both on lesioned and non-lesioned neural circuitries.
In the context of PD, a predominant deleterious role has been attributed to microglial activation. In analogue to peripheral macrophages, the microglial polarization to a destructive state was labeled as M1 phenotype. In sharp contrast, microglia may be a permissive cellular component to promote regeneration by secretion of soluble factors like growth factors and anti-inflammatory cytokines, e.g., brain derived neurotrophic factor (BDNF) and IL-10, respectively. This neurotrophic patterning of microglia is also referred to as M2. M2 polarized microglia remove dysfunctional and apoptotic cells, promote axonal sprouting and synaptogenesis, and confer axonal and neuronal protection (see review by Lucin and Wyss-Coray, 2009). Thus, in our experimental set up, the observed regenerative effect involving axonal sprouting and/or TH upregulation within the contralateral, non-lesioned striatum may involve a site-specific patterning of microglia into M2 phenotype.
For the compensatory effect within the contralateral, non-lesioned striatum after acute lesioning of the contralateral side may implicate (1) increased axonal sprouting may be fostered by an increased number of microglia providing trophic support; (2) molecular and biochemical signatures generated within a lesioned site may alternatively activate microglia within the contralateral striatum via interhemispheric connections. In turn, these activated microglia may promote compensatory effects, i.e., increased axonal sprouting and TH upregulation. In addition, activated microglia may promote even a permissive environment for increased adult neurogenesis in the SVZ or even in the striatum.
However, one has to take into account that PD is an age-associated disease. Microglia may be affected by aging implying that they are in a less plastic state and thereby are limited in their propensity to support or even induce regenerative events (see review by Lucin and Wyss-Coray, 2009). Therefore, the role of aging on microglial functioning may hold pivotal clues for possible disease pathogenesis in PD.
Have compensatory effects been detected in human PD? Positron emission tomography studies also suggest compensatory changes within the brain of PD as demonstrated by detection of increased dopamine synthesis in the putamen of patients in the early stage of the disease (Nandhagopal et al., 2011). As the disease progresses, however, these compensatory effects diminish. It is tempting to speculate that increased microglial activation dampens possible functional reorganization during the course of PD.
One current major drawback to elucidate the role of microglia in PD is the limited number of studies performed in human. A neurotoxic role of microglia in the context of PD is based on our knowledge in rodent models. Accordingly, anti-inflammatory treatment strategies were so far only successful in rodent models. One opportunity to compare primary rodent and human microglia will arise from recent advancements in induced pluripotent stem (IPS) cell technology. Characterizing differences between microglia derived from human control and PD IPS cells may further provide important insights into the neuroinflammatory signature of PD.
To conclude, we hypothesize that microglial site specific and directed polarization into a M2 phenotype is a potential target to successfully promote axonal and neuronal regeneration as well as neuronal differentiation and survival of newly generated neurons. However, there are unmet needs in translational neuroscience, particularly to decipher the microglial inflammatory signature in PD. For example, the microglial activation state in the pre-motor stage of PD is not clear. Assessing and consecutively modulating neuroinflammation during early disease stages of PD may have the potential to slow down disease progression. To successfully modify the disease it would be favorable to define a targeted approach that not only dampens an inflammatory response evoked by microglia but also to utilize microglia for neuroregeneration and -restoration, i.e., shifting microglia from a pro-inflammatory state to an anti-inflammatory, tissue-regenerating phenotype. Thus, a better understanding of the interplay of neuronal as well as glial components within the striatum may help to restore local neural circuits in PD.
This work is supported by the ELAN-Fonds (program No. 10.08.18.1) and the Interdisciplinary Center for Clinical Research (IZKF E9 and N3) of the Medical Faculty, FAU Erlangen-Nürnberg.
References
- Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, Possnert G, Druid H, Frisen J. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156:1072–1083. doi: 10.1016/j.cell.2014.01.044. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marxreiter F, Regensburger M, Winkler J. Adult neurogenesis in Parkinson's disease. Cell Mol Life Sci. 2013;70:459–473. doi: 10.1007/s00018-012-1062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, McKenzie J, McCormick S, Ruth TJ, Sossi V, de la Fuente-Fernandez R, Stoessl AJ. Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson's disease. Brain. 2011;134:3290–3298. doi: 10.1093/brain/awr233. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlachetzki JC, Marxreiter F, Regensburger M, Kulinich A, Winner B, Winkler J. Increased tyrosine hydroxylase expression accompanied by glial changes within the non-lesioned hemisphere in the 6-hydroxydopamine model of Parkinson's disease. Restor Neurol Neurosci. 2014;32:447–462. doi: 10.3233/RNN-130371. [DOI] [PubMed] [Google Scholar]
- Winner B, Geyer M, Couillard-Despres S, Aigner R, Bogdahn U, Aigner L, Kuhn G, Winkler J. Striatal deafferentation increases dopaminergic neurogenesis in the adult olfactory bulb. Exp Neurol. 2006;197:113–121. doi: 10.1016/j.expneurol.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Winner B, Couillard-Despres S, Geyer M, Aigner R, Bogdahn U, Aigner L, Kuhn HG, Winkler J. Dopaminergic lesion enhances growth factor-induced striatal neuroblast migration. J Neuropathol Exp Neurol. 2008;67:105–116. doi: 10.1097/nen.0b013e3181630cff. [DOI] [PubMed] [Google Scholar]
- Winner B, Desplats P, Hagl C, Klucken J, Aigner R, Ploetz S, Laemke J, Karl A, Aigner L, Masliah E, Buerger E, Winkler J. Dopamine receptor activation promotes adult neurogenesis in an acute Parkinson model. Exp Neurol. 2009;219:543–552. doi: 10.1016/j.expneurol.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]