

Abstract
Dendritic cells (DCs) are promising therapeutic agents in the field of cancer immunotherapy due to their intrinsic immune-priming capacity. The potency of DCs, however, is readily attenuated immediately after their administration in patients as tumours and various immune cells, including DCs, produce various immunosuppressive factors such as interleukin (IL)-10 and transforming growth factor (TGF)-β that hamper the function of DCs. In this study, we used small interfering RNA (siRNA) to silence the expression of endogenous molecules in DCs, which can sense immunosuppressive factors. Among the siRNAs targeting various immunosuppressive molecules, we observed that DCs transfected with siRNA targeting IL-10 receptor alpha (siIL-10RA) initiated the strongest antigen-specific CD8+ T cell immune responses. The potency of siIL-10RA was enhanced further by combining it with siRNA targeting TGF-β receptor (siTGF-βR), which was the next best option during the screening of this study, or the previously selected immunoadjuvant siRNA targeting phosphatase and tensin homologue deleted on chromosome 10 (PTEN) or Bcl-2-like protein 11 (BIM). In the midst of sorting out the siRNA cocktails, the cocktail of siIL-10RA and siTGF-βR generated the strongest antigen-specific CD8+ T cell immunity. Concordantly, the knock-down of both IL-10RA and TGF-βR in DCs induced the strongest anti-tumour effects in the TC-1 P0 tumour model, a cervical cancer model expressing the human papillomavirus (HPV)-16 E7 antigen, and even in the immune-resistant TC-1 (P3) tumour model that secretes more IL-10 and TGF-β than the parental tumour cells (TC-1 P0). These results provide the groundwork for future clinical development of the siRNA cocktail-mediated strategy by co-targeting immunosuppressive molecules to enhance the potency of DC-based vaccines.
Keywords: dendritic cell, IL-10RA, immunotherapy, siRNA, TGF-β
Introduction
Dendritic cells (DCs) serve as a fascinating target in cancer immunotherapy due to their capacity to act as antigen-presenting cells (APCs), which play a critical role in the induction and regulation of immune responses 1,2. The immunological efficacy of DC-based cancer vaccines has been demonstrated successfully in numerous murine tumour models 3,4. However, the clinical outcomes of DC-based vaccination against cancer have not been very promising. One of the limitations in the clinical application of DC-based cancer vaccine may be the presence of various immunosuppressive factors produced by established tumours.
Tumours produce a variety of immunosuppressive cytokines and metabolites, including interleukin (IL)-10, transforming growth factor (TGF)-β, prostaglandin E2 (PGE2), cyclooxygenase-2 (COX2) and nitric oxide synthase 2 (NOS2) 5–7. These immunosuppressive molecules not only inhibit the immune response by inducing regulatory T cells (Tregs) and tolerogenic DCs, but also constrain DC differentiation and function. Among these, IL-10, also known as human cytokine synthesis inhibitory factor, converts immature DCs into tolerogenic APCs through decreased expression of co-stimulatory molecules, such as major histocompatibility complex (MHC)-II, CD80 and CD86. IL-10 promotes DC apoptosis and inhibits DC migration to the secondary lymphoid organs 8,9. Conversely, TGF-β shows pleiotropic features, acting as both a tumour suppressor and a tumour promoter according to the physiological setting. In the tumour microenvironment a high level of TGF-β, however, causes evasion of immune surveillance 10. TGF-β reduces the expression of co-stimulatory molecules and chemokine receptors, leading to the inhibition of tumour-specific T cell responses and migration of DCs to the draining lymph nodes 5,11. In addition, IL-1R-associated kinase 3 (IRAK-3), suppressors of cytokine signalling 1 (SOCS-1) and src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP-1) were reported as negative regulators of various immune responses, including lipopolysaccharide (LPS) tolerance, and consequently they are responsible for the limited efficacy of DC-based vaccines 12–15. Thus, tumour-derived immunosuppression could be the main obstacle in the clinical application of DC-based vaccines, depending on the local micro milieu and cytokine environment.
RNA interference (RNAi) technology is used widely to silence a variety of genes in vitro and in vivo due to its ability to specifically destroy the target mRNA. Our study and other studies have demonstrated successfully that silencing of endogenous enzymes, cytokines or receptors involved in DC apoptosis and immunosuppressive signalling improves antigen-specific CD8+ cytotoxic T lymphocyte (CTL) responses against a tumour antigen in vitro and in vivo 16–22. We have demonstrated that the siRNA targeting Bcl-2-like protein 11 (BIM) or phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a potent immunoadjuvant siRNA for DC-based vaccines, considering its ability to generate antigen-specific CD8+ T cell immune responses. These siRNAs render DCs resistant to apoptotic cell death and activate protein kinase B (Akt) survival signalling, respectively 20,21. Although the potency of the siRNA-treated DC vaccination therapy was profoundly improved in a tumour-bearing mouse model, the overall outcome was not satisfactory and an improvement in the outcome is needed.
In the current study, we tried to further increase the potency of DCs using cocktail siRNA to silence the expression of endogenous molecules in the DCs that can sense the presence of tumour-derived immunosuppressive factors which constrain the function of DCs. The knock-down of the immunosuppression-related targets substantially enhanced anti-tumour CD8+ T cell responses in tumour-bearing mice. In particular, although single silencing of a leading immunosuppressive molecule, IL-10 receptor alpha (hereafter IL-10RA), generated the strongest antigen-specific CD8+ T cell immunity, co-silencing of immunosuppressive signalling receptors with the cocktail, which was made by mixing siIL-10RA and siTGF-βR, enhanced more effectively the frequency of antigen-specific CD8+ CTLs and anti-tumour effects in the TC-1 (P0) tumour model, a cervical cancer model expressing the human papillomavirus (HPV)-16 E7 antigen. More importantly, we also observed that the siRNA cocktail-treated DCs exhibited profound anti-tumour effects against highly immunosuppressive tumour cells (TC-1 P3) that secrete more IL-10 and TGF-β than the parental tumour cells, TC-1 (P0) 16–18. These findings would support future clinical development of DC-based cancer immunotherapy.
Materials and methods
Tumour cell culture
The HPV-16 E7-expressing murine tumour model, TC-1, was generated as described previously 16. In brief, HPV-16 E6, E7 and H-Ras oncogenes were introduced to transform primary C57BL/6 mice lung epithelial cells to generate TC-1 (P0). Immune-resistant TC-1 (P3) cells were established previously through in-vivo selection of a susceptible cell line (TC-1 P0) in the mice immunized with a vaccinia virus encoding an endosome-targeted E7, sightless (Sig)/E7/lysosomal-associated membrane protein 1 (LAMP-1) 16–18. Cells were grown in RPMI-1640 medium supplemented with 5% fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 units/ml penicillin, 100 units/ml streptomycin and 100 mM non-essential amino acids at 37 °C in 5% CO2.
In-vitro stimulation of bone marrow-derived dendritic cells (BMDCs)
BMDCs were generated from bone marrow progenitor cells as described previously 20. For the stimulation of BMDCs, 2 × 105 cells/ml isolated BMDCs were cultured in the presence of LPS at a concentration of 1 μg/ml for 18 h. After stimulation, the LPS-containing medium was changed with fresh media.
Preparation of siRNAs and transfection
siRNA targeting cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), IRAK-3, mothers against decapentaplegic homologue 2 (SMAD2), SMAD3, SOCS-1, SHIP-1, IL-10, IL-10RA, TGF-β, TGF-βR, BIM, PTEN and green fluorescent protein (GFP) were synthesized by Bioneer (Daejeon, Korea). The sense strand of each siRNA duplex consisted of an 18–23 nt target sequence followed by a dTdT 3′ overhang. The anti-sense strand was composed of nucleotides that are complementary to the target sequence and the dTdT 3′ overhang. The siRNA sequences are summarized in the Supporting information, Table S1. The RNAs were deprotected and annealed according to the manufacturer's instructions. GFP-targeting siRNA (siGFP) was used as an irrelevant non-specific control. DCs on a six-well vessel (2 × 105 cell/well) were transfected twice with 300 pmol of the synthesized siRNA using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions at an interval of 1 day. The transfected cells were used for subsequent experiments 2 days later. We used fluorescein isothiocyanate (FITC)-labelled siRNA (Bioneer) to demonstrate the transfection efficiency of the DCs using flow cytometry analysis. More than 95% of the DCs were transfected successfully with siRNAs (data not shown). The rate of cell survival was also measured by the trypan blue staining method. More than 95% of the DCs were alive up to 3 days after the transfection of the siRNAs. The siGFP treatment did not alter the expression of surface molecules on the transfected DCs compared to that on the non-transfected DCs, as reported previously 19,20.
Reverse transcription–polymerase chain reaction (RT–PCR) analysis
To assess the mRNA expression of each target gene, total RNA was isolated from the DCs using a Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instructions. RNA concentration was determined by a spectrophotometer. Then, 1 ug of RNA from each sample was reverse-transcribed with SuperScript II (Invitrogen, Frederick, MD, USA). For PCR amplification, the primer sets mentioned in the Supporting information, Table S2 were used. To ensure equal loading of all lanes, samples were subjected to RT–PCR amplification of the constitutively expressed β-actin gene. Amplified PCR products were measured quantitatively using scanning densitometry.
Immunization with DCs
Six–8-week-old female C57BL/6 mice were acquired from Daehan Biolink (Chungbuk, Korea). All animal procedures were performed according to the protocol approved by the Korea University Institutional Animal Care and Use Committee (KUIACUC-2010-98 and KUIACUC-2013-210). BMDCs were generated from bone marrow progenitor cells, as described previously 20. The DCs were transfected with the synthesized siRNAs, as described previously 19,20. Two days after transfection, the DCs transfected with various siRNAs were pulsed with HPV-16 E7 aa49-57 peptide (aa49-57, RAHYNIVTF) (10 μg/ml) at 37 °C for 2 h. IL-10RA siRNA transfected BMDCs that were not pulsed with E7 peptide were used as the negative control. The cells were then washed with RPMI-1640, supplemented with 10% fetal calf serum (FCS) and Hanks's balanced salt solution (HBSS) and resuspended in HBSS at a final concentration of 1 × 107 cells/ml. BMDCs were injected into mice via the footpad at a rate of 2 × 105 cells/mouse. One week later, the mice were boosted once with the same dose. Several chemical inhibitors were used to analyse the expression level of proteins involved in signal transduction. PD98059, an extracellular-regulated kinase (ERK) inhibitor, API2, a protein kinase B (AKT) inhibitor and SB203580, a p38 inhibitor, were purchased from Calbiochem (Merck, Darmstadt, Germany). MG132, a ubiquitin inhibitor, was purchased from Sigma Chemical Co. (St Louis, MO, USA).
Intracellular cytokine staining and flow cytometry analysis
Splenocytes were harvested from mice (five mice per group) 1 week after the last vaccination. Prior to intracellular cytokine staining, 5 × 106 pooled splenocytes from each vaccination group were incubated overnight with 1 μg/ml of E7 peptide or ovalbumin (OVA) peptide containing a major histocompatibility complex (MHC) class I epitope for the detection of E7 or OVA-specific CD8+ T cell precursors. Interferon (IFN)-γ staining and flow cytometry analysis were conducted as described previously 23. Analysis was conducted on a Becton Dickinson fluorescence activated cell sorter (FACS)can with the CellQuest software (Becton Dickinson Immunocytometry Systems, Mountain View, CA, USA). The number of E7 or OVA-specific IFN-γ-secreting CD8+ T cells was determined using intracellular cytokine staining and FACScan analysis, as described previously 23.
Cytokine analysis using cytometric bead array (CBA)
Cytokine levels were determined using CBA (CBA™; BD Biosciences, San Jose, CA, USA). The procedure was carried out according to the manufacturer's instructions. Briefly, 10 μl of each mouse capture bead suspension was mixed in 50 μl of each cell culture supernatant sample from TC-1 (P0) or TC-1 (P3) cell line or in 50 μl of serum derived from TC-1 (P0) or TC-1 (P3) tumour-bearing mice, and the mixed bead suspensions were transferred to the assay tubes. Plain cell culture medium was used as a negative control to demonstrate the background level of cytokines. Standard dilutions or test samples were added to the appropriate tubes (50 μl/tube), and phycoerythrin (PE) detection reagent (50 μl) was added. After 2 h incubation in the dark at room temperature (RT), the samples were washed and analysed on FACSCalibur (BD Biosciences) using the supplied cytometer set-up beads and the CellQuest™ software.
In-vivo tumour treatment
TC-1 (P0) and immune-resistant TC-1 (P3) cells were used for evaluating the immunoadjuvant efficacy of siIL-10RA and siTGF-βR. C57BL/6 mice (five per group) were inoculated subcutaneously (s.c.) with 1 × 105 TC-1 (P0) or TC-1 (P3) cells/mouse into the armpit of the right limb at 7 days (P0) or 1 day (P3) prior to immunization with E7 or OVA peptide-pulsed BMDCs that were transfected with siRNA targeting various immune suppressors or GFP, and after another week mice were boosted with the E7 peptide-pulsed BMDCs. Mice were monitored twice a week for evidence of tumour growth by palpation and inspection. For the determination of tumour volume, the size of each individual tumour was measured with a caliper and the tumour volume was calculated using the following equation: tumour volume (mm3) = width × length2/2. To investigate further the therapeutic potential of the DCs modified with siRNAs targeting IL-10RA, TGF-βR or IL-10RA+TGF-βR, we evaluated the anti-tumour effect using a TC-1 lung metastasis model. Mice were injected with 1 × 105 TC-1 tumour cells/mouse into the tail vein to simulate haematogeneous tumour spread 24. Mice were treated with DCs 1 day after tumour challenge, and boosted a week later. Mice were monitored twice a week and killed on day 56 after the vaccination. The mean number of pulmonary nodules in each mouse was counted by experimenters blinded to sample identity. In-vivo tumour experiments were performed at least three times to generate reproducible data.
Statistical analysis
All data are representative of at least two independent experiments. Non-parametric one-way or two-way analysis of variance (anova) was performed with spss version 12·0 software (IBM, Chicago, IL, USA). Comparisons between individual data-points were assessed with Student's t-test. A P-value of P < 0·05 was considered statistically significant in all cases.
Results
IL-10RA is the leading immunosuppressive target molecule expressed on DCs whose silencing enhances the antigen-specific CD8+ T cell immune responses
We performed RT–PCR analysis to determine whether transfection of BMDCs with the siRNA targeting immunosuppressive factors (COX-2, NOS2, SMAD2, SMAD3, SOCS1, SHIP1, IL-10, IL-10RA, TGF-β and TGF-βR) would down-regulate the expression of the target mRNA in the transfected cells. As shown in Fig. 1a, the mRNA expression target gene was decreased substantially to an undetectable level 48 h after transfection. The expression of the immunosuppressive genes was suppressed up to 5 days after transfection and then it was first detected below the normal levels on day 6, but was restored to normal levels by day 7 after transfection (data not shown). In contrast, the expression of the target genes was unchanged in the BMDCs transfected with control siRNA targeting GFP (siGFP), and the expression levels were similar to those in BMDCs which were not transfected with siRNA (data not shown). Thus, we confirmed that the siCOX-2, siNOS, siSMAD2, siSMAD3, siSOCS1, siSHIP1, siIL-10, siIL-10RA, siTGF-β and siTGF-βR used in the current study could silence successfully the target gene expression in our DC system.
Figure 1.
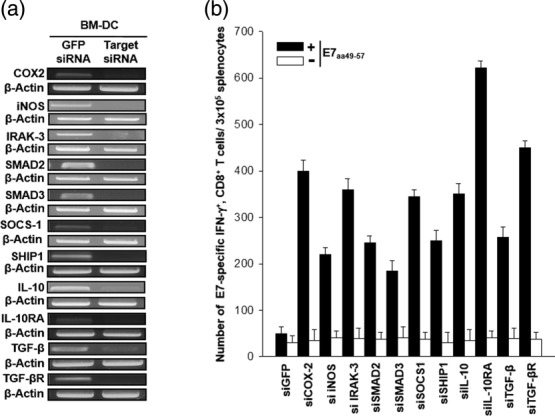
Vaccination with dendritic cells (DCs) transfected with small interfering RNA (siRNA) targeting immunosuppressive molecules causes a significant increase in the number of antigen-specific CD8+ T cells. (a) Reverse transcriptase–polymerase chain reaction (RT–PCR) analysis to detect the expression of cyclooxygenase-2 (COX-2), mothers against decapentaplegic homologue 2 (SMAD2), SMAD3, interleukin (IL)-10, IL-10RA, inducible nitric oxide synthase (iNOS), IL-1R-associated kinase 3 (IRAK3), suppressors of cytokine signalling 1 (SOCS-1), src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP-1), transforming growth factor (TGF)-β or TGF-βR mRNA in DCs transfected with various siRNA constructs. DCs were transfected with siRNAs targeting green fluorescent protein (GFP), COX-2, SMAD2, SMAD3, IL-10, IL-10RA, iNOS, IRAK3, SOCS-1, SHIP-1, TGF-β or TGF-βR. RT–PCR analysis was conducted with 2 μg of the total RNA and specific primer, 48 h after transfection. β-actin was used as a loading control. (b) Intracellular cytokine staining and flow cytometry analysis were performed to determine the number of interferon (IFN)-γ-producing E7-specific CD8+ T cells in mice after immunization with E7 peptide-pulsed DCs transfected with various siRNA constructs. Mice (five per group) were vaccinated twice with E7 peptide-pulsed DCs transfected with siRNAs targeting GFP, COX-2, SMAD2, SMAD3, IL-10, IL-10RA, iNOS, IRAK3, SOCS-1, SHIP-1, TGF-β or TGF-βR. There was a 1-week interval between injections of the transfected DCs for the purpose of vaccination. Splenocytes were harvested 1 week after the last vaccination, stained for CD8+ and IFN-γ, and analysed by flow cytometry to detect activated E7-specific CD8+ T cells. Representative flow cytometry data for splenocytes harvested from the vaccinated mice and stimulated with E7 aa49-57 peptide or without peptide stimulation. The bar graph depicts the number of IFN-γ-expressing E7-specific CD8+ T cells per 3 × 105 splenocytes from vaccinated mice (mean ± standard deviation). The data presented in this figure are representative of two independent experiments.
To examine whether the transfection of DCs with the siRNA would influence their ability to generate E7-specific IFN-γ+CD8+ T cell precursors in vaccinated mice, we next performed intracellular cytokine staining and flow cytometry analysis using splenocytes from mice vaccinated with siRNA-transfected DCs that were pulsed with MHC class I epitopic E7 peptide (aa49-57). As shown in Fig. 1b, vaccination with the BMDCs transfected with siRNAs targeting all the above immunosuppressive molecules led to an increase in the number of E7-specific CD8+ T cell responses. Among all mice, those vaccinated with DCs transfected with siIL-10RA exhibited the highest frequency of E7-specific IFN-γ+ CD8+ T cell precursors (628·7 ± 13·5/3 × 105 splenocytes), and this finding was consistent with the result of our previous study 22; a 12-fold increase compared to the number of T cells in mice that were vaccinated with the siGFP-transfected DCs (50·9 ± 14·3/3 × 105 splenocytes) (P < 0·0003). The next greatest increase in E7-specific IFN-γ+ CD8+ T cell precursors was observed in mice vaccinated with siTGF-βR-transfected DCs (427·7 ± 17·2/3 × 105 splenocytes), which enhanced the E7-specific CD8+ T cell responses. In contrast, vaccination with BMDCs transfected with siIL-10RA without peptide pulsing failed to generate significant E7-specific CD8+ T cell responses (26·7 ± 5·5/3 × 105 splenocytes), thereby suggesting that the E7-specific CD8+ T cell responses induced by immunization with the E7 peptide-pulsed BM-DCs transfected with siIL-10RA are specific to the E7 antigen. Taken together, these data demonstrate that immunization with DCs transfected with siRNAs silencing key immunosuppressive molecules can cause a marked induction of antigen-specific CD8+ T cell immune responses. Among the siRNAs targeting various immunosuppressive molecules, siIL-10RA generated the strongest antigen-specific CD8+ T cell immune responses. Therefore, siIL-10RA was selected as the main component of immunoadjuvant siRNA cocktail for DC-based vaccination therapy.
TGF-βR is reciprocally independent of IL-10RA in DCs transfected with various cocktails of the IL-10RA-based immunoadjuvant siRNAs
We have previously reported siBIM and siPTEN as the potent immunoadjuvant siRNAs for DC-based vaccines, considering their ability to generate antigen-specific CD8+ T cell immune responses, by rendering DCs resistant to apoptotic cell death and activating Akt signalling, respectively 20,21. Along with the second-best adjuvant siTGF-βR in this study, siBIM and siPTEN were therefore chosen as components of the immunoadjuvant siRNA cocktail, and they were co-transfected in turn with siIL-10RA, designated as DC/siIL-10RA+siGFP, DC/siIL-10RA+siPTEN, DC/siIL-10RA+siBIM and DC/siIL-10RA+TGF-βR. In this setting, siGFP was used as a negative control (DC/siGFP+siGFP). We first investigated whether knock-down of the selected immunosuppressive molecules with siRNA could cause cross-interference in their expression. For this purpose, we analysed the mRNA and protein levels of the target molecules in DCs transfected with the cocktails of siRNAs targeting immunosuppressive molecules. The results of RT–PCR analysis showed that the target gene mRNA expression was decreased substantially to an almost undetectable level, as shown in Fig. 2a. In contrast, the expressions of the target genes were unchanged in the DCs transfected with control siGFP, and the levels of expression were similar to those in the DCs that were not transfected with the siRNAs (data not shown). For analysing the target protein expression, we conducted Western blotting in the siRNA cocktail-transfected DCs. Protein expressions of TGF-βR and PTEN were not influenced reciprocally by IL-10RA in DCs transfected with these siRNA cocktails, whereas protein expression of BIM was regulated negatively by IL-10RA (Fig. 2b). Collectively, the expressions of TGF-βR and PTEN were independent of IL-10RA at both mRNA and protein levels in DCs transfected with the siRNA cocktails, but the expression of BIM was not independent of IL-10RA. Hence, the results suggest that it is feasible to target IL-10RA and TGF-βR or PTEN simultaneously using the siRNA cocktail without any interference in their expression.
Figure 2.
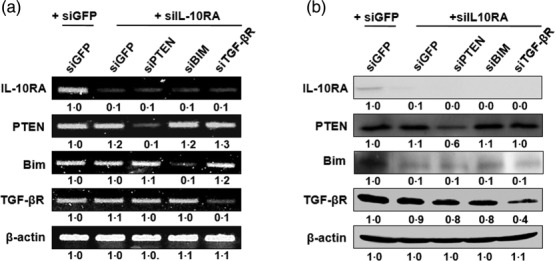
The expression of interleukin (IL)-10RA, phosphatase and tensin homologue deleted on chromosome 10 (PTEN), Bcl-2-like protein 11 (BIM) and transforming growth factor (TGF)-βR in dendritic cells (DCs) transfected with various small interfering RNA (siRNA) constructs. DCs were transfected with siRNA cocktails co-targeting green fluorescent protein (GFP), PTEN, BIM or TGF-β with IL-10RA. (a) Reverse transcriptase–polymerase chain reaction (RT–PCR) analysis to detect the expression of IL-10RA, PTEN, BIM and TGF-βR mRNA in DCs transfected with various siRNA constructs. RT–PCR analysis was conducted with 2 μg of the total RNA and specific primer, 48 h after transfection. β-actin was used as a loading control. (b) Western blot analysis for expression of IL-10RA, PTEN, BIM and TGF-βR in DCs transfected with the siRNAs. Equal amounts of protein (50 µg) were loaded and separated by sodium dodecyl sulphate-polymerase gel electrophoresis (SDS-PAGE). Western blot analysis was performed with the cell lysate and monoclonal antibodies against IL-10RA, PTEN, BIM and TGF-β. Numbers represent fold changes in expression under various experimental conditions, which is normalized to β-actin. The data presented in this figure are representative of three independent experiments.
IL-10RA regulates negatively a pro-apoptotic protein, BIM, via an ERK-dependent pathway in DCs
Contrary to our expectation, blocking IL-10RA caused interference in the BIM expression, as described above. Therefore, we investigated further the underlying mechanism for the siIL-10RA-mediated interference in BIM expression. As it has been reported that activation of BIM is related to the AKT, ERK and p38 pathways, we first examined the activity of these kinases in LPS-stimulated DC/siIL-10RA. Kinase activation was determined by Western blotting using antibodies that specifically recognize both the non-phosphorylated and phosphorylated forms of these kinases, implying inactive and active status, respectively 25–27. The results demonstrated that ERK was activated in DC/siIL-10RA, as shown in Fig. 3a. To investigate further whether the activation of ERK was responsible for the down-regulation of BIM induced by silencing of IL-10RA in DCs, we applied kinase inhibitors such as an ERK inhibitor PD98059, an AKT inhibitor API2 and a p38 inhibitor SB203580 to DC/siIL-10RA stimulated with LPS, and protein levels of BIM were analysed 24 h after treatment. The result showed that inhibition of ERK led to an increase in the BIM protein level, but inhibition of AKT and p38 did not induce an increase in the BIM protein level, indicating that the activation of ERK caused by siIL-10RA treatment regulates BIM expression negatively in DCs (Fig. 3b). There have been reports suggesting that BIM was degraded via the proteasome pathway when it was phosphorylated by ERK 28. Therefore, we applied a ubiquitin inhibitor, MG132, for 24 h in DC/siIL-10RA and DC/siGFP as a negative control, to confirm that the activation of ERK and the decrease in BIM protein level are involved in the ubiquitination. As shown in Fig. 3c, the BIM protein expression level was not decreased in MG132-treated DC/siIL-10RA, unlike that in MG132-untreated DC/siIL-10RA. Overall, in DC/siIL-10RA, ERK was activated and it consequently facilitated the ubiquitin-mediated degradation of BIM. These data suggest that targeting IL-10RA leads to pERK-dependent degradation of a key pro-apoptotic molecule, BIM, in DCs.
Figure 3.
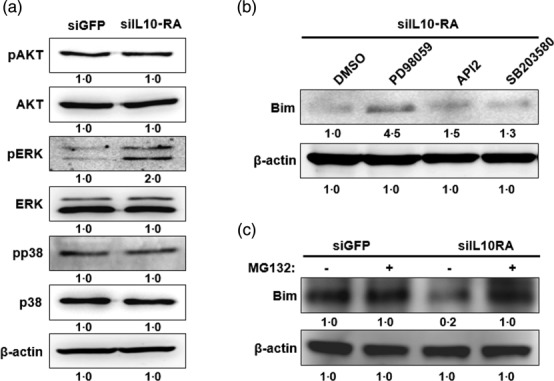
Interleukin (IL)-10RA down-regulates a pro-apoptotic protein, Bcl-2-like protein 11 (BIM), via an extracellular-regulated kinase (ERK)-dependent pathway in dendritic cells (DCs). DCs were transfected with small interfering RNA (siRNA) targeting green fluorescent protein (GFP) or IL-10RA. (a) Western blot analysis for expression of protein kinase B (AKT), p-AKT, ERK, pERK, p38, pp38 and BIM in DCs transfected with the siRNA. Equal amounts of protein were loaded and separated by sodium dodecyl sulphate-polymerase gel electrophoresis (SDS-PAGE). Western blot analysis was performed with the cell lysate and the identical monoclonal antibodies. β-actin was used as a loading control. (b) Western blot analysis of BIM in lysates of DCs transfected with siIL-10RA after overnight treatment with various kinase inhibitors such as an ERK inhibitor PD98059, an AKT inhibitor API2 and a p38 inhibitor SB203580. (c). Western blot analysis of BIM in lysates of DCs transfected with siGFP or siIL-10RA after 4 h of treatment with MG132 (a ubiquitin inhibitor). The data presented in this figure are representative of three independent experiments.
Vaccination with DCs transfected with the cocktail of siRNA targeting IL-10RA and TGF-βR leads to a significant increase in the number of E7-specific CD8+ T cells
To assess whether transfection of DCs with a variety of siRNA constructs and their cocktails increased their ability to produce E7-specific IFN-γ+ CD8+ T cell precursors in vaccinated mice, we injected DCs transfected with each set of siRNA cocktails into the footpad after pulsing with the E7 peptide. For immunization, the DCs were injected twice at an interval of 1 week. The number of E7-specific IFN-γ-secreting CD8+ T cells generated by the DCs was counted using a flow cytometer, as described previously 23. As shown in Fig. 4, although all the tested siRNAs substantially increased the frequency of E7-specific IFN-γ+ CD8+ T cell precursors, DC/siIL-10RA+siTGF-βR generated the highest frequency of E7-specific IFN-γ+ CD8+ T cell precursors (980 ± 45/3 × 105 splenocytes) (P < 0·04), a 17-fold increase compared to the number of T cells in mice vaccinated with the DC/siGFP+siGFP (58·0 ± 11·5/3 × 105 splenocytes) (P < 0·0003). Taken together, these data demonstrate that co-targeting IL-10RA and TGF-βR can synergistically augment the immune-priming capacity of IL-10RA-silenced DCs.
Figure 4.
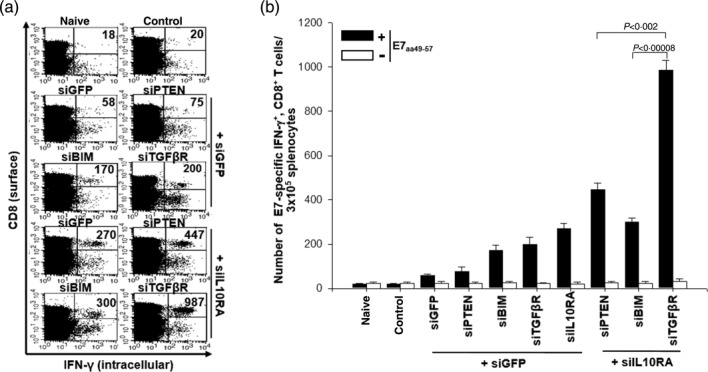
Vaccination with dendritic cells (DCs) transfected with various small interfering RNA (siRNA) increases the number of E7-specific CD8+ T cells. (a) Intracellular cytokine staining and flow cytometry analysis to determine the number of interferon (IFN)-γ-producing E7-specific CD8+ T cells in mice after immunization with E7 peptide-pulsed DCs transfected with various siRNA constructs. Mice (five per group) were vaccinated twice with E7 peptide-pulsed DCs transfected with siRNA targeting green fluorescent protein (GFP), phosphatase and tensin homologue deleted on chromosome 10 (PTEN), Bcl-2-like protein 11 (BIM) interleukin (IL)-10RA, transforming growth factor (TGF)-βR, IL-10RA+PTEN, IL-10RA+BIM or IL-10RA+TGF-βR. There was a 1-week interval between injections of the transfected DCs for the purpose of vaccination. Splenocytes were harvested 1 week after the last vaccination, stained for CD8+ and IFN-γ, and analysed by flow cytometry to detect activated E7-specific CD8+ T cells. Representative flow cytometry data for splenocytes harvested from the vaccinated mice and stimulated with E7 aa49-57 peptide or without peptide stimulation. The naive group has non-transfected DCs without E7 peptide pulsing, while the control group has siGFP transfected DC without E7 peptide pulsing. (b) The bar graph indicates the number of IFN-γ-expressing E7-specific CD8+ T cells per 3 × 105 splenocytes from vaccinated mice (mean ± standard deviation). The data presented in this figure are representative of two independent experiments.
DCs transfected with the cocktail of siRNA targeting IL-10RA and TGF-βR express high levels of MHC-I, -II, CD40, CD80, CD86 and CCR7
To evaluate further the immunoadjuvant effects of the siRNA cocktail, we compared the difference in maturation phenotype among DC/siIL-10RA, DC/siTGF-βR and DC/siIL-10RA+siTGF-βR by measuring the expression levels of MHC-I, -II, CD40, CD80, CD86 and CCR7, and DC/siGFP+siGFP was used as a negative control. The results showed that the expression of all phenotype factors was increased significantly in the DC/siIL-10RA compared to a negative control, as shown in Fig. 5. Notably, the expression levels of MHC-II, CD40, CD80, CD86 and CCR7 in DC/siIL-10RA+siTGF-βR were higher than those in DC/siIL-10RA and DC/siTGF-βR. Thus, these data suggest that the profound immune-enhancing effects of the siRNA cocktails might be due to an increase in the expression of molecules involved in antigen presentation such as CD40, CD80, CD86, MHC-I and -II and molecules involved in the migration of T cells to secondary lymphoid organs such as CCR7 in the DCs.
Figure 5.
Surface expression of CD40, CD80, CD86, major histocompatibility complex (MHC)-I, -II, and CCR7 on dendritic cells (DCs) transfected with small interfering RNA (siRNA) targeting green fluorescent protein (GFP), interleukin (IL)-10RA, transforming growth factor (TGF)-βR, or IL-10RA+TGF-βR. DCs transfected with siGFP, siIL-10RA, siTGF-β, or siIL-10RA+siTGF-βR were treated with 1 mg/ml lipopolysaccharide (LPS) for 18 h and then collected for analysis of expression of each molecule to characterize the maturation phenotypes. Bar graphs describing the mean fluorescence intensity (MFI) indicate MHC-I, -II, CD40, CD80, CD86 and CCR7 expression, respectively. The data presented in the figure are from one representative experiment out of three.
Silencing of IL-10RA and TGF-βR in DCs provoked Th1-favouring but T helper type 2 (Th2)-non-favouring effects
It is well established that the Th1 response is crucial for priming of antigen-specific CD8+ T cells. Type 1 (Th1-like) cytokines include IL-2, IL-12, IFN-γ and tumour necrosis factor (TNF)-α. In contrast, type 2 (Th2-like) cytokines include IL-4, IL-5, IL-6, IL-10 and TGF-β. Th2 cytokines, however, counteract the Th1 responses. For instance, IL-10 inhibits differentiation of Th1 cells and function of DCs 29, whereas IL-12, a prototypical DC-derived cytokine, was considered as a key factor for the development of Th1 responses 30. A high level of TGF-β in the tumour microenvironment plays a major role in evasion from immune surveillance 10. Our previous studies demonstrated that silencing of IL-10 and IL-10RA induced a significant increase in IL-12 secretion from DCs, indicating their Th1-favouring effect 22. In the current study, we determined whether silencing of IL-10RA and TGF-βR affected the secretion of cytokines such as IL-10, TGF-β, IL-12 and TNF-α from BMDCs. Supernatants from all modified BMDCs were collected to analyse the expression level by flow cytometry using cytometric bead array. As shown in Fig. 6, the concentration of IL-10 in supernatants from DC/siIL-10RA and the concentration of TGF-β in DC/siTGF-βR were decreased, compared to that in supernatants from negative control DC/siGFP, indicating that silencing of the IL-10 and TGF-β receptors caused a decrease in the IL-10 and TGF-β secretion from DCs, respectively. In contrast, the concentration of the Th1 cytokine, TNF-α, in both DC/siIL-10RA and DC/siTGF-βR was increased more than 1·5-fold and the concentration of the Th1 cytokine, TNF-α, in DC/siIL-10RA+siTGF-βR was increased more than fourfold, respectively, compared to that in DC/siGFP. Consistently, the expression level of IL-12 protein in DC/siIL-10RA+siTGF-βR was also increased compared to that in DC/siGFP. Hence, we demonstrated that Th2 cytokines were secreted less and Th1 cytokines were secreted more from DCs transfected with the siRNA cocktail targeting IL-10RA and TGF-βR than from a negative control. These findings imply that silencing of IL-10RA and TGF-βR in DCs triggered Th1-favouring effects.
Figure 6.
Secretion of cytokines from dendritic cells (DCs) transfected with small interfering RNA (siRNA) targeting green fluorescent protein (GFP), interleukin (IL)-10RA, transforming growth factor (TGF)-βR or IL-10RA+TGF-βR. Expression of secreted cytokines such as IL-10, TGF-β, tumour necrosis factor (TNF)-α and IL-12 in DCs transfected with siRNAs targeting GFP, IL-10RA, TGF-βR or IL-10RA+TGF-βR. Transfected DCs were stimulated with lipopolysaccharide (LPS) for 18 h, and then the culture medium was collected for flow cytometry analysis with cytometric bead array (CBA). The bar graph represents the concentration of each secreted cytokine in the medium from DCs transfected with siGFP, siIL-10RA or siTGF-βR. The fluorescence activated cell sorter (FACS) data are presented as the mean ± standard deviation of three independent experiments.
Vaccination with E7 peptide-loaded BM-DCs transfected with the cocktail of siIL-10RA and siTGF-βR dramatically enhanced anti-tumour effects
To determine if the enhancement in E7-specific CD8+ T cell immune responses induced by DC/siIL-10RA+siTGF-βR can cause an increase in E7-specific anti-tumour effects, we conducted an in-vivo tumour treatment experiment using the previously described E7-expressing tumour model (TC-1 P0 cells). Mice were first injected s.c. with 1 × l05 TC-1 cells/mouse, followed by treatment with the DCs 7 days after tumour challenge, and 1 week later the mice were boosted as described in Fig. 4. Tumour volume was monitored for up to 34 days after tumour challenge. As shown in Fig. 7a, mice treated with the DCs transfected with the cocktail of siIL-10RA and siTGF-βR demonstrated the smallest tumour volume compared to mice treated with the DCs transfected with siGFP, siGFP+siIL-10RA (P < 0·0006) or siGFP+siTGF-βR (P < 0·0005). Next, we investigated the anti-tumour effect of siRNA cocktail targeting IL-10RA and TGF-βR using a TC-1 lung metastasis model. Mice were challenged by intravenous (i.v.) injection of 1 × 105 TC-1 tumour cells/mouse to simulate haematogeneous tumour spread 24 and immunized as described in Fig. 7. Mice were monitored twice a week and killed on day 56 after the vaccination. Mice immunized with DC/siIL-10RA+siTGF-βR had a remarkably low mean number of pulmonary nodules (2 ± 1) compared to mice immunized with DC/siGFP+siIL-10RA (22 ± 12; P < 0·03), DC/siGFP+siTGF-βR (32 ± 12; P < 0·01) or DC/siGFP+siGFP (63 ± 11; P < 0·002), as shown in Fig. 7b,c. Together with the results in Fig. 4, these data indicate that the cocktail of siIL-10RA and siTGF-βR could be a suitable candidate as an immunoadjuvant for DC-mediated vaccine immunotherapy.
Figure 7.
In-vivo TC-1 (P0) tumour treatment experiments in mice vaccinated with dendritic cells (DCs) transfected with small interfering RNA (siRNA) targeting green fluorescent protein (GFP), interleukin (IL)-10RA, transforming growth factor (TGF)-βR or IL-10RA+TGF-βR. Mice were inoculated with 1 × 105 TC-1 (P0) cells/mouse into the right limb armpits and then treated with E7 peptide-pulsed DCs transfected with GFP, IL-10RA, TGF-βR or IL-10RA+TGF-βR 7 days after inoculation via subcutaneous injection (a), and 1 day after inoculation via intravenous injection (b). Bone marrow (BM)DCs transfected with siIL-10RA without peptide pulsing were used as control. Mice were boosted with the same dose and regimen of E7 peptide-loaded DCs 1 week later. (a) Mice were monitored for evidence of tumour growth by palpation and inspection twice a week. For the determination of tumour volume, each individual tumour size was measured with a caliper. Line and scatterplot graphs depicting the tumour volume (mm3) are presented. Data are expressed as the tumour volumes (mm3) from each group recorded for 34 days after tumour innoculation and (b) the number of metastatic lung nodules in mice at 56 days after the last vaccination. Error bars represent the mean ± standard deviation. The data presented in this figure (c) are from one representative experiment out of three.
Immune-resistant TC-1 (P3) tumour is more immunosuppressive than the parental TC-1 (P0) tumour due partially to an increase in IL-10 and TGF-β secretion
In most of the clinical cases, the recurrent tumours after immunotherapy are frequently refractory to other immunotherapeutic agents, including DC-based vaccines, due presumably to immunosuppression 5. To characterize the immune suppressive phenotypes observed in recurrent tumours, we used an immune-resistant cancer cell line, called TC-1 (P3), which was established previously by in-vivo selection of a susceptible cell line (TC-1 P0) in the mice immunized with a vaccinia virus encoding an endosome-targeted E7, Sig/E7/LAMP-1 17,18,27. To examine if the TC-1 (P3) tumour has greater immunosuppressive capacity than the TC-1 P0 tumour, we immunized non-tumour-bearing mice, TC-1 (P0) tumour-bearing mice or TC-1 (P3) tumour-bearing mice with DCs pulsed with MHC class I epitope OVA aa 257-264 peptide. In this experimental setting, we used non-tumour OVA antigen instead of E7 peptide to distinguish them from spontaneous E7 antigen-specific CD8+ T cell immune responses initiated by TC-1 tumour models that express E7 antigen. Using flow cytometry, the number of OVA-specific IFN-γ-secreting CD8+ T cells was counted, as described previously 23. As shown in Fig. 8a,b, DCs from the TC-1 (P3) tumour-bearing mice almost failed to generate the number of OVA-specific CD8+ T cells compared with DCs from the TC-1 (P0) tumour-bearing mice (a fivefold decrease, 104·3 ± 14·2 versus 20·9 ± 5·66/3 × 105 splenocytes). To elucidate the relationship between TC-1 (P3)-mediated immunosuppression and IL-10 and TGF-β secretion, we measured the concentration of IL-10 in cell culture medium and in the serum from TC-1 (P0) or TC-1 (P3) tumour-bearing mice. As shown in Fig. 8c,d, the amount of IL-10 and TGF-β secretion in TC-1 (P3) was increased definitively compared with that in TC-1 (P0), in both in-vitro cell culture media (10·5 ± 0·7 pg/ml versus 7·8 ± 0·3 pg/ml for IL-10; 14·0 ± 1·2 pg/ml versus 11·8 ± 0·3 pg/ml for TGF-β) and in-vivo serum (1400 ± 30 pg/ml versus 890 ± 10 pg/ml for IL-10; 95 ± 2 pg/ml versus 71 ± 1 pg/ml for TGF-β). Collectively, these data indicate that TC-1 P3 tumour is a highly immunosuppressive tumour model that secretes immunosuppressive cytokines such as IL-10 and TGF-β.
Figure 8.
Intracellular cytokine staining and flow cytometry analysis to determine the number of interferon (IFN)-γ-producing ovalbumin (OVA)-specific CD8+ T cells in TC-1 (P0) or TC-1 (P3) tumour-bearing mice after immunization with OVA peptide-pulsed dendritic cells (DCs) and secretion of cytokines from TC-1 (P0) or TC-1 (P3). OVA peptide-pulsed bone marrow dendritic cells (BMDCs) were administered 7 days after TC-1(P0) or TC-1(P3) tumour challenge (1 × 105 cells/mouse) via the subcutaneous route. One week later, mice were boosted with the same dose and regimen of OVA peptide-loaded BMDCs. (a) Representative flow cytometry data for splenocytes harvested from the vaccinated mice and stimulated with OVA aa257-264 peptide or without peptide stimulation, and (b) the bar graph depicts the number of IFN-γ-expressing OVA-specific CD8+ T cells per 3 × 105 splenocytes from vaccinated mice (mean ± standard deviation). Proinflammatory cytokines were secreted by tumours in vitro and in vivo. (c) Concentration of interleukin (IL)-10 (pg/ml) in TC-1 (P0) or TC-1 (P3) culture media as measured by cytometric bead array (CBA). Culture media was used as a control. (d) Concentration of IL-10 (pg/ml) in plasma of naive, TC-1 (P0) or TC-1 (P3) tumour-bearing mice as measured by CBA. The data presented are representative of two independent experiments.
Vaccination with DC/siIL-10RA+siTGF-βR causes a significant increase in the number of antigen-specific CD8+ T cells and anti-tumour effects in mice bearing a highly immunosuppressive TC-1 (P3) tumour
To determine if the immune adjuvant effects of siIL-10RA+siTGF-βR in TC-1 (P0) tumour-bearing mice subsist in immunosuppressive TC-1 (P3) tumour-bearing mice, we immunized mice with OVA peptide-pulsed DC/siGFP+siGFP and OVA peptide-pulsed DC/siIL-10RA+siTGF-βR. After peptide pulsing, each group of DCs was injected into the footpad of TC-1 P3 tumour-bearing mice four times at a 4-day interval. DC/siIL-10RA+siTGF-βR that were not pulsed with OVA peptide were used as control DCs. As shown in Fig. 9a,b, vaccination with DC/siIL-10RA+siTGF-βR increased the number of OVA-specific CD8+ T cells drastically compared to that after vaccination with DC/siGFP+siGFP (405·5 ± 12.8 versus 72 ± 32·3/3 × 105 splenocytes). Thus, blocking IL-10RA and TGF-βR in DCs using the siRNA technology can significantly enhance antigen-specific CD8+ T cell immune responses in the presence of an immunosuppressive tumour.
Figure 9.
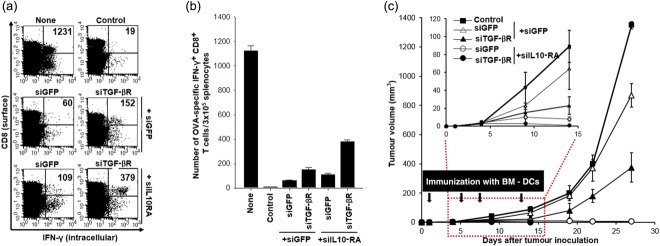
Intracellular cytokine staining and flow cytometry analysis to determine the number of interferon (IFN)-γ-producing ovalbumin (OVA)-specific CD8+ T cells in TC-1 (P3) tumour-bearing mice after immunization with dendritic cells (DCs) transfected with small interfering RNA (siRNA) targeting interleukin (IL)-10RA and transforming growth factor (TGF)-βR and in-vivo TC-1 (P3) tumour treatment experiments. (a) Intracellular cytokine staining and flow cytometry analysis to determine the number of IFN-γ-producing OVA-specific CD8+ T cells in tumour-bearing mice after immunization with DCs transfected with siRNA targeting IL-10RA and TGF-βR. Mice were inoculated with 5 × 105 TC-1 (P3) tumour cells/mouse into the armpits of the right limb and then immunized with OVA peptide- pulsed DCs at 7 days after inoculation. There was a 1-week interval between injections of the DCs for the purpose of vaccination. Splenocytes were harvested 1 week after the last vaccination, stained for CD8+ and IFN-γ and analysed by flow cytometry to detect activated OVA-specific CD8+ T cells. Naive mice served as a negative control. Representative flow cytometry data for splenocytes harvested from the vaccinated mice and stimulated with OVA aa257-264 peptide or without peptide stimulation. None indicates a non-tumour-bearing group with DC vaccination, while the control indicates a tumour-bearing group without DC vaccination. (b) The bar graph depicts the number of IFN-γ-expressing OVA-specific CD8+ T cells per 3 × 105 splenocytes from vaccinated mice (mean ± standard deviation). (c) Mice were inoculated with 1 × 105 TC-1 (P3) cells/mouse into the right limb armpits and then treated with the DCs transfected with GFP, IL-10RA, or TGF-βR on the day after inoculation via subcutaneous injection. BMDCs transfected with siIL-10RA without peptide pulsing were used as control. Mice were boosted with the same dose and regimen of E7 peptide-loaded DCs 4 days later, and there was a 4-day interval on three occasions between injections of the DCs for the purpose of vaccination. Mice were monitored for evidence of tumour growth by palpation and inspection twice a week. For the determination of tumor volume, each individual tumour size was measured with a caliper. Line and scatter plot graphs depicting the tumour volume (mm3) are presented. Data are expressed as the tumour volumes (mm3) from each group recorded for 27 days after tumour inoculation. Error bars represent the mean ± standard deviation. The data presented in this figure (c) are from one representative experiment out of three.
To determine whether the enhancement in antigen-specific CD8+ T cell immune responses induced by DC/siIL-10RA+siTGF-βR could lead to an increase in the tumour antigen-specific anti-tumour effects, we conducted an in-vivo tumour treatment experiment with the TC-1 (P3) model. Mice were first injected s.c. with 1 × l05 TC-1 (P3) cells/mouse, followed by quadruple treatment with the DCs as described in Fig. 9c. The tumour volume was monitored for up to 27 days after tumour challenge. As shown in Fig. 9c, treatment with E7 peptide-pulsed DC/siGFP+siIL-10RA and DC/siGFP+TGF-βR suppressed the TC-1 (P3) tumour growth, due more to silencing of IL-10RA. In contrast, treatment with E7 peptide-pulsed DC/siGFP+siGFP led to rapid tumour growth. As a consequence, mice treated with DC/siGFP+siIL-10RA and DC/siGFP+siTGF-βR demonstrated the smallest tumour volume compared to that in mice treated with DC/siGFP+siGFP (P < 0·002) or mice that did not receive any treatment (P < 0·003). More importantly, treatment with E7 peptide-pulsed DC/siGFP+siIL-10RA+siTGF-βR hindered the TC-1 (P3) tumour growth until the completion of the study. Thus, the potent immunoadjuvant effects of the siRNA cocktail targeting IL-10RA and TGF-βR were reproduced even in an immune-refractory tumour model.
Discussion
DCs are considered to be the most effective APCs that have the ability to prime tumour antigen-specific T cells and in turn initiate immune responses against tumours 31. Although DC-based anti-cancer therapy is promising, due to fewer side effects, the efficacy needs to be improved for clinical applications. One of the limitations is an intrinsic immunosuppressive mechanism, which restrains mature DCs in a negative feedback manner after treatment with Toll-like receptor (TLR) ligands such as lipopolysaccharide (LPS). In addition, there are many studies suggesting that tumour-infiltrating DCs lose their ability to activate anti-tumour T cell responses in a variety of human cancers due to the generation of anti-tumour immune responses, such as the increase in tumour-derived immunosuppressive cytokines in the tumour microenvironment 32–34. RNA interference (RNAi) has been considered to be one of the most potent biological therapeutics based on molecular-targeted therapy 35,36. Recently, siRNA cocktails or combinatorial RNAi therapeutics are an emerging technique that inhibits cancer growth and migration more efficiently by silencing multiple target genes based on consideration of their interference effects 37–39.
In our previous studies, we attempted to enhance DC efficacy by blocking negative regulators including pro-apoptotic Bcl-2 family members such as BAK/BAX and BIM 19,20, PTEN 21, immunosuppressive cytokines including IL-10 and IL-10RA 22 via a siRNA-based technology. Along with IL-10, TGF-β serves as a target immunosuppressive cytokine. In particular, tumour-derived TGF-β has been shown to immobilize DCs and prevent migration to tumour-draining lymph nodes in mouse and human skin cancers 40,41.
In the current study, we set priorities for these selected targets based on the in-vitro and in-vivo tests. Among the siRNAs targeting various immunosuppressive molecules, siIL-10RA and siTGF-βR showed the strongest antigen-specific CD8+ T cell immune responses (Fig. 1). Moreover, the expression of TGF-βR was not influenced by IL-10RA knock-down at both mRNA and protein levels in DCs transfected with the selected immunoadjuvant siRNA cocktail as shown in Fig. 2, implying that it is a practicable strategy to target IL-10RA and TGF-βR simultaneously by immunoadjuvant siRNAs without interferences in the signal to improve further the anti-tumour effects of DCs by in-vitro transfection of various cocktails of the selected immunoadjuvant siRNAs, with siIL-10RA as the essential component. As a result, the DCs modified with IL-10RA and TGF-βR (DC/siIL-10RA+siTGF-βR) generated more E7-specific CD8+ T cells and stronger anti-tumour effects in vaccinated mice than the DCs modified with other siRNA cocktails. DC/siIL-10RA+siTGF-βR showed significant up-regulation of major histocompatibility complex (MHC) I, co-stimulatory molecule CD40, CD80, CD86 and chemokine CCR7 after LPS stimulation compared to DC/siGFP (control DC), DC/siIL-10RA or DC/siTGF-βR. More importantly, DC/siIL-10RA+siTGF-βR exhibited significant anti-tumour effects against highly immune resistant tumour cells (TC-1 P3) that secrete more IL-10 and TGF-β than the parental tumour cells, TC-1 P0, in mice bearing the immune-resistant cell line TC-1 (P3) and in turn showed significant in-vivo anti-tumour effects against TC-1 (P3) tumour.
Thus, we have demonstrated successfully that the siRNA technology can be employed to block the IL-10RA and TGF-βR-mediated immunosuppressive axis in DCs, and consequently to increase the potency of DC-based immunotherapeutics against immune-refractory or immune-resistant tumours.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF-2014R1A2A1A10054205 and 2013M3A9D3045881) and the Korea Healthcare Technology R&D Project (HI11C-0052-030013).
Disclosure
The authors declare no competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Table S1. List of small interfering RNA (siRNA) sequences for genes targeted in this study.
Table S2. List of primer sequences for reverse transcriptase–polymerase chain reaction (RT–PCR).
References
- Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobie JJ, Wu RS, Kurt RA, et al. Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003;63:1860–4. [PubMed] [Google Scholar]
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–25. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- Goh C, Narayanan S, Hahn YS. Myeloid-derived suppressor cells: the dark knight or the joker in viral infections? Immunol Rev. 2013;255:210–21. doi: 10.1111/imr.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–52. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- Chen L, Qiu M, He W, Huang A, Liu J. Functional study of immature dendritic cells co-transfected with IL-10 and TGF-beta 1 genes in vitro. Mol Biol Rep. 2012;39:6633–9. doi: 10.1007/s11033-012-1468-4. [DOI] [PubMed] [Google Scholar]
- Wojtowicz-Praga S. Reversal of tumor-induced immunosuppression by TGF-beta inhibitors. Invest New Drugs. 2003;21:21–32. doi: 10.1023/a:1022951824806. [DOI] [PubMed] [Google Scholar]
- Sato K, Kawasaki H, Nagayama H, et al. TGF-beta 1 reciprocally controls chemotaxis of human peripheral blood monocyte-derived dendritic cells via chemokine receptors. J Immunol. 2000;164:2285–95. doi: 10.4049/jimmunol.164.5.2285. [DOI] [PubMed] [Google Scholar]
- Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–76. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- Rauh MJ, Sly LM, Kalesnikoff J, et al. The role of SHIP1 in macrophage programming and activation. Biochem Soc Trans. 2004;32:785–8. doi: 10.1042/BST0320785. [DOI] [PubMed] [Google Scholar]
- Turnis ME, Song XT, Bear A, et al. IRAK-M removal counteracts dendritic cell vaccine deficits in migration and longevity. J Immunol. 2010;185:4223–32. doi: 10.4049/jimmunol.0903507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yu J, Yang L, et al. Enhanced activation of human dendritic cells by silencing SOCS1 and activating TLRs simultaneously. Cancer Immunol Immunother. 2012;61:1653–61. doi: 10.1007/s00262-012-1218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KY, Guarnieri FG, Staveley-O'Carroll KF, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–6. [PubMed] [Google Scholar]
- Wu TC. The role of vascular cell adhesion molecule-1 in tumor immune evasion. Cancer Res. 2007;67:6003–6. doi: 10.1158/0008-5472.CAN-07-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TC, Guarnieri FG, Staveley-O'Carroll KF, et al. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci USA. 1995;92:11671–5. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Lee JH, Noh KH, et al. Enhancing dendritic cell vaccine potency by combining a BAK/BAX siRNA-mediated antiapoptotic strategy to prolong dendritic cell life with an intracellular strategy to target antigen to lysosomal compartments. Int J Cancer. 2007;120:1696–703. doi: 10.1002/ijc.22377. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kang TH, Noh KH, et al. Enhancement of dendritic cell-based vaccine potency by anti-apoptotic siRNAs targeting key pro-apoptotic proteins in cytotoxic CD8(+) T cell-mediated cell death. Immunol Lett. 2009;122:58–67. doi: 10.1016/j.imlet.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kang TH, Noh KH, et al. Enhancement of DC vaccine potency by activating the PI3K/AKT pathway with a small interfering RNA targeting PTEN. Immunol Lett. 2010;134:47–54. doi: 10.1016/j.imlet.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kang TH, Noh KH, et al. Blocking the immunosuppressive axis with small interfering RNA targeting interleukin (IL)-10 receptor enhances dendritic cell-based vaccine potency. Clin Exp Immunol. 2011;165:180–9. doi: 10.1111/j.1365-2249.2011.04410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Hung CF, Ling M, et al. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. J Clin Invest. 2003;112:109–17. doi: 10.1172/JCI17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Lee JH, He L, et al. Modification of professional antigen-presenting cells with small interfering RNA in vivo to enhance cancer vaccine potency. Cancer Res. 2005;65:309–16. [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–6. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem. 2006;281:813–23. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- Lu J, Quearry B, Harada H. p38-MAP kinase activation followed by BIM induction is essential for glucocorticoid-induced apoptosis in lymphoblastic leukemia cells. FEBS Lett. 2006;580:3539–44. doi: 10.1016/j.febslet.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–93. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–69. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Garra A, Hosken N, Macatonia S, Wenner CA, Murphy K. The role of macrophage- and dendritic cell-derived IL12 in Th1 phenotype development. Res Immunol. 1995;146:466–72. doi: 10.1016/0923-2494(96)83017-3. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Pulendran B, Steinman R, Palucka K. Will the making of plasmacytoid dendritic cells in vitro help unravel their mysteries? J Exp Med. 2000;192:F39–44. doi: 10.1084/jem.192.12.f39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoitzner P, Green LK, Jung JY, et al. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol Immunother. 2008;57:1665–73. doi: 10.1007/s00262-008-0487-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot I, Blanchard D, Freymond N, et al. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol. 2007;178:2763–9. doi: 10.4049/jimmunol.178.5.2763. [DOI] [PubMed] [Google Scholar]
- Gigante M, Blasi A, Loverre A, et al. Dysfunctional DC subsets in RCC patients: ex vivo correction to yield an effective anti-cancer vaccine. Mol Immunol. 2009;46:893–901. doi: 10.1016/j.molimm.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Ge Q, Xu JJ, Evans DM, Mixson AJ, Yang HY, Lu PY. Leveraging therapeutic potential of multi-targeted siRNA inhibitors. Future Med Chem. 2009;1:1671–81. doi: 10.4155/fmc.09.131. [DOI] [PubMed] [Google Scholar]
- Ashley CE, Carnes EC, Phillips GK, et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10:389–97. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Chen H, You Q, Shi H, Wang Z. The siRNA cocktail targeting VEGF and HER2 inhibition on the proliferation and induced apoptosis of gastric cancer cell. Mol Cell Biochem. 2014;386:117–24. doi: 10.1007/s11010-013-1850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Byrne SN, Le S, et al. Transforming growth factor-beta1 immobilises dendritic cells within skin tumours and facilitates tumour escape from the immune system. Cancer Immunol Immunother. 2005;54:898–906. doi: 10.1007/s00262-004-0652-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, Le S. Transforming growth factor-beta produced by progressor tumors inhibits, while IL-10 produced by regressor tumors enhances, Langerhans cell migration from skin. Int Immunol. 2001;13:1147–54. doi: 10.1093/intimm/13.9.1147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of small interfering RNA (siRNA) sequences for genes targeted in this study.
Table S2. List of primer sequences for reverse transcriptase–polymerase chain reaction (RT–PCR).