

Abstract
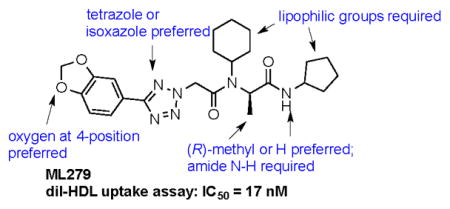
A new series of potent inhibitors of cellular lipid uptake from HDL particles mediated by Scavenger Receptor, Class B, Type I (SR-BI) was identified. The series was identified via a high-throughput screen of the National Institutes of Health Molecular Libraries Small Molecule Repository (NIH MLSMR) that measured the transfer of the fluorescent lipid DiI from HDL particles to CHO cells overexpressing SR-BI. The series is characterized by a linear peptidomimetic scaffold with two adjacent amide groups, as well as an aryl-substituted heterocycle. Analogs of the initial hit were rapidly prepared via Ugi 4-component reaction, and select enantiopure compounds were prepared via a stepwise sequence. Structure-activity relationship (SAR) studies suggest an oxygenated arene is preferred at the western end of the molecule, as well as highly lipophilic substituents on the central and eastern nitrogens. Compound 5e, with (R)-stereochemistry at the central carbon, was designated as probe ML279. Mechanistic studies indicate that ML279 stabilizes the interaction of HDL particles with SR-BI, and its effect is reversible. It shows good potency (IC50 = 17 nM), is non-toxic, plasma stable, and has improved solubility over our alternative probe ML278.
Keywords: ML279, SR-BI inhibitor, HDL receptor, cholesterol transport, HCV
Graphical Abstract
The trafficking of lipids (e.g., cholesterol and its esters) between tissues is critical for lipid homeostasis as well as steroidogenesis. The key players in this transport system include lipoprotein particles (e.g., LDL and HDL) that carry the water-insoluble lipids through the bloodstream, the enzymes that modify the lipid cargos and/or help to transfer them from one particle to another (including LCAT and CETP), and the receptors for the lipoprotein particles that serve to transfer the lipids into and out of cells (i.e., uptake and efflux). The cellular receptor for high-density lipoprotein (HDL) particles, Scavenger Receptor class B, type I (SR-BI),1–2 has been studied in detail in recent years,3–5 however a complete picture of its mode of action is still incomplete, despite related structural data.6 SR-BI has important effects on cardiovascular physiology,7 as well as pathogen entry (e.g., Hepatitis C virus),8–10 immune response,11–13 and female fertility.7
We herein report our discovery of a second class of SR-BI inhibitors that shows distinct advantages over those previously discovered in our labs (e.g., BLT-1 and BLT-3).14 Concurrent with this work was our discovery of the indoline-thiazole ML278,15–16 followed soon after by the discovery of the benzofused lactams represented by ML312,17 described in the companion paper in this journal.18 Several other inhibitors of SR-BI have been reported, including HDL376,19–20 ITX-5061,21–24 R-138329, and R-154716 (Figure 1).25 Recently, researchers at iTherX reported additional HCV entry inhibitors, including ITX-4520, which is postulated to be an inhibitor of SR-BI.26–27 Our discovery of the bisamide inhibitors described herein was undertaken as part of the NIH Molecular Libraries Program (MLP).28
Figure 1.
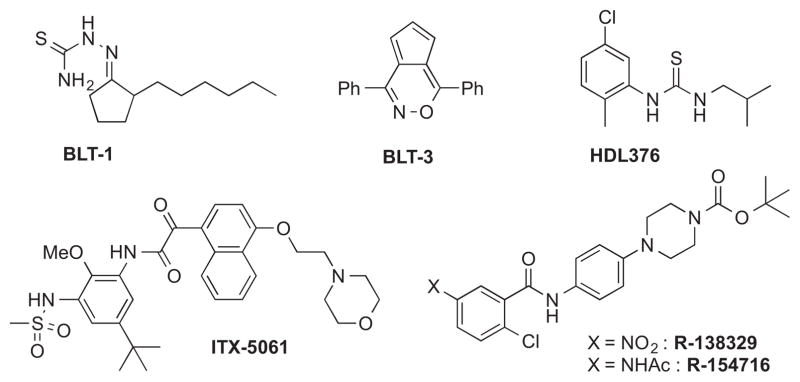
Known inhibitors of SR-BI.
To elucidate more details about the mechanism of lipid uptake and efflux via SR-BI, and to potentially identify less toxic and more potent small molecule probes, we undertook a high-throughput screening (HTS) campaign measuring the uptake of the fluorescent lipid surrogate 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) from HDL particles into CHO cells overexpressing mouse SR-BI (ldlA[mSR-BI]).14 3,046 compounds (0.96%) were classified as inhibitors of DiI-HDL uptake out of 319,533 compounds tested, with inhibition at 12.5 μM of ≥ 70% relative to 1 μM BLT-1 as positive control. Hit compounds were omitted that were on plates with Z′ < 0.3 or that were active in 10% or more of the HTS assays listed in PubChem. A counterscreen was also performed that rejected hit compounds that quenched the fluorescence of DiI-HDL in a dose-dependent manner.
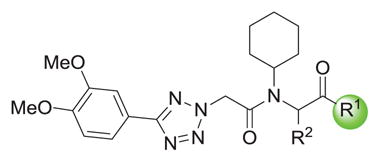
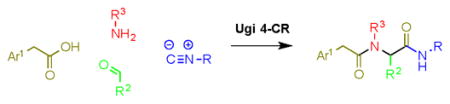
Of the numerous scaffolds verified to inhibit DiI uptake with IC50 of < 1 μM, we focused our efforts in part on a bisamide-tetrazole series characterized by the commercially-available compound 1a,29 with IC50 of 0.055 μM in the primary assay (Table 1). We deduced that such compounds could be made rapidly via Ugi 4-component coupling reactions (eq. 1),30–32 and thus would be amenable to rapid structure-activity relationship (SAR) studies. 1a also showed no measurable cytotoxicity in the cell line used for our assays (ldlA[mSR-BI]) after incubation for 24 h. Additionally, 1a lacked non-selective behavior according to data published in PubChem.33 Finally, in contrast to our hit compounds in the indoline-thiazole series,16–17 members of the bisamide-tetrazole series showed improved aqueous solubility (114 μM for 1a in PBS with 1% DMSO).
Table 1.
Amide analogs
| ||
|---|---|---|
| Cmp | R1 | IC50 (μM)a |
| 1a |
|
0.055±0.014 |
| 1c |
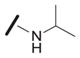
|
0.81 |
| 1e |
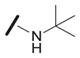
|
17 |
| 1g |
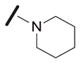
|
7.1 |
| 1b |
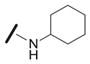
|
0.20 |
| 1d |
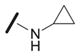
|
0.42 |
| 1f |
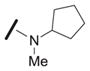
|
1.6 |
Average of at least two measurements in DiI uptake assay, ± standard error of mean when n > 2.
 |
(1) |
We explored the SAR of the dipeptide scaffold by varying each of the components of the Ugi reaction. Reactions were typically performed using a 1 : 1 : 1 : 1 ratio of aldehyde, amine, carboxylic acid, and isonitrile in methanol, and stirring for 18 h. Variation of the isonitrile component allowed us to explore the eastern amide of the scaffold (Table 1). The SAR in this area was very sensitive to modifications, as slight changes in structure from N-cyclopentyl diminished the activity substantially, such as with N-cyclohexyl 1b (IC50 = 0.20 μM) and N-isopropyl 1c (0.81 μM). The amide N-H is required for high activity, as the tertiary amide 1f had low activity. Compound 1f was prepared via a sequential peptide synthesis, as selective methylation of 1a proved to be problematic.
Our results exploring tetrazole replacements are summarized in Table 2. Replacement of the aryl tetrazole with a simple phenyl group (2a) abrogated activity. Several heterocyclic replacements for the tetrazole were prepared from commercially available arylacetic acids: oxazole 2b showed approximately two-fold decreased activity (IC50 = 0.13 μM), thiazole 2c possessed poor activity (IC50 = 6.3 μM), and isoxazole 2d demonstrated excellent potency (IC50 = 0.023 μM). One possible explanation for these results is that a hydrogen-bond acceptor at the 4-position (numbering of tetrazole ring system) is required for optimal activity. Despite the excellent potency with isoxazole 2d, we elected to continue our studies with the tetrazole analogs, for which we had a larger collection of building blocks.
Table 2.
Tetrazole replacements
| ||
|---|---|---|
| Cmp | Ar1 | IC50 (μM)a |
| 1a |
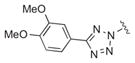
|
0.055±0.014 |
| 2b |
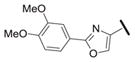
|
0.13 |
| 2d |
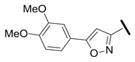
|
0.023 |
| 2a |
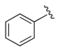
|
37 |
| 2c |
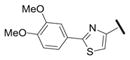
|
6.3 |
Average of at least two measurements in DiI uptake assay, ± standard error of mean when n > 2.
Next, the western end of the scaffold was examined in detail by varying the aromatic substituent (Table 3). Removal of the 4-methoxy group of 1a led to a significant drop in potency (3b, IC50 = 0.18 μM). Several 2- and 3-substituted arenes were examined with both electron-withdrawing and donating groups; these were uniformly inferior to the 3,4-dimethoxyarene of 1a.
Table 3.
Arene SAR
| ||
|---|---|---|
| Cmp | Ar2 | IC50 (μM)a |
| 1a |
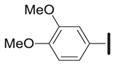
|
0.055±0.014 |
| 3b |
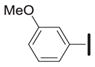
|
0.18 |
| 3d |
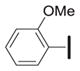
|
>25 |
| 3f |
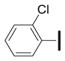
|
3.8 |
| 3h |
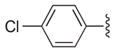
|
0.17 |
| 3j |
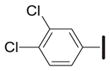
|
0.18 |
| 3l |
|
0.23 |
| 3a |
|
0.37 |
| 3c |
|
0.38 |
| 3e |
|
0.62 |
| 3g |
|
0.49 |
| 3i |
|
0.13 |
| 3k |
|
0.027±0.008 |
Average of at least two measurements in DiI uptake assay, ± standard error of mean when n > 2.
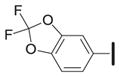
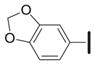
Several 4-substituents were tolerated to some extent, including chlorine (3h, IC50 = 0.17 μM) and fluorine (3i, IC50 = 0.13 μM). The best results were obtained with the 3,4-methylenedioxy group (3k, IC50 = 0.027 μM), which is more potent than 1a. We also examined the difluoromethylene analog of 3k (3l, IC50 = 0.23 μM). Its decreased activity relative to 1a, along with the decreased activity of the 4-methyl analog (3g, IC50 = 0.49 μM), suggests that an appropriate 4-aryl substituent may act as a hydrogen bond acceptor with the target.
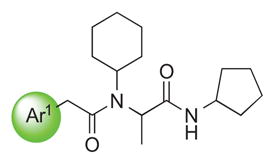
We examined the effects of varying internal positions of the structure while maintaining the tetrazole along with either a terminal 3,4-dimethoxyphenyl or 3,4-methylenedioxyphenyl group at the western end of the scaffold. A variety of amines were screened in the Ugi reaction, leading to different substituents at R4 (Table 4). The cyclohexyl substituent of 1a again proved optimal. Conservative changes were tolerated to some degree (such as 4c, R4 = cyclopentyl, IC50 = 0.11 μM), but clearly a bulky, lipophilic group is optimal. Inserting a carbon atom between the cyclohexyl group and the backbone nitrogen decreased activity by greater than 20-fold (4d, IC50 = 1.6 μM). Introducing a nitrogen atom expected to be protonated at physiological pH abrogated all activity (4e).
Table 4.
SAR of central amide N-substituent
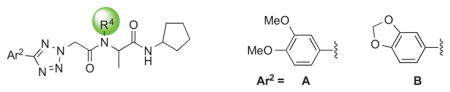
| ||
|---|---|---|
| Cmp | R4 | IC50 (μM)a |
|
1a Ar2=A |
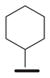
|
0.055±0.014 |
|
4b Ar2=A |
|
7.5 |
|
4d Ar2=A |
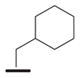
|
1.6 |
|
4f Ar2=B |
|
0.84 |
|
4a Ar2=A |
|
19 |
|
4c Ar2=A |
|
0.11 |
|
4e Ar2=A |
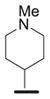
|
>25 |
Average of at least two measurements in DiI uptake assay, ± standard error of mean when n > 2.
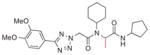
Next, substitution at the alpha carbon atom of the eastern amide was investigated (Table 5). Introducing a second methyl group gave a drastic drop in activity (5a), as did the replacement of the methyl group of 1a with a bulkier isopropyl group (5b). Compound 5c, lacking the methyl group of the parent compound 3k, showed slightly improved activity (IC50 = 0.027 μM).
Table 5.
SAR analysis of central carbon substituents
| Cmp | Structure | IC50 (μM)a |
|---|---|---|
| 1a |
|
0.055±0.014 |
| 5a |
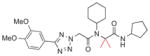
|
19 |
| 5b |
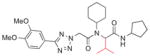
|
>25 |
| 3k |
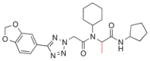
|
0.027±0.008 |
| 5c |
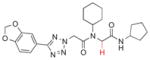
|
0.026±0.012 |
| 5d |
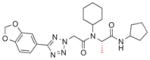
|
1.6 |
| 5e |
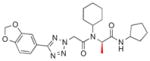
|
0.017±0.004 |
Average of at least two measurements in DiI uptake assay, ± standard error of mean when n > 2.
Finally, each enantiomer of 3k was prepared in a stepwise fashion from (R)- or (S)-alanine (Scheme 1). The required tetrazole acetic acid building block was made by cycloaddition between benzonitrile 6 and sodium azide, then alkylation with methyl bromoacetate and hydrolysis of the resulting ester to give 8. (R)-N-Boc alanine (9) was then coupled with aminocyclopentane using standard peptide coupling conditions (EDC, HOBt, (i-Pr)2NEt). The resulting amide 10 was treated with TFA to liberate the amine, then subjected to reductive alkylation with cyclohexanone and sodium cyanoborohydride to generate the N-cyclohexylamine 12. Peptide coupling reactions with this relatively hindered secondary amine were sluggish, so 12 was reacted with the acid chloride of tetrazole acetic acid 8 to generate the desired bisamide 5e. The enantiomer 5d was prepared analogously from (S)-N-Boc-alanine. The (R)-isomer 5e (IC50 = 0.017 μM) was 100-fold more potent than its enantiomer 5d.
Scheme 1.

Synthesis of ML279
Additional studies with 5e were performed to study its mode of action. Its inhibitory action is reversible. In experiments where cells were pre-treated with ML279 for 2 hours, washed extensively with PBS and then incubated with DiI-HDL, no inhibition was observed. In addition to measuring the uptake of DiI from HDL in the presence of 5e, the uptake of 3H-labeled cholesteryl oleate ester ([3H]CE) from [3H]CE–HDL was measured with an IC50 of 0.005 μM (Supporting Information Figure 2), and is significantly more potent than the clinical compound ITX-5061 in a head-to-head study. The binding of Alexa-488-labeled HDL particles to ldlA[mSR-BI] cells via SR-BI was also measured. As with BLT-1 and other inhibitors, including our recently disclosed probe ML278, 5e enhanced the level of HDL binding to SR-BI, with a measured EC50 of 0.27 μM (Supporting Information Figure 3). One possibility is that such compounds act to inhibit lipid transport by slowing the turnover (release) of the bound HDL particles. Finally, we tested to see if 5e was a general inhibitor of receptor-mediated endocytosis by examining its effects on the endocytosis of Alexa-594-labeled transferrin by ldlA[mSR-BI] cells. 5e showed no inhibition of this process, at concentrations up to 35 μM. This result is consistent with the previous studies that indicated that SR-BI does not mediate lipid uptake via receptor-mediated endocytosis.34
In summary, potent inhibitors of SR-BI-mediated lipid uptake were discovered as part of the NIH Molecular Libraries Probe Production Centers Network (MLPCN) initiative. Profiling of several top compounds led to the nomination of the bisamide tetrazole 5e (ML279) as a probe compound. ML279 has superior solubility to ML278 (28 μM vs. 0.57 μM), though it is slightly less potent than ML278 (IC50 = 17 vs. 6 nM in the diI-uptake assay). It is also not cytotoxic, has no significant chemical liabilities, shows reversible inhibition, and appears to be selective, as determined by inspection of PubChem assay results. ML279 is plasma stable, with >99% remaining after incubation with human or mouse plasma, though it suffers from a lack of metabolic stability as determined in a microsomal stability assay (<1% remaining after 1 h with mouse or human microsomes). Altogether, ML279 represents a promising lead compound for the blocking of SR-BI.
Supplementary Material
Acknowledgments
We thank Stephen Johnston, Carrie Mosher, Travis Anthoine, and Mike Lewandowski for analytical chemistry support.
Footnotes
General protocol for Ugi reactions, preparation and characterization of 5e (ML279), compound profiling protocols, representative dose-response curves of ML279 in DiI-HDL, [3H]CE uptake, and HDL binding assays, and assay protocols can be found at http://dx.doi.org/10.1016/j.bmcl.2015.XX.XXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Science. 1996;271:518. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 2.Rigotti A, Miettinen HE, Krieger M. Endocr Rev. 2003;24:357. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 3.Yu M, Romer KA, Nieland TJ, Xu S, Saenz-Vash V, Penman M, Yesilaltay A, Carr SA, Krieger M. Proc Nat Acad Sci USA. 2011;108:12243. doi: 10.1073/pnas.1109078108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papale GA, Hanson PJ, Sahoo D. Biochemistry. 2011;50:6245. doi: 10.1021/bi2005625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaidukov L, Nager AR, Xu S, Penman M, Krieger M. J Biol Chem. 2011;286:18452. doi: 10.1074/jbc.M111.229872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S, Dhe-Paganon S. Nature. 2013;504:172. doi: 10.1038/nature12684. [DOI] [PubMed] [Google Scholar]
- 7.Trigatti B, Rayburn H, Viñals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Proc Nat Acad Sci USA. 1999;96:9322. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voisset C, Callens N, Blanchard E, Op De Beeck A, Dubuisson J, Vu-Dac N. J Biol Chem. 2005;280:7793. doi: 10.1074/jbc.M411600200. [DOI] [PubMed] [Google Scholar]
- 9.Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, Vitelli A, Nicosia A. J Virol. 2007;81:8063. doi: 10.1128/JVI.00193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, Nicosia A. J Virol. 2010;84:34. doi: 10.1128/JVI.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fioravanti J, Medina-Echeverz J, Berraondo P. Immunotherapy. 2011;3:395. doi: 10.2217/imt.10.104. [DOI] [PubMed] [Google Scholar]
- 12.Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, Bernard P, Daugherty A, Huang B, Li XA. J Biol Chem. 2009;284:19826. doi: 10.1074/jbc.M109.020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu P, Liu X, Treml LS, Cancro MP, Freedman BD. J Biol Chem. 2009;284:22878. doi: 10.1074/jbc.M109.018580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nieland TJ, Penman M, Dori L, Krieger M, Kirchhausen T. Proc Nat Acad Sci USA. 2002;99:15422. doi: 10.1073/pnas.222421399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. [accessed February 4, 2015]; http://www.ncbi.nlm.nih.gov/books/NBK133420/
- 16.Dockendorff C, Faloon PW, Yu M, Youngsaye W, Penman M, Nieland TJF, Nag PP, Lewis TA, Pu J, Bennion M, Negri J, Paterson C, Lam G, Dandapani S, Perez JR, Munoz B, Palmer MA, Schreiber SL, Krieger M. ACS Med Chem Lett. 2015 doi: 10.1021/ml500154q. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. [accessed February 4, 2015]; http://www.ncbi.nlm.nih.gov/books/NBK143554/
- 18.Dockendorff C, Faloon PW, Pu J, Yu M, Johnston S, Bennion M, Penman M, Nieland TJF, Dandapani S, Perez JR, Munoz B, Palmer MA, Schreiber SL, Krieger M. Bioorg. Med. Chem. Lett. 2015 doi: 10.1016/j.bmcl.2015.03.073. this issue (insert details here) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppola GM, Damon RE, Eskesen B, France DS, Paterniti JR. Bioorg Med Chem Lett. 2006;16:113. doi: 10.1016/j.bmcl.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 20.Nieland TJ, Shaw JT, Jaipuri FA, Maliga Z, Duffner JL, Koehler AN, Krieger M. J Lipid Res. 2007;48:1832. doi: 10.1194/jlr.M700209-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Masson D, Koseki M, Ishibashi M, Larson CJ, Miller SG, King BD, Tall AR. Arterioscler Thromb Vasc Biol. 2009;29:2054. doi: 10.1161/ATVBAHA.109.191320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong-Staal F, Syder AJ, McKelvy JF. Viruses. 2010;2:1718. doi: 10.3390/v2081718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, McKelvy J, Wong-Staal F. J Hepatol. 2011;54:48. doi: 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 24.Sulkowski MS, Kang M, Matining R, Wyles D, Johnson VA, Morse GD, Amorosa V, Bhattacharya D, Coughlin K, Wong-Staal F, Glesby MJ. J Infect Dis. 2014;209:658. doi: 10.1093/infdis/jit503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitayama K, Nishizawa T, Abe K, Wakabayashi K, Oda T, Inaba T, Amemiya Y. J Pharm Pharmacol. 2006;58:1629. doi: 10.1211/jpp.58.12.0010. [DOI] [PubMed] [Google Scholar]
- 26.Mittapalli GK, Jackson A, Zhao F, Lee H, Chow S, McKelvy J, Wong-Staal F, Macdonald JE. Bioorg Med Chem Lett. 2011;21:6852. doi: 10.1016/j.bmcl.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 27.Mittapalli GK, Zhao F, Jackson A, Gao H, Lee H, Chow S, Pal Kaur M, Nguyen N, Zamboni R, McKelvy J, Wong-Staal F, Macdonald JE. Bioorg Med Chem Lett. 2012;22:4955. doi: 10.1016/j.bmcl.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 28. [accessed February 4, 2015]; http://www.ncbi.nlm.nih.gov/books/NBK133438/
- 29.Purchased from Asinex Ltd.
- 30.Ugi I, Meyr R, Fetzer U, Steinbrückner C. Angew Chem. 1959;71:386. [Google Scholar]
- 31.Dömling A. Chem Rev. 2006;106:17. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 32.Marcaccini S, Torroba T. Nature Protocols. 2007;2:632. doi: 10.1038/nprot.2007.71. [DOI] [PubMed] [Google Scholar]
- 33.It was listed as a hit in 4 assays not involving SR-BI out of 614 bioassays listed on December 10, 2011, and a lack of dose-response data on any of these hits means that none had apparently been confirmed as actives.
- 34.Nieland TJ, Ehrlich M, Krieger M, Kirchhausen T. Biochim Biophys Acta. 2005:1734, 44. doi: 10.1016/j.bbalip.2005.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.