

Abstract
We report a new series of 8-membered benzo-fused lactams that inhibit cellular lipid uptake from HDL particles mediated by Scavenger Receptor, Class B, Type I (SR-BI). The series was identified via a high-throughput screen of the National Institutes of Health Molecular Libraries Small Molecule Repository (NIH MLSMR), measuring the transfer of the fluorescent lipid DiI from HDL particles to CHO cells overexpressing SR-BI. The series is part of a previously reported diversity-oriented synthesis (DOS) library prepared via a build-couple-pair approach. Detailed structure-activity relationship (SAR) studies were performed with a selection of the original library, as well as additional analogs prepared via solution phase synthesis. These studies demonstrate that the orientation of the substituents on the aliphatic ring have a critical effect on activity. Additionally, a lipophilic group is required at the western end of the molecule, and a northern hydroxyl group and a southern sulfonamide substituent also proved to be optimal. Compound 2p was found to possess a superior combination of potency (avg. IC50 = 0.10 μM) and solubility (79 μM in PBS), and it was designated as probe ML312.
Keywords: ML312, SR-BI inhibitor, HDL receptor, cholesterol transport, DOS macrocycle
Graphical Abstract
Cholesterol has been widely recognized to have a critical impact on cardiovascular health. Our understanding of the connection between plasma cholesterol levels and adverse cardiovascular events has benefited tremendously from the identification of the proteins and macromolecular complexes involved in cholesterol biosynthesis and transport. This basic research has supported the development of numerous pharmacological interventions, including the development of HMG-CoA reductase inhibitors (statins), which lower LDL-C (low-density lipoprotein cholesterol) and decrease substantially the risk of adverse events and death in patients with high levels of LDL-C.
Despite the identification of the key players involved in cholesterol biosynthesis and transport, our mechanistic understanding of how these pathways are regulated is lacking in some important details. For example, it is now apparent that simply elevating HDL-C (high density lipoprotein cholesterol) levels may not necessarily be beneficial, despite a positive clinical correlation between HDL-C levels and cardiovascular outcomes. The receptor for HDL particles, Scavenger Receptor class B, type I (SR-BI),1–2 is one of many Pattern Recognition Receptors and is important for the uptake of HDL-C into liver cells (hepatocytes).3 Inhibition of SR-BI has been explored in animal models as a means to boost plasma HDL-C levels, but early results suggest that this approach is counterproductive as a means to improve cardiovascular health.3–4 Conversely, SR-BI is a co-receptor for the entry of pathogens into hepatocytes, including Hepatitis C virus (HCV)5–7 and malaria (plasmodium) parasites8–9 and its inhibition has recently been explored as a strategy for HCV patients requiring liver transplant.10–11
Though it is now recognized as an important target for the study of lipid metabolism and infectious disease, as well as immune response12–14 and female fertility,3 the structure and mechanism of SR-BI is not understood in full detail.15–18 In an effort to identify novel small molecule modulators of this receptor that may exert unique effects on lipid transport, as well as more potent and less toxic compounds, we embarked on a high-throughput screen (HTS) supported by the NIH Molecular Libraries Program (MLP). We recently reported two classes of inhibitors from this screen with distinct advantages over prior inhibitors identified in our laboratories.19 This work culminated in the nomination of ML27820–21 and ML27922–23 as Molecular Libraries probes. Other small molecule inhibitors of SR-BI have been disclosed, including those from the labs of Sankyo4 and iTherX.24–28 We report here a third class of inhibitors identified from a diversity-oriented synthesis (DOS) library, some members of which form a subset of the NIH Molecular Libraries Small Molecule Repository. Screening and assay data for this project are also publicly available in the PubChem database (http://pubchem.ncbi.nlm.nih.gov/). The primary assay involved measuring the uptake of the fluorescent lipid 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) from DiI-labeled HDL particles into CHO cells overexpressing mouse SR-BI (ldlA[mSR-BI]).22 3,046 compounds (0.96%) were classified as inhibitors of DiI-HDL uptake out of 319,533 compounds tested, with inhibition at 12.5 μM of ≥ 70% of the level inhibition with 1 μM BLT-1 (positive control). Hit compounds were omitted that were on plates with Z′ < 0.3 or that were active in 10% or more of the HTS assays listed in PubChem. A counterscreen was also performed that rejected hit compounds that quenched the fluorescence of DiI-HDL in a dose-dependent manner.
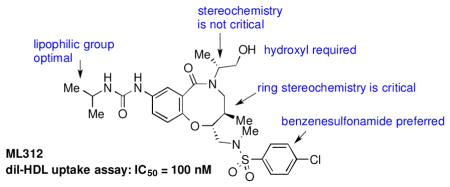
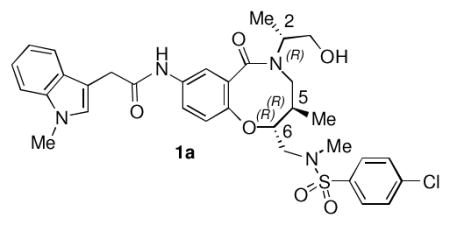
One of the more promising scaffolds that emerged from this screen was the benzo-fused lactam represented by compound 1a (Table 1). 1a and related compounds had been prepared as part of a diversity-oriented synthesis (DOS) library, with a SNAr reaction as the key ring-forming step (Scheme 1).29 Since only a selection of the original DOS library had been included in our primary assay, we began our follow-up studies by testing DMSO stock solutions of each of the stereoisomers of 1a for activity. The 2R, 5R, 6R stereochemistry of 1a provided the best results (Table 1); compounds with different stereochemistry at the carbons of the 8-membered ring (carbons 5 and 6 according to our convention29) suffered from a sizable drop in potency. Inversion of stereochemistry at the 2-position caused a less substantial drop in activity (compound 1ad).
Table 1.
SSAR of lead compound 1a
| ||
|---|---|---|
| Cmp | Stereochem. | IC50(μM)a |
| 1a | 2R, 5R, 6R | 0.18 |
| 1ab | 2R, 5R, 6S | 11.5 |
| 1ac | 2R, 5S, 6R | >25 |
| 1ad | 2S, 5R, 6R | 0.91 |
| 1ae | 2S, 5S, 6S | 17.4 |
| 1af | 2S, 5S, 6R | 12.3 |
| 1ag | 2S, 5R, 6S | 10.0 |
| 1ah | 2R, 5R, 6S | 8.9 |
Average of at least two measurements in DiI uptake assay with DMSO stock solutions.
Scheme 1.
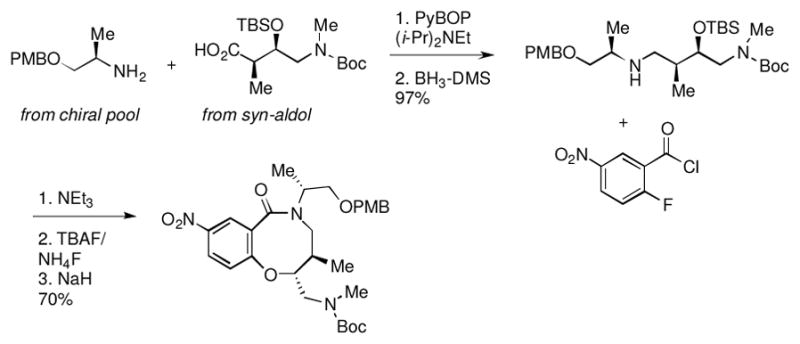
Previous synthesis of core scaffold for library synthesis via SNAr reaction
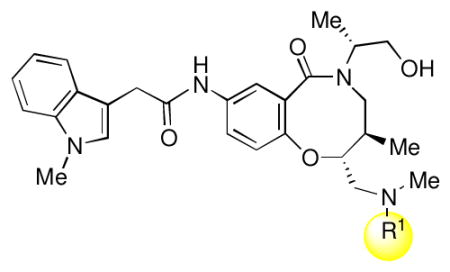
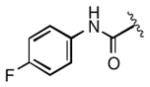
Full SAR studies continued with reverse-phase HPLC repurification of a large number analogs of 1a with the 2R, 5R, 6R stereochemistry. First, the substituents of the southern amine were examined. A selection of these results is given in Table 2. Our lead compound, 4-chlorophenylsulfonamide 1a, inhibited DiI-HDL uptake with IC50 = 0.46 μM when retested as a purified dry powder. The 4-fluorophenyl analog 1b was comparable in potency (IC50 = 0.56 μM), but the larger and more electron-rich 4-methoxy analog 1c suffered a significant decrease in potency (IC50 = 2.0 μM). The unsubstituted benzenesulfonamide 1d also showed good potency, but in early assays it was not superior to the 4-chloro analog 1a. Interestingly, thiophene 1e was not a useful isostere (IC50 = 2.4 μM). One liability of 1a is its low solubility (0.1 μM in PBS with 1% DMSO). The imidazole analog 1f possessed good solubility (79 μM), but was only weakly potent (IC50 = 6.4 μM). The truncated methylsulfonamide 1g also showed good solubility but weak potency. A series of ureas from the original DOS library (including 1h and 1i) were mediocre inhibitors. Many amine compounds with both small (1j, 1k) and larger (1l, 1m, 1n, 1p, 1r) N-alkyl substituents were also mediocre, though the 4-CF3 benzyl analog 1o showed moderate levels of inhibition (IC50 = 0.65 μM).
Table 2.
SAR of southern amine substituent
| ||
|---|---|---|
| Cmp | R1 | IC50(μM)a |
| 1a |
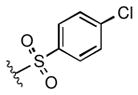
|
0.46 |
| 1b |
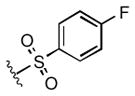
|
0.56 |
| 1c |
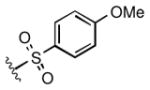
|
2.0 |
| 1d |
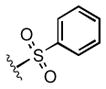
|
0.22 |
| 1e |
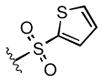
|
2.4 |
| 1f |
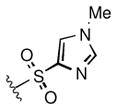
|
6.4 |
| 1g |
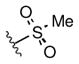
|
9.9 |
| 1h |
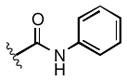
|
4.9 |
| 1i |
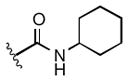
|
9.2 |
| 1j |
|
>25 |
| 1k |
|
18 |
| 1l |
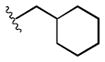
|
9.2 |
| 1m |
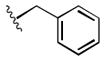
|
1.6 |
| 1n |
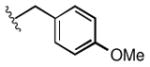
|
2.6 |
| 1o |
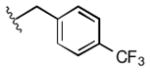
|
0.65 |
| 1p |
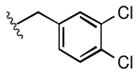
|
>25 |
| 1q |
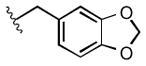
|
0.89 |
| 1r |
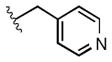
|
1.3 |
Average of at least two measurements in DiI uptake assay with resynthesized or repurified compounds.
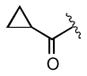
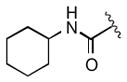
Keeping the southern 4-chlorophenylsulfonamide of 1a in place, we proceeded to test analogs with variable substitutions at the western nitrogen (Table 3). This region is very sensitive to modification, as moving from the cyclohexyl amide 2e (IC50 = 0.054 μM), to the less bulky and lipophilic cyclopropyl amide 2g (IC50 = 2.1 μM), gave a nearly 40-fold drop in potency. It is evident that lipophilic, uncharged substituents are highly preferred at this location. The trifluoromethyl substituted amide 2b showed moderate inhibition (IC50 = 0.26 μM), but molecules with terminal oxygens or nitrogens that can be protonated or accept hydrogen bonds were poor inhibitors (2c, 2d, 2f, 2h, 2n). The lone exceptions observed were the isoxazole sulfonamide 2t (IC50 = 0.19 μM), which has a unique steric and electronic environment about its terminal nitrogen, and the 4-methoxyphenyl urea 2m (IC50 = 0.30 μM). Several other ureas also showed good activity, including 2l (IC50 = 0.092 μM), 2o (IC50 = 0.35 μM), and the isopropyl urea 2p (IC50 = 0.10 μM). Several sulfonamides at this position that were tested showed weak inhibition (2q to 2s). Interestingly, the highly simplified N,N-dimethylaniline 8 (Table 4) gave modest inhibition (IC50 = 0.63 μM).
Table 3.
SAR of western amine substituent
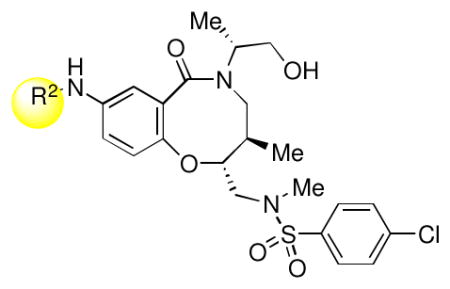
| ||
|---|---|---|
| Cmp | R1 | IC50(μM)a |
| 1a |
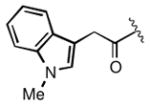
|
0.46 |
| 2b |
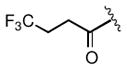
|
0.26 |
| 2c |
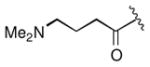
|
>25 |
| 2d |
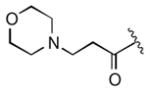
|
5.0 |
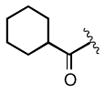
| 2e |
|
0.054 |
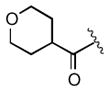
| 2f |
|
>25 |
| 2g |
|
2.1 |
| 2h |
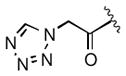
|
10 |
| 2i |
|
>25 |
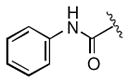
| 2l |
|
0.092 |
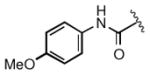
| 2m |
|
0.30 |
| 2n |
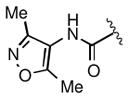
|
7.1 |
| 2o |
|
0.35 |
| 2p |
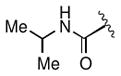
|
0.10 ± 0.02b |
| 2q |
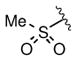
|
1.6 |
| 2r |
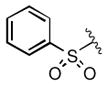
|
3.2 |
| 2s |
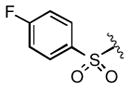
|
14 |
| 2t |
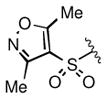
|
0.19 |
Average of at least two measurements in DiI uptake assay with resynthesized or repurified compounds.
Standard error of mean, N = 3.
Table 4.
Various analogs
| Cmp | Structure | IC50(μM)a |
|---|---|---|
| 3 |
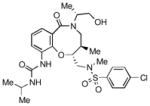
|
>25 |
| 4 |
|
>25 |
| 5 |
|
>25 |
| 6 |
|
0.80 |
| 7 |
|
0.18 |
| 8 |
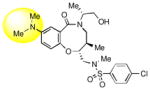
|
0.63 |
| 9 |
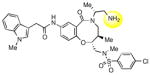
|
>25 |
| 10 |
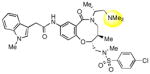
|
>25 |
| 11 |
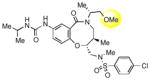
|
8.0 |
| 12 |
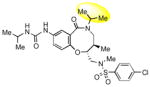
|
9.9 |
Average of at least two measurements in DiI uptake assay.
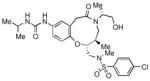
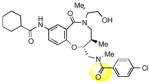
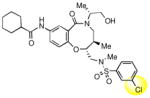
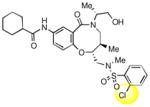
To obtain additional insights into the functionality and structural requirements for inhibition of SR-BI, several additional analogs were prepared and tested during the course of this project (Table 4). Changing the position on the benzene ring of the western amine substituent gave an inactive analog (3) of isopropyl urea 2p. Expansion of the lactam scaffold from an 8-membered ring to a 9-membered ring (4) also abrogated the activity. Further investigation of the southern nitrogen substituent indicated that a sulfonamide is optimal, as the southern amide analog 5 of sulfonamide 2e was inactive. The position of the chlorine substituent was also examined with respect to the western cyclohexyl amide 2e. 3-Chloro (6, IC50 = 0.80 μM) and 2-chlorophenyl (7, IC50 = 0.18 μM) analogs showed decreased inhibition relative to 2e.
The hydroxyl group present in all compounds reported thus far served as an anchor for attachment of the DOS scaffolds to a solid support for library preparation,30 and also for attachment of library compounds to solid surfaces for small molecule microarray screens.31 Therefore, the SAR around this region remained unexplored. Treatment of 1a with DPPA and DBU, followed by Staudinger reduction with triphenylphosphine in THF/water, provided primary amine 9, which proved to be inactive. Displacement of the intermediate azide with dimethylamine gave the tertiary amine 10, which was also inactive. The methyl ether analog of 2p was also prepared and found to be only weakly active (11). Limited attempts at reductive deoxygenation of 2p were unsuccessful, so the deoxy analog 12 was prepared via a de novo route (see Supporting Information) and also found to have weak activity, suggesting that the hydroxyl group is required for substantial inhibition of SR-BI with this scaffold.
The water solubility of the lead compound in this series (1a, 0.1 μM in PBS) was reminiscent of the poor water solubility of one of our previous probes (ML278, 0.57 μM in PBS). Fortunately, several of the potent analogs screened in these studies showed much higher solubility. In particular, the diisopropyl urea 2p exhibited the best combination of potency (average IC50 = 0.10 μM, Supporting Information Figure S1) and solubility (79 μM in PBS). As this compound was present in our original DOS library along with all its stereoisomers, we were able to confirm that the 2R, 5R, 6R stereochemistry of 2p is optimal, in line with the results with 1a. 2p was nominated as Molecular Libraries Probe ML312. Its re-synthesis is outlined in Scheme 2.
Scheme 2.
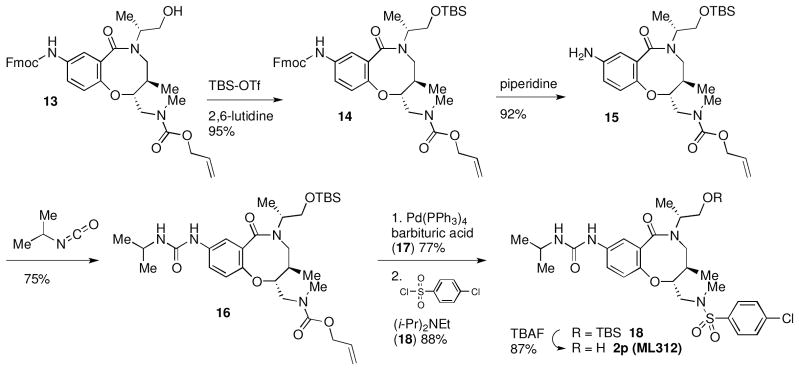
Synthesis of 2p (ML312)
Our most promising compounds in this project were profiled in a series of secondary assays to gain insights into their mode of action and potential for further development. None of the compounds in Tables 1 to 4 showed any measurable cytotoxicity after incubation with the cell line used in our assays (ldlA[mSR-BI]) for 24 h, using a CellTiter-Glo assay (Promega) to measure cellular ATP levels. ML312 also exhibited excellent plasma stability, with >99% remaining after 5 h incubation with human or mouse plasma. Plasma protein binding (PPB) studies showed that it was 94.5% bound in human plasma, and 94.7% bound in mouse plasma. ML312 has limited metabolic stability, with 25% and 10% remaining after incubation with mouse and human liver microsomes for 1 h.
Additional studies with ML312 were performed to study its mode of action. In addition to measuring the uptake of DiI from DiI-HDL in the presence of ML312, the uptake of [3H]-labeled cholesteryl oleate ester ([3H]CE) was measured. ML312 inhibited [3H]CE uptake with a measured IC50 of 0.25 μM (Supporting Information Figure S2), a result superior to the clinical compound ITX-5061, but inferior to our alternative probes ML278 and ML279. The binding of Alexa-488-labeled HDL particles to ldlA[mSR-BI] cells via SR-BI was also measured. As with our prior probes, ML312 enhanced the level of HDL binding to SR-BI, with an EC50 of 0.4 μM (Supporting Information Figure S3), a phenomenon that we have observed previously.21 One possibility is that such compounds act to inhibit lipid transport by preventing release of the bound HDL particles from the receptor. Finally, to determine if ML312 might have inhibited SR-BI-mediated lipid uptake by blocking receptor-mediated endocytosis, the endocytosis of Alexa-594-labeled transferrin was monitored. ML312 showed no inhibition of endocytosis by ldlA[mSR-BI] cells at concentrations up to 35 μM. Additionally, binding studies were performed with a panel of 67 different receptors and secondary targets (see Supporting Information for details). At a concentration of 10 μM, no targets were inhibited by 20% or more, with the highest level of inhibition observed with the L-type Ca2+ channel (19% inhibition). It thus appears to have better selectivity than our previously reported probe ML278, perhaps due to its multiple stereocenters and more complex 3D structure.32
In summary, potent inhibitors of SR-BI-mediated lipid (DiI) uptake were discovered as part of the NIH Molecular Libraries Probe Production Centers Network (MLPCN) initiative. Profiling and SAR analyses of several top compounds led to the nomination of the benzo-fused lactam 2p (ML312) as a probe compound for SR-BI.33 ML312 has superior solubility to the other probes identified in this project, is not cytotoxic, has no significant chemical liabilities, and appears to act selectively at SR-BI, though it is less potent than ML278 (IC50 = 6 nM) or ML279 (IC50 = 17 nM) in the primary assay.
Supplementary Material
Acknowledgments
We thank Eamon Comer for providing compound 13 and Carrie Mosher, Travis Anthoine, and Mike Lewandowski for analytical chemistry support.
Footnotes
Preparation and characterization of 2p (ML312), 11, and 12; compound profiling protocols; representative dose-response curves of ML312 in DiI-HDL uptake assay, [3H]CE uptake assay, and HDL binding assay; assay protocols; and secondary target screening data can be found at http://dx.doi.org/10.1016/j.bmcl.2015.XX.XXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Science. 1996;271:518. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 2.Rigotti A, Miettinen HE, Krieger M. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 3.Trigatti B, Rayburn H, Viñals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Proc Nat Acad Sci USA. 1999;96:9322. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitayama K, Nishizawa T, Abe K, Wakabayashi K, Oda T, Inaba T, Amemiya Y. J Pharm Pharmacol. 2006;58:1629. doi: 10.1211/jpp.58.12.0010. [DOI] [PubMed] [Google Scholar]
- 5.Voisset C, Callens N, Blanchard E, Op De Beeck A, Dubuisson J, Vu-Dac N. J Biol Chem. 2005;280:7793. doi: 10.1074/jbc.M411600200. [DOI] [PubMed] [Google Scholar]
- 6.Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, Vitelli A, Nicosia A. J Virol. 2007;81:8063. doi: 10.1128/JVI.00193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, Nicosia A. J Virol. 2010;84:34. doi: 10.1128/JVI.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues CD, et al. Cell Host & Microbe. 2008;4:271–282. doi: 10.1016/j.chom.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Yalaoui S, Huby T, Franetich JF, Gego A, Rametti A, Moreau M, Collet X, Siau A, van Gemert GJ, Sauerwein RW, Luty AJ, Vaillant JC, Hannoun L, Chapman J, Mazier D, Froissard P. Cell Host & Microbe. 2008;4:283. doi: 10.1016/j.chom.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Wong-Staal F, Lee H, Syder A, McKelvy J, Schooley RT, Wyles DL. J Infect Dis. 2012;205:656. doi: 10.1093/infdis/jir802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulkowski MS, Kang M, Matining R, Wyles D, Johnson VA, Morse GD, Amorosa V, Bhattacharya D, Coughlin K, Wong-Staal F, Glesby MJ. J Infect Dis. 2014;209:658. doi: 10.1093/infdis/jit503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fioravanti J, Medina-Echeverz J, Berraondo P. Immunotherapy. 2011;3:395. doi: 10.2217/imt.10.104. [DOI] [PubMed] [Google Scholar]
- 13.Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, Bernard P, Daugherty A, Huang B, Li XA. J Biol Chem. 2009;284:19826. doi: 10.1074/jbc.M109.020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu P, Liu X, Treml LS, Cancro MP, Freedman BD. J Biol Chem. 2009;284:22878. doi: 10.1074/jbc.M109.018580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu M, Romer KA, Nieland TJ, Xu S, Saenz-Vash V, Penman M, Yesilaltay A, Carr SA, Krieger M. Proc Nat Acad Sci USA. 2011;108:12243. doi: 10.1073/pnas.1109078108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papale GA, Hanson PJ, Sahoo D. Biochemistry. 2011;50:6245. doi: 10.1021/bi2005625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaidukov L, Nager AR, Xu S, Penman M, Krieger M. J Biol Chem. 2011;286:18452. doi: 10.1074/jbc.M111.229872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S, Dhe-Paganon S. Nature. 2013;504:172. doi: 10.1038/nature12684. [DOI] [PubMed] [Google Scholar]
- 19.Nieland TJ, Penman M, Dori L, Krieger M, Kirchhausen T. Proc Nat Acad Sci USA. 2002;99:15422. doi: 10.1073/pnas.222421399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. [accessed February 4, 2015]; See http://www.ncbi.nlm.nih.gov/books/NBK133420/
- 21.Dockendorff C, Faloon PW, Yu M, Youngsaye W, Penman M, Nieland TJF, Nag PP, Lewis TA, Pu J, Bennion M, Negri J, Paterson C, Lam G, Dandapani S, Perez JR, Munoz B, Palmer MA, Schreiber SL, Krieger M. ACS Med Chem Lett. 2015 doi: 10.1021/ml500154q. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. [accessed February 4, 2015]; http://www.ncbi.nlm.nih.gov/books/NBK143554/
- 23.Dockendorff C, Faloon PW, Germain A, Yu M, Youngsaye W, Nag PP, Bennion M, Nieland TJF, Dandapani S, Perez JR, Munoz B, Palmer MA, Schreiber SL, Krieger M. Bioorg. Med. Chem. Lett. 2015 doi: 10.1016/j.bmcl.2015.03.074. submitted (insert details here) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masson D, Koseki M, Ishibashi M, Larson CJ, Miller SG, King BD, Tall AR. Arterioscler Thromb Vasc Biol. 2009;29:2054. doi: 10.1161/ATVBAHA.109.191320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong-Staal F, Syder AJ, McKelvy JF. Viruses. 2010;2:1718. doi: 10.3390/v2081718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, McKelvy J, Wong-Staal F. J Hepatol. 2011;54:48. doi: 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Mittapalli GK, Jackson A, Zhao F, Lee H, Chow S, McKelvy J, Wong-Staal F, Macdonald JE. Bioorg Med Chem Lett. 2011;21:6852. doi: 10.1016/j.bmcl.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 28.Mittapalli GK, Zhao F, Jackson A, Gao H, Lee H, Chow S, Pal Kaur M, Nguyen N, Zamboni R, McKelvy J, Wong-Staal F, Macdonald JE. Bioorg Med Chem Lett. 2012;22:4955. doi: 10.1016/j.bmcl.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 29.Marcaurelle LA, et al. J Am Chem Soc. 2010;132:16962. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryba TD, Depew KM, Marcaurelle LA. J Comb Chem. 2009;11:110. doi: 10.1021/cc8000986. [DOI] [PubMed] [Google Scholar]
- 31.Miao H, Tallarico JA, Hayakawa H, Munger K, Duffner JL, Koehler AN, Schreiber SL, Lewis TA. J Comb Chem. 2007;9:245. doi: 10.1021/cc060135m. [DOI] [PubMed] [Google Scholar]
- 32.Lovering F, Bikker J, Humblet CJ. Med Chem. 2009;52:6752. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 33. [accessed February 4, 2015]; http://www.ncbi.nlm.nih.gov/books/NBK143554/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.