

Abstract
Subarachnoid hemorrhage is frequently associated with poor prognoses. Three different hemodynamic phases were identified during subarachnoid hemorrhage: oligemia, hyperemia, and vasospasm. Each phase is associated with brain metabolic changes. In this review, we correlated the hemodynamic phases with brain metabolism and potential treatment options in the hopes of improving patient prognoses.
Keywords: hemodynamic phases, cerebral subarachnoid hemorrhage, metabolic crises
Introduction
Patients with subarachnoid hemorrhage (SAH) exhibit a high incidence of specific complications that are associated with a poor prognosis. Severe brain edema, high intracranial pressure, and ischemia may be associated both with cerebral blood flow modifications and brain metabolic changes (Seule et al., 2009; de Lima Oliveira et al., 2014c). Brain hemodynamic phases are classically described in traumatic brain injury (Martin et al., 1997); however, these phases may also be applicable to SAH (Figure 1) (Martin et al., 1997; de Lima Oliveira et al., 2014c). Few articles in the literature have presented correlations between brain blood flow changes and changes in cerebral metabolism during SAH. The objective of this review is to correlate the hemodynamic phases during SAH with brain metabolism, brain metabolic crises, and the main causes of cerebral ischemia.
Figure 1.
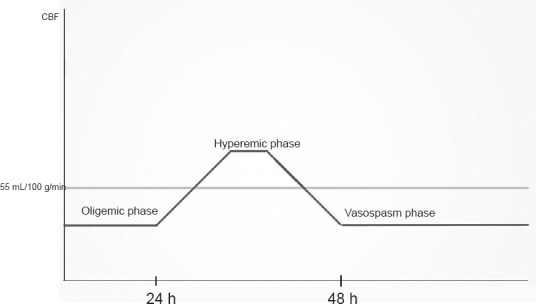
Cerebral hemodynamic abnormalities, which are classically described in traumatic brain injury.
Brain Hemodynamic Phases Associated with SAH
There are three phases associated with SAH: oligemia, hyperemia, and vasospasm (Martin et al., 1997; de Lima Oliveira et al., 2014c). These phases are also collectively known as triphasic phases and will be presented here.
Oligemia pattern, oligemic and non-oligemic metabolic crises
A decrease in cerebral blood flow (CBF) associated with high intracranial pressure (HIP) and low cerebral perfusion pressure (CPP) can be observed in the first 24 hours after SAH (acute phase) (de Lima Oliveira et al., 2014c). The CBF decrease may also be associated with intense microvascular vasoconstriction related to low oxide nitric concentrations (Goodman and Robertson, 2009; Westermaier et al., 2011; de Lima Oliveira et al., 2014c). These cited events lead to a critical CBF (brain flow under 40 mL/100 g/min) associated with O2 decreases in brain tissues and consequently ischemia. As result, the anaerobic path (Figure 2) is used for brain energy synthesis with one glucose molecule producing 2 adenosine triphosphate (ATP) and high lactate amounts. Microdialysis studies have revealed a high lactate/pyruvate relationship (LPR). The relationship is also known as oligemic metabolic crisis and involves high lactate concentrations and low O2 tissue levels (de Lima Oliveira et al., 2014c; de Lima Oliveira et al., 2014a). Reductions in blood glucose concentrations are associated with the collapse of brain energy syntheses in ischemia, as glucose becomes the primary substrate for energy synthesis. A prolonged oligemic phase may be correlated with a poorer neurological prognosis (Soehle et al., 2007; de Lima Oliveira et al., 2014c).
Figure 2.
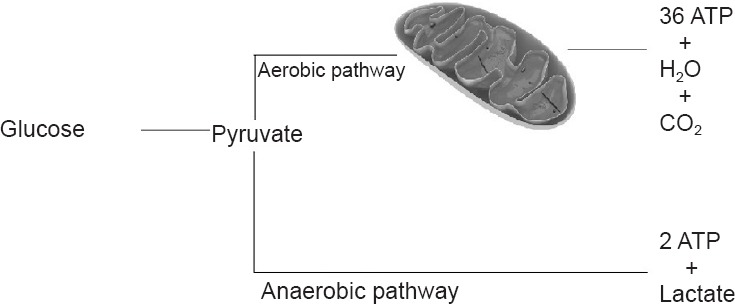
Aerobic and anaerobic pathway for energy synthesis.
In the aerobic pathway glucose is converted to pyruvate, which is converted to 36 adenosine triphosphate (ATP), water, and CO2 in the mitochondria. In the anaerobic pathway glucose molecule is converted to pyruvate, resulting in 2 ATP and lactate in the cytoplasm.
It is logical to assume that the establishment of adequate brain blood flow would normalize ATP synthesis via aerobic mechanisms (36 ATP produced by 1 glucose molecule), thus reestablishing brain function. However, various events following intense oligemic states, such as Ca2+ cellular influx linked with Na/K pump impairment, free radicals, and brain tissue acidosis, harm mitochondrial energy synthesis and therefore hinder appropriate energy synthesis. This latter condition, known as mitochondrial dysfunction (Bor-Seng-Shu et al., 2010; de Lima Oliveira et al., 2014c), is characterized by persistent ATP synthesis via anaerobic mechanisms. This condition is recognized as non-oligemic brain metabolic crisis, which exhibits increased lactate and O2 amounts (Bor-Seng-Shu et al., 2010; de Lima Oliveira et al., 2014a, c). Persistent higher cerebral PtiO2 may also be linked with unfavorable outcome since there is no consumption of tissue O2 due to mitochondrial harm (Bor-Seng-Shu et al., 2010; de Lima Oliveira et al., 2014a, c). An experimental study (Westermaier et al., 2009) has demonstrated that cerebral PtiO2 declined 57% from the basal levels in SAH hyper-acute phase, recovered after the second hour, and reached ≥ 140% after 6 hours. The authors also observed disparity between low PtiO2 and CPP possibly because of microvascular vasoconstriction. They concluded that acute vasoconstriction and incapacity in oxygen use could be additional factors that cause secondary brain damage after SAH, regardless of acute intracranial pressure (Westermaier et al., 2009).
In general, oligemic metabolic crisis is clinically managed by providing intracranial hypertension management and treating brain hypermetabolic states; adequate CPP is also important during this phase. However, the restoration of aerobic energy synthesis in non-oligemic metabolic crises is a challenge. Specific treatments for mitochondrial dysfunction are not well established. Although various studies demonstrated positive results with cyclosporin A, which decreases the mitochondrial pore permeability, no evidence regarding its use in clinical practice is available (Bor-Seng-Shu et al., 2010; de Lima Oliveira et al., 2014c; Gajavelli et al., 2015). Experimental studies have shown a reduction in brain ischemia after SAH with intravenous and intra-cisternal hydrogen infusion (Takeuchi et al., 2014). Hydrogen can reduce hydroxyl radicals and peroxynitrates, both of which can be associated with DNA fragmentation, lipid peroxidation, and protein inactivation. Hydrogen has also antioxidant, anti-apoptotic, anti-inflammatory and cytoprotective properties. Due to its high diffusion, hydrogen can reach cells subcomponents such as mitochondria and nuclei. Such structures are the primary sites of oxygen species generation and DNA damage (Takeuchi et al., 2014). Progesterone and Magnesium Sulfate may reduce the intensity of mitochondrial dysfunction by blocking NMDA channels receptors, while estrogen has antioxidant effects (de Lima Oliveira et al., 2014a). For both oligemic and non-oligemic metabolic crises, it is necessary to maintain appropriate brain blood flow and glucose levels because brain tissues are sensitive to hypoflux and hypoglycemic states (de Lima Oliveira et al., 2014c). Spare energy is also important to prevent the disruption between flow and metabolism; hypermetabolic states, such as fever, epileptic or non-epileptic seizures, and cortical spread depression, must be identified and treated (de Lima Oliveira et al., 2014c). It is important to reinforce that cortical spread depression is associated with increased brain metabolism and sustained microvascular vasoconstriction, which may increase brain oligemia (Brennan et al., 2007; de Lima Oliveira et al., 2014c).
Hyperemic pattern
It is known that brain tissue acidosis causes microvascular bed dilatation, leading to cerebral hyperemic states, which reflects the impaired vascular coupling (Bor-Seng-Shu et al., 2012b; de Lima Oliveira et al., 2014c). This non-physiological hyperemia is potentially associated with high lactate amounts as a result of brain tissue acidosis. On the other side, physiological hyperemia can lead to hyperemic states to restore the equilibrium of damaged tissue. Thus, various metabolic events are associated with the hyperemia phase in SAH as pointed below:
-
1)
Persistent non-oligemic metabolic crisis, which starts during SAH oligemic phase (Westermaier et al., 2009), may be associated with mitochondrial dysfunction, consequently leading to impaired energy production from this organelle. Excessive lactate levels are produced (Figure 1), leading to a prolonged hyperemic brain state as a consequence of cerebral tissue acidosis (Bor-Seng-Shu et al., 2010; de Lima Oliveira et al., 2014a, c).
-
2)
Elevated lactate levels are also observed during hyperglycolysis states, which are necessary to provide energy during recovery after ischemia. This energy demand is necessary to decrease interstitial glutamate levels, restore ion pump homeostasis, increase pentose phosphate pathway activity to remove free radicals, and provide energy to infiltrated inflammatory cells (de Lima Oliveira et al., 2014c).
-
3)
Hypermetabolic states related to fever, seizures, and cortical spread depression require high-energy demands, leading to uncoupled flow metabolism and consequently increased lactate levels (de Lima Oliveira et al., 2014c).
-
4)
Increased lactate levels in brain tissue may be attributed to transitory intense oligemia states. After reestablishing normal flow and energy synthesis, lactate is available to produce energy by other mechanisms. Thus, lactate is quickly removed from cerebral tissues (de Lima Oliveira et al., 2014c). This condition is likely related to fast and transitory hyperemia.
-
5)
Hypercapnia, which increases H+ protons in brain tissues, is also associated with microvascular vasodilatation and CBF augment. Thus, hypercapnia increases cerebral blood volume and intracranial pressure. On the other hand, hypocapnia may decrease CBF (Bor-Seng-Shu et al., 2012b; de Lima Oliveira et al., 2014c).
-
6)
Prostacyclin and its analogues are metabolites produced by inflammatory cells. These substances produce arterial relaxation, which increases CBF during inflammation associated with SAH (Bor-Seng-Shu et al., 2012b).
Cerebral pressure autoregulation is defined by brain intrinsic blood vessels to maintain a relatively constant CBF over a wide range of arterial blood pressure (ABP) levels. There are three main mechanisms, which may be related to cerebral autoregulation (CA): metabolic, myogenic, and neurogenic (Bor-Seng-Shu et al., 2012b, 2013; Nogueira et al., 2013). Assessment of CA is crucial to adequately CBF control during the hyperemic phase (Bor-Seng-Shu et al., 2013). This condition can be assessed by two methods: static and dynamic evaluation. In the static method, CBF is estimated from basal ABP levels followed by an additional CBF estimation after pharmacologic ABP manipulation. This technique should be applied with caution especially in critical patients whose wide ABP changes could increase the brain damage. The dynamic method assesses the temporal response of autoregulatory mechanisms, which promote fast restoration of CBF after sudden ABP changes. Studies have unveiled that the dynamic methods allow the evaluation of CA using spontaneous ABP fluctuations in critical patients (Bor-Seng-Shu et al., 2012b). Intracranial pressure catheter can provide both continuous CA (PRx) and CPP, although its use is restricted once it is an invasive method. Transcranial Doppler is a non-invasive method that can also supply CA (Mx) and CPP (eCPP) through the intermittent blood flow velocities measurement. Budohoski et al. have described moderate correlation between PRx and Mx (Budohoski et al., 2012). Critical closing pressure (CrCP) of the cerebral circulation indicates the value of ABP at which CBF approaches zero (Panerai, 2003). Transcranial Doppler also estimates CrCP adding some physiological parameters to eCPP, such as heart rate and measures of resistance and compliance of the cerebrovascular bed (Varsos et al., 2015). However Transcranial Doppler sensibility to detect high intracranial pressure is limited (Bellner et al., 2004).
SAH may impair cerebral pressure autoregulation, which is caused in part by disarrangement of metabolic and myogenic mechanism of CA. In this situation, cerebral blood volume and ICP will increase or decrease passively in response to changes in ABP (Bor-Seng-Shu et al., 2013). The impairment of cerebral pressure autoregulation related to metabolic mechanisms may be attributed to brain acidosis, which was discussed above. In the neural mechanism, brain flow is regulated by astrocytes as well as parasympathetic and sympathetic nerves (Salinet et al., 2012; Nogueira et al., 2013). The impairment of CA can also be attributed to disturb of neurogenic mechanism due to brain damage that occurs in SAH.
Increased CPP values (70 mmHg) are acceptable in patients with intact cerebral pressure autoregulation. On the other hand, when cerebral autoregulation is compromised, CPP values should not be greater than 60 mmHg because brain edema is potentially aggravated by cerebral hyperemia (Bor-Seng-Shu et al., 2006, 2012a, 2013). Therefore, all efforts should be made to maintain adequate CBF to ensure adequate brain hemodynamics during hyperemia (Bor-Seng-Shu et al., 2006, 2012a). Another key point in hyperemic phase treatments involves the identification of hypermetabolic states, such as fever and seizures. These states may be associated with hyperemia, which provides glucose and O2 to meet the increased cerebral metabolic demand (de Lima Oliveira et al., 2014c).
Persistent non oligemic metabolic crises may also be identified by microdialysis method during the hyperemia phase; however, no evidence is available regarding specific treatments.
Vasospasm phase
Cerebral vasospasm remains the main cause linked with delayed ischemia. Various mechanisms are associated with brain ischemia as follows:
-
1)
Brain vasospasm can lead to cerebral oligemia. The intensity in narrow arteries, multiple intracranial arteries with intense vasospasm, sequential vasospasm (“Tandem”), and vasospasm intensity with rapid progression are frequently associated with brain ischemia (Bor-Seng-Shu et al., 2011). Proximal narrowing in basal cerebral arteries is potentially associated with distal microcirculatory vasodilatation caused by autoregulatory mechanisms. Thus, small reduction in systemic blood pressure may intensify distal oliguemia to the vessel afflicted with vasospasm because of decrease in CrCP (Soehle et al., 2004).
-
2)
Hypermetabolism can also occur during vasospasm. Various microdialysis studies indicated that glutamate, an excitatory amino acid, is an early ischemia marker. Glutamate reuptake in synapses is an active process, which involves energy consumption. In oligemic metabolic crises, glutamate reuptake may be disrupted. As consequence, increased glutamate levels in the synapses are associated with seizures, which lead to uncoupled flow metabolism (Bellander et al., 2004; de Lima Oliveira et al., 2014a, b, c, b). Other high metabolic states, such as fever and cortical spread depression, also occur during vasospasm and lead to uncoupled flow metabolism (de Lima Oliveira et al., 2014c).
-
3)
Mitochondrial dysfunction, which is associated with non-oligemic metabolic crises, can occur during the vasospasm phase (de Lima Oliveira et al., 2014c). On the other hand, hypermetabolic states, such as seizures, fever, and cortical spread depression, can simultaneously appear during mitochondrial dysfunction. The high metabolic demands of these conditions are associated with low energy synthesis during mitochondrial dysfunction, thus enhancing the ischemic events (de Lima Oliveira et al., 2014c).
Patients with severe vasospasm associated with mitochondrial dysfunction and hypermetabolic states may progress to malignant edema (de Lima Oliveira et al., 2014c); high intracranial pressure is a common complication observed in the first week after SAH and is associated with early brain injury and mortality (Zoerle et al., 2015). Various authors describe decreased glutamate levels when SAH patients are cooled, suggesting that brain temperature reductions offer neuroprotection (Peerdeman et al., 2000; de Lima Oliveira et al., 2014c). Hypothermia also reduces other cellular distress markers, such as lactate. The lactate/pyruvate relationship is potentially associated with decreased brain metabolism (de Lima Oliveira et al., 2014c). Wang et al. (2011) showed that mild hypothermia reduced brain vasospasm intensity by potentially regulating increases in endothelin-1 and nitric oxide in cerebral spine fluid and plasma. Anei et al. (2010) demonstrated brain swelling in hyperthermia patients after therapeutic hypothermia; these results support the maintenance of normothermia rather than interventional hypothermia for aggressive brain protection during SAH. Fever occurs in 70% of all patients with SAH; this condition potentially results as a reaction to hemoglobin molecules. Fever is also associated with the development of vasospasm. Various investigators associate fever with increased death or severe disability; in these studies, therapeutic normothermia was associated with an improvement in 12-month outcomes (Badjatia, 2009).
There is a consensus that oral nimodipine reduces delayed cerebral ischemia and improves the outcome of patients with SAH, although there is no correlation with vasospasm incidence decrease. The main role of nimodipine may be to generate neuroprotective effect by blocking Ca2+ channels cells, rather than having vasodilatory properties (Siasios et al., 2013). Magnesium Sulfate may provide some advantages such as vasodilatation of vessels, attenuation of cells death through blockage of the voltage-dependent calcium channels, inhibition of glutamate release, and interference with N-methy-D-aspartate cells receptor (Gajavelli et al., 2015). Nevertheless, randomized control trials and meta-analyses studies have failed to demonstrate the benefits of intravenous administration of magnesium sulfate in patients with SAH. This fact is likely associated with limited increases of magnesium in cerebral spinal fluid after intravenous infusion and/or late onset of the magnesium sulfate infusion. An interesting alternative could be to apply intra-cisternal magnesium sulfate, which some studies have shown precocious high magnesium levels in cerebral spinal fluid (Gajavelli et al., 2015). Erithropoetin is also tied with several neuroprotective effects such as inflammation limitation, apoptosis inhibition, oxidative damage limitation, and neurogenese upregulation (Siasios et al., 2013). Endovenous erithropoetin can also reduce vasospasm incidence and enhance CA (Siasios et al., 2013). Helbok et al. (2012) have stated PtbO2 improvement in patients with severe vasospasm after venous eritropoethin treatment. They have stressed the erithropoetin neuroprotective effect, due to the fact PtbO2 is a marker of oxygen delivery, diffusion and tissue consumption.
Statins are related with oxide nitric pathway and arterial vasodilatation. An analysis of four high quality studies have demonstrated lower delayed cerebral ischemia and mortality in patients with vasospasm (Tseng, 2011; Siasios et al., 2013). Another useful treatment is to insert prolonged-release intra-cisternal nicardipine pallets to prevent or reverse cerebral vasospasm. Some authors have noted important decrease in the angiographic vasospasm and delayed cerebral ischemia in this technique (Siasios et al., 2013). Intraterial Dantrole infusion may lessen cerebral vasoconstriction, though its administration might also be related with lower systemic arterial pressure. Milrinone has the same effect and maintains suitable level of CBF (Siasios et al., 2013).
Electrographic seizures leading to increased brain metabolism and cellular stress, but without clinical signals, are detected in 50% of seizures occurring in intensive care, Therefore, it is important to monitor patients to identify and treat this condition using continuous electroencephalogram (EEG) (de Lima Oliveira et al., 2014c). Such technique also identifies severe cerebral vasospasm in advance through the reduction of alpha power wave (sensibility 89% and specificity 77%) and indicates imminent delayed cerebral ischemia earlier than any other widely used technique (Gollwitzer et al., 2014). Unfortunately, cortical spread depression, which can increase brain metabolism by 200%, is diagnosed through electrocorticography, an invasive method with limited availability (de Lima Oliveira et al., 2014c). Specific treatments to brain metabolic crises (de Lima Oliveira et al., 2014a, c) are limited and not validated as previously mentioned in this review.
Classically, systemic hypertension induced by normovolemia is recommended to improve cerebral perfusion during severe vasospasm. Some authors demonstrated that high systemic blood pressure is related with reversions of symptomatic vasospasm and augmentation of cerebral PtiO2 (Vath et al., 2002; Connolly et al., 2012; Siasios et al., 2013); arterial angioplasty may be recommended to patients with symptomatic vasospasm who do not respond to clinical treatment (Siasios et al., 2013). Severe vasospasm is associated with decreases in CrCP and consequently may result in oligemia during decreases in systemic arterial pressure (Panerai, 2003). Experimental research in rats demonstrated improved brain autoregulation after the induction of hypothermia during SAH; cellular markers of distress, such as lactate, glucose depletion, and glutamate, also improved (Schubert et al., 2008).
Conclusion
It is important to emphasize that brain metabolic changes during SAH involve continuous processes that exhibit strict correlations with brain flow. Therefore, each treatment step requires the identification of brain metabolic status and provides adequate blood flow to each hemodynamic phase.
Footnotes
Conflicts of interest: None declared.
References
- Anei R, Sakai H, Iihara K, Nagata I. Effectiveness of brain hypothermia treatment in patients with severe subarachnoid hemorrhage: comparisons at a single facility. Neurol Med Chir (Tokyo) 2010;50:879–883. doi: 10.2176/nmc.50.879. [DOI] [PubMed] [Google Scholar]
- Badjatia N. Hyperthermia and fever control in brain injury. Crit Care Med. 2009;37:S250–257. doi: 10.1097/CCM.0b013e3181aa5e8d. [DOI] [PubMed] [Google Scholar]
- Bellander BM, Cantais E, Enblad P, Hutchinson P, Nordstrom CH, Robertson C, Sahuquillo J, Smith M, Stocchetti N, Ungerstedt U, Unterberg A, Olsen NV. Consensus meeting on microdialysis in neurointensive care. Intensive Care Med. 2004;30:2166–2169. doi: 10.1007/s00134-004-2461-8. [DOI] [PubMed] [Google Scholar]
- Bellner J, Romner B, Reinstrup P, Kristiansson KA, Ryding E, Brandt L. Transcranial Doppler sonography pulsatility index (PI) reflects intracranial pressure (ICP) Surg Neurol. 2004;62:45–51. doi: 10.1016/j.surneu.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Bor-Seng-Shu E, Hirsch R, Teixeira MJ, De Andrade AF, Marino R., Jr Cerebral hemodynamic changes gauged by transcranial Doppler ultrasonography in patients with posttraumatic brain swelling treated by surgical decompression. J Neurosurg. 2006;104:93–100. doi: 10.3171/jns.2006.104.1.93. [DOI] [PubMed] [Google Scholar]
- Bor-Seng-Shu E, de Lima Oliveira M, Teixeira MJ. Traumatic brain injury and metabolism. J Neurosurg. 2010;112:1351–1353. doi: 10.3171/2009.10.JNS091426. [DOI] [PubMed] [Google Scholar]
- Bor-Seng-Shu E, de-Lima-Oliveira M, Teixeira MJ, Panerai RB. Predicting symptomatic cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2011;69:E501–502. doi: 10.1227/NEU.0b013e31821b7ed1. [DOI] [PubMed] [Google Scholar]
- Bor-Seng-Shu E, Figueiredo EG, Amorim RL, Teixeira MJ, Valbuza JS, de Oliveira MM, Panerai RB. Decompressive craniectomy: a meta-analysis of influences on intracranial pressure and cerebral perfusion pressure in the treatment of traumatic brain injury. J Neurosurg. 2012a;117:589–596. doi: 10.3171/2012.6.JNS101400. [DOI] [PubMed] [Google Scholar]
- Bor-Seng-Shu E, Kita WS, Figueiredo EG, Paiva WS, Fonoff ET, Teixeira MJ, Panerai RB. Cerebral hemodynamics: concepts of clinical importance. Arq Neuropsiquiatr. 2012b;70:352–356. doi: 10.1590/s0004-282x2012000500010. [DOI] [PubMed] [Google Scholar]
- Bor-Seng-Shu E, Figueiredo EG, Fonoff ET, Fujimoto Y, Panerai RB, Teixeira MJ. Decompressive craniectomy and head injury: brain morphometry, ICP, cerebral hemodynamics, cerebral microvascular reactivity, and neurochemistry. Neurosurg Rev. 2013;36:361–370. doi: 10.1007/s10143-013-0453-2. [DOI] [PubMed] [Google Scholar]
- Brennan KC, Beltran-Parrazal L, Lopez-Valdes HE, Theriot J, Toga AW, Charles AC. Distinct vascular conduction with cortical spreading depression. J Neurophysiol. 2007;97:4143–4151. doi: 10.1152/jn.00028.2007. [DOI] [PubMed] [Google Scholar]
- Budohoski KP, Czosnyka M, de Riva N, Smielewski P, Pickard JD, Menon DK, Kirkpatrick PJ, Lavinio A. The relationship between cerebral blood flow autoregulation and cerebrovascular pressure reactivity after traumatic brain injury. Neurosurgery. 2012;71:652–660. doi: 10.1227/NEU.0b013e318260feb1. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke. 2012;43:1711–1737. doi: 10.1161/STR.0b013e3182587839. [DOI] [PubMed] [Google Scholar]
- de Lima Oliveira M, Paiva W, Teixeira MJ, Bor-Seng-Shu E. Brain metabolic crisis in traumatic brain injury: what does it mean? J Neurotrauma. 2014a;31:1750–1751. doi: 10.1089/neu.2014.3386. [DOI] [PubMed] [Google Scholar]
- de Lima Oliveira M, de Azevedo MK, Machado MF, Teixeira MJ, Bor-Seng-Shu E. The role of metabolism in the ischemia associated with vasospasm following brain tumor resection. J Neurointerv Surg. 2014b doi: 10.1136/neurintsurg-2014-011205. doi: 10.1136/neurintsurg-2014-011205. [DOI] [PubMed] [Google Scholar]
- de Lima Oliveira M, Kairalla AC, Fonoff ET, Martinez RC, Teixeira MJ, Bor-Seng-Shu E. Cerebral microdialysis in traumatic brain injury and subarachnoid hemorrhage: state of the art. Neurocrit Care. 2014c;21:152–162. doi: 10.1007/s12028-013-9884-4. [DOI] [PubMed] [Google Scholar]
- Gajavelli S, Sinha VK, Mazzeo AT, Spurlock MS, Lee SW, Ahmed AI, Yokobori S, Bullock RM. Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J Bioenerg Biomembr. 2015;47:133–148. doi: 10.1007/s10863-014-9589-1. [DOI] [PubMed] [Google Scholar]
- Gollwitzer S, Groemer T, Rampp S, Hagge M, Olmes D, Huttner HB, Schwab S, Madzar D, Hopfengaertner R, Hamer HM. Early prediction of delayed cerebral ischemia in subarachnoid hemorrhage based on quantitative EEG: A prospective study in adults. Clin Neurophysiol. 2014 doi: 10.1016/j.clinph.2014.10.215. doi: 10.1016/j.clinph.2014.10.215. [DOI] [PubMed] [Google Scholar]
- Goodman JC, Robertson CS. Microdialysis: is it ready for prime time. Curr Opin Crit Care. 2009;15:110–117. doi: 10.1097/MCC.0b013e328325d142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbok R, Shaker E, Beer R, Chemelli A, Sojer M, Sohm F, Broessner G, Lackner P, Beck M, Zangerle A, Pfausler B, Thome C, Schmutzhard E. High dose erythropoietin increases brain tissue oxygen tension in severe vasospasm after subarachnoid hemorrhage. BMC Neurol. 2012;12:32. doi: 10.1186/1471-2377-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NA, Patwardhan RV, Alexander MJ, Africk CZ, Lee JH, Shalmon E, Hovda DA, Becker DP. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion hyperemia, and vasospasm. J Neurosurg. 1997;87:9–19. doi: 10.3171/jns.1997.87.1.0009. [DOI] [PubMed] [Google Scholar]
- Nogueira RC, Bor-Seng-Shu E, Santos MR, Negrao CE, Teixeira MJ, Panerai RB. Dynamic cerebral autoregulation changes during sub-maximal handgrip maneuver. PLoS One. 2013;8:e70821. doi: 10.1371/journal.pone.0070821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panerai RB. The critical closing pressure of the cerebral circulation. Med Eng Phys. 2003;25:621–632. doi: 10.1016/s1350-4533(03)00027-4. [DOI] [PubMed] [Google Scholar]
- Peerdeman SM, Girbes AR, Vandertop WP. Cerebral microdialysis as a new tool for neurometabolic monitoring. Intensive Care Med. 2000;26:662–669. doi: 10.1007/s001340051230. [DOI] [PubMed] [Google Scholar]
- Salinet AS, Panerai RB, Robinson TG. Effects of active, passive and motor imagery paradigms on cerebral and peripheral hemodynamics in older volunteers: a functional TCD study. Ultrasound Med Biol. 2012;38:997–1003. doi: 10.1016/j.ultrasmedbio.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Schubert GA, Poli S, Mendelowitsch A, Schilling L, Thome C. Hypothermia reduces early hypoperfusion and metabolic alterations during the acute phase of massive subarachnoid hemorrhage: a laser-Doppler-flowmetry and microdialysis study in rats. J Neurotrauma. 2008;25:539–548. doi: 10.1089/neu.2007.0500. [DOI] [PubMed] [Google Scholar]
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery. 2009;64:86–92. doi: 10.1227/01.NEU.0000336312.32773.A0. [DOI] [PubMed] [Google Scholar]
- Siasios I, Kapsalaki EZ, Fountas KN. Cerebral vasospasm pharmacological treatment: an update. Neurol Research Int 2013. 2013 doi: 10.1155/2013/571328. 571328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehle M, Czosnyka M, Pickard JD, Kirkpatrick PJ. Critical closing pressure in subarachnoid hemorrhage: effect of cerebral vasospasm and limitations of a transcranial Doppler-derived estimation. Stroke. 2004;35:1393–1398. doi: 10.1161/01.STR.0000128411.07036.a9. [DOI] [PubMed] [Google Scholar]
- Soehle M, Chatfield DA, Czosnyka M, Kirkpatrick PJ. Predictive value of initial clinical status, intracranial pressure and transcranial Doppler pulsatility after subarachnoid haemorrhage. Acta Neurochir. 2007;149:575–583. doi: 10.1007/s00701-007-1149-6. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Mori K, Arimoto H, Fujii K, Nagatani K, Tomura S, Otani N, Osada H, Wada K. Effects of intravenous infusion of hydrogen-rich fluid combined with intra-cisternal infusion of magnesium sulfate in severe aneurysmal subarachnoid hemorrhage: study protocol for a randomized controlled trial. BMC Neurol. 2014;14:176. doi: 10.1186/s12883-014-0176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng MY. Summary of evidence on immediate statins therapy following aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2011;15:298–301. doi: 10.1007/s12028-011-9596-6. [DOI] [PubMed] [Google Scholar]
- Varsos GV, Kolias AG, Smielewski P, Brady KM, Varsos VG, Hutchinson PJ, Pickard JD, Czosnyka M. A noninvasive estimation of cerebral perfusion pressure using critical closing pressure. J Neurosurg. 2015:1–11. doi: 10.3171/2014.10.JNS14613. [DOI] [PubMed] [Google Scholar]
- Vath A, Kunze E, Roosen K, Meixensberger J. Therapeutic aspects of brain tissue pO2 monitoring after subarachnoid hemorrhage. Acta Neurochir Suppl. 2002;81:307–309. doi: 10.1007/978-3-7091-6738-0_78. [DOI] [PubMed] [Google Scholar]
- Wang ZP, Chen HS, Wang FX. Influence of plasma and cerebrospinal fluid levels of endothelin-1 and NO in reducing cerebral vasospasm after subarachnoid hemorrhage during treatment with mild hypothermia, in a dog model. Cell Biochem Biophys. 2011;61:137–143. doi: 10.1007/s12013-011-9170-z. [DOI] [PubMed] [Google Scholar]
- Westermaier T, Jauss A, Eriskat J, Kunze E, Roosen K. Time-course of cerebral perfusion and tissue oxygenation in the first 6 h after experimental subarachnoid hemorrhage in rats. J Cereb Blood Flow Metab. 2009;29:771–779. doi: 10.1038/jcbfm.2008.169. [DOI] [PubMed] [Google Scholar]
- Westermaier T, Jauss A, Eriskat J, Kunze E, Roosen K. The temporal profile of cerebral blood flow and tissue metabolites indicates sustained metabolic depression after experimental subarachnoid hemorrhage in rats. Neurosurgery. 2011;68:223–229. doi: 10.1227/NEU.0b013e3181fe23c1. [DOI] [PubMed] [Google Scholar]
- Zoerle T, Lombardo A, Colombo A, Longhi L, Zanier ER, Rampini P, Stocchetti N. Intracranial pressure after subarachnoid hemorrhage. Crit Care Med. 2015;43:168–176. doi: 10.1097/CCM.0000000000000670. [DOI] [PubMed] [Google Scholar]