

Abstract
A 33-year-old woman with past history of Sjögren's syndrome and systemic lupus erythematosus presented with dyspnea and syncope secondary to pulmonary hypertension. After progressive symptoms over 4 years, she received bilateral lung transplantation. Histopathology of the explanted lungs showed isolated pulmonary amyloid light-chain amyloidosis and pulmonary cysts. No evidence of systemic amyloidosis was found at the time of transplantation. Seven years post lung transplantation, she remains well with no evidence of systemic amyloidosis recurrence.
Keywords: Amyloidosis, pulmonary hypertension, Sjögren's syndrome
Introduction
Amyloidosis causing pulmonary hypertension (PH) is a rare condition, with 10 cases reported 1–3. Most cases of pulmonary amyloidosis have been found in the context of systemic disease (e.g. myeloma). A few cases of isolated pulmonary amyloid light-chain (AL) amyloidosis secondary to systemic lupus erythematosus (SLE) and Sjögren's syndrome (SS) have been described; however, none reported PH as a complication.
Case Report
A 33-year-old woman presented with syncope on a background of progressive dyspnea over 12 months at the New York Heart Association–World Health Organization Functional Class (NYHA-FC) III at presentation.
Past history was of an autoimmune disorder diagnosed 7 years prior – presenting as large joint arthropathy and sicca symptoms – with positive autoantibodies, double-stranded DNA, antinuclear antibody, and extractable nuclear antibody (SS-A/Ro and SS-B/LA). A diagnosis of SLE was made and symptoms were well controlled on hydroxychloroquine. She had two episodes of pulmonary embolism (PE) 5 years before presentation, the second despite daily prophylactic dose of 40 mg of enoxaparin. Her prothrombotic risk factors were autoimmune disease, long haul flights (both occasions), and oral contraceptive pill; she had no lupus anticoagulant and was a lifelong non-smoker.
An echocardiogram showed normal left ventricular size with normal systolic and diastolic function, a dilated right ventricle with moderately reduced function, and a right ventricular systolic pressure of 85 mmHg. A ventilation perfusion scan was normal (excluding chronic thromboembolic disease). Pulmonary function testing showed normal spirometry with severely reduced gas transfer. Computer tomography (CT) pulmonary angiogram previously performed at the time of PE diagnosis was reviewed and showed enlarged pulmonary arteries and some basal cystic changes, but no other parenchymal abnormalities.
A right heart catheterization confirmed PAH with a mean pulmonary arterial pressure of 46 mmHg and pulmonary capillary wedge pressure of 2 mmHg (cardiac index, transpulmonary gradient, and pulmonary vascular resistance were unavailable), thought secondary to connective tissue disease.
Bosentan was commenced with improvement in NYHA-FC class III to II; however, over 24 months she deteriorated, despite addition of sildenafil. Given her deterioration, she began assessment for lung transplantation. A chest CT showed numerous well-defined bilateral pulmonary nodules of 4–5 mm in size and thin-walled cysts (Fig. 1A and one year later Fig. 1B). CT abdomen/pelvis had previously been performed in the setting of lower abdominal pain and showed a uterine fibroid. This was investigated and found to have no evidence of malignancy on serial follow-up. A fluorodeoxyglucose positron emission tomography scan demonstrated moderate tracer uptake in the cervical nodes of the posterior triangle of the neck, axillary and mediastinal lymph nodes, and pulmonary nodules with moderate tracer activity. An axillary lymph node core biopsy was performed and had no malignant cells or granuloma, likely reactive lymphadenopathy. Given the absence of malignancy and worsening symptoms, she was listed for lung transplantation.
Figure 1.
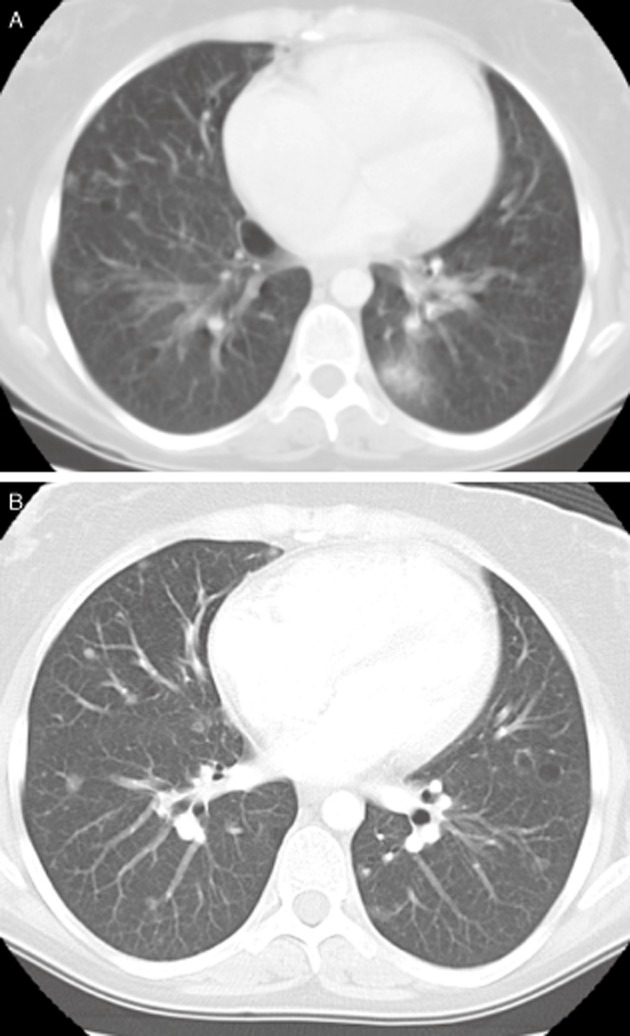
(A) Computed tomography (CT) of the chest at time of initial transplant work up showing cystic lung disease and diffuse fine nodules. (B) CT of the chest showing nodules 6 months following initial work up.
Ultimately 10 months following listing, she received bilateral sequential lung transplantation. Explanted lungs showed no evidence of malignancy, with pulmonary amyloidosis (congo red positive with apple-green birefringence) concentrated around bronchovascular tissues, surrounded by lymphocytes and monotypic plasma cells, and within arterioles (Fig. 2). This was in association with localized cystic structure of 20 mm in diameter with localized amyloid deposits. Amyloid was classified as AL amyloid type by lambda in situ hybridization; there was no transthyretin or serum A amyloid. There were vessel features of PH, but no evidence of vasculitis, pulmonary veno-occlusive disorder, or thromboembolic disease. Amyloidosis secondary to plasma cell dyscrasia was excluded with a normal full blood film, serum protein electrophoresis, serum-free light chains, and 24-h urine collection with no evidence of paraproteins. A bone marrow aspirate was considered but not performed due to an advice from hematology that there was no evidence of a plasma cell dyscrasia on screening tests. The final diagnosis was PH and pulmonary AL amyloidosis both thought secondary to a connective tissue disease (overlap of SLE and SS).
Figure 2.
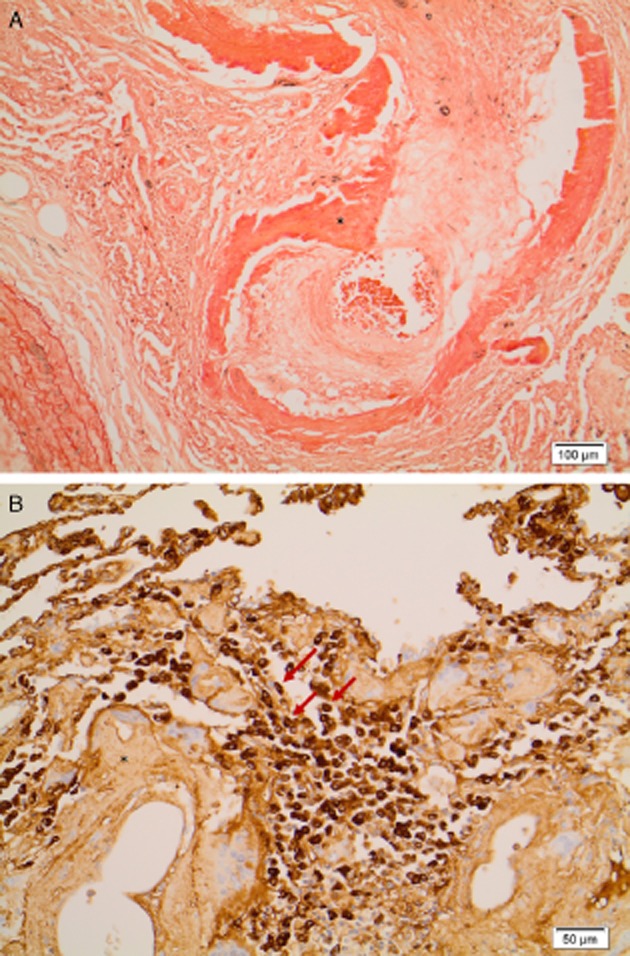
(A) Pulmonary vascular amyloid (*) showing luminal restriction (congo red ×100). (B) Lambda light chain restriction within interstitial plasma cells (arrows) present adjacent to amyloid (*) (lambda light chain immunoperoxidase).
Seven years post transplantation our patient is well, with no evidence of PH on serial echocardiograms and an excellent improvement in functional state. Her ongoing autoimmune disease appears well controlled with negative autoantibodies and only ongoing mild sicca symptoms. There has been no evidence of recurrent amyloidosis or cystic lung disease on subsequent routine imaging or transbronchial lung biopsies.
Discussion
This patient presented with symptoms and auto-antibodies consistent with SLE, a condition known to cause pulmonary vasculitis and PH but rarely associated with pulmonary amyloidosis (usually AA type). Over time our patient evolved symptoms more consistent with SS and ongoing relevant auto-antibodies. The association between SS and cystic interstitial lung disease is establish [4]; however, unlike other connective tissue diseases SS is rarely associated with PH. Baqir et al. [4] described eight cases with SS and thin-walled cysts with a lymphoid infiltrate and light chain-restricted plasma cells +/− amyloid on open lung biopsy similar to our case, however, without PH.
PH due to pulmonary amyloidosis is a rare condition, with only 10 cases described. In a series of nine cases reviewed by Eder et al. [2], the most common type was AL amyloid (78% cases) and all bar one there was evidence of systemic disease such as myeloma or familial Mediterranean fever (see Figure 3 for a summary of the cases).
Figure 3.
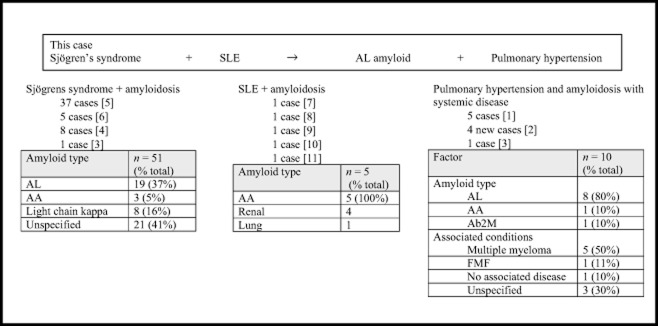
Summary of previous cases reporting pulmonary hypertension, amyloid and autoimmune diseases.
We suggest that this case may represent a rare complication of an autoimmune connective tissue disease. We hypothesize that in secondary SS, a monotypic plasma cell population producing localized AL amyloid developed and caused cystic lung disease plus AL amyloid. The AL amyloid then contributed to PH with severe symptoms necessitating lung transplantation. Unlike the other cases of PH and amyloid where patients had time to death of 73 days, our patient has responded to lung transplantation [2]. We postulate that this difference in prognosis is due to the isolated involvement in an otherwise well young woman, compared with patients with systemic disease driving amyloidosis and that the immunosuppression treatment post transplantation has controlled the underlying autoimmune disease.
This case adds to the small cohort of patients with PH from amyloidosis, however, highlights a different pattern with isolated pulmonary involvement and no evidence of hematologic malignancy. In this case, there has been an excellent response 7 years post transplant, and highlights how screening for systemic disease is important once amyloidosis has been found as it aids in prognostication.
Disclosure Statements
No conflict of interest declared.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
Acknowledgments
Dr. C. M. Ellender's fellowship was partly funded by Actelion Pharmaceuticals.
References
- Dingli D, Utz JP. Gertz MA. Pulmonary hypertension in patients with amyloidosis. Chest. 2001;120:1735–1738. doi: 10.1378/chest.120.5.1735. [DOI] [PubMed] [Google Scholar]
- Eder L, Zisman D, Wolf R, et al. Pulmonary hypertension and amyloidosis – an uncommon association: a case report and review of the literature. J. Gen. Intern. Med. 2007;22:416–419. doi: 10.1007/s11606-006-0052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen J. Kettunen P. Pulmonary hypertension as a dominant clinical picture in a case of amyloidosis and smoldering multiple myeloma. Int. J. Cardiol. 2007;115:e29–e30. doi: 10.1016/j.ijcard.2006.07.059. [DOI] [PubMed] [Google Scholar]
- Baqir M, Kluka EM, Aubry MC, et al. Amyloid-associated cystic lung disease in primary Sjögren's syndrome. Respir. Med. 2013;107:616–621. doi: 10.1016/j.rmed.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Rajagopala S, Singh N, Gupta K, et al. Pulmonary amyloidosis in Sjögren's syndrome: a case report and systematic review of the literature. Respirology. 2010;15:860–866. doi: 10.1111/j.1440-1843.2010.01772.x. [DOI] [PubMed] [Google Scholar]
- Jeong YJ, Lee KS, Chung MP, et al. Amyloidosis and lymphoproliferative disease in Sjögren's syndrome: thin-section computed tomography findings and histopathologic comparisons. J. Comput. Assist. Tomogr. 2004;28:776–781. doi: 10.1097/00004728-200411000-00008. [DOI] [PubMed] [Google Scholar]
- Al-Hoqail I, Naddaf H, Al-Rikabi A, et al. Systemic lupus erythematosus and amyloidosis. Clin. Rheumatol. 1997;16:422–424. doi: 10.1007/BF02242463. [DOI] [PubMed] [Google Scholar]
- Ridley MG, Maddison PJ. Tribe CR. Amyloidosis and systemic lupus erythematosus. Ann. Rheum. Dis. 1984;43:649–650. doi: 10.1136/ard.43.4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queffeulou G, Berenbaum F, Michel C, et al. AA amyloidosis in systemic lupus erythematosus: an unusual complication. Nephrol Dial. Transplant. 1998;13:1846–1848. doi: 10.1093/ndt/13.7.1846. [DOI] [PubMed] [Google Scholar]
- Singh NP, Prakash A, Sridhara GS, et al. Renal and systemic amyloidosis in systemic lupus erythematosus. Ren. Fail. 2003;25:671–675. doi: 10.1081/jdi-120022561. [DOI] [PubMed] [Google Scholar]
- Nomura S, Kumagai N, Kanoh T, et al. Pulmonary amyloidosis associated with systemic lupus erythematosus. Arthritis Rheum. 1986;29:680–682. doi: 10.1002/art.1780290514. [DOI] [PubMed] [Google Scholar]