

Abstract
An understanding of the pathophysiologic mechanisms of post-renal transplant (PRT) bone disease is of important clinical significance. Although bone disease occurs after all solid organ transplantation, the cumulative skeletal fracture rate remains high in PRT subjects while reaching a plateau with other transplantations. One major difference in the pathophysiology of PRT bone disease is, perhaps, due to persistent renal phosphorus (Pi) wasting. Novel phosphaturic agents have recently been suggested to participate in the development of bone disease in PRT subjects. However, it is unclear as of yet whether these factors alone or in conjunction with excess parathyroid hormone (PTH) secretion play a key role in the development of negative Pi balance and consequent bone disease in this population. In this review, I present a natural history of PRT hypophosphatemia and persistent renal Pi leak, provide pathophysiologic insight into these developments, and discuss the difficulty in diagnosing these phenotypes in both adult and pediatric populations.
Keywords: Bone disease, Hypophosphatemia, Phosphorus wasting, Post-renal transplantation
Introduction
Over the past 50 years, there has been significant progress in the field of pediatric kidney transplantation [1]. These improvements have largely been due to the development of new immunomodulatory agents used for the treatment of this population [2–5], which has resulted in the number of pediatric recipients of any organ increasing by over 20%. The greatest increase occurred between 1997 and 2006 in recipients 11–17 years of age [1]. The improvement of these treatments has increased both patient and graft survival. However, despite the development of new immunomodulatory agents, which has limited the use of glucocorticoids, post-renal transplant (PRT) bone disease remains a major complication in both pediatric [6, 7] and adult populations [8–10].
The pathophysiologic mechanism responsible for bone disease following solid organ transplantation has mainly been linked to the use of immunomodulatory agents [11–13]. However, PRT bone disease has its own specific presentation [10] since renal osteodystrophy is commonly encountered in patients with chronic kidney disease prior to transplantation. One unique phenotypic characteristic in PRT patients is persistent hypophosphatemia, which usually does not occur with other solid organ transplantation [14, 15].
Normal phosphorus homeostasis
Normal total body phosphorus content is roughly 10 g/kg body weight. Nearly 85% of inorganic phosphorus (Pi) is contained in the skeleton, with approximately 15% in the soft tissue, and only 1% present in the extracellular fluid. Three major organs, the kidney, bone, and intestine, participate in the tight regulation of Pi homeostasis [16]. Pi is freely filtered and tightly reabsorbed by the kidney to accommodate Pi need. Approximately 70% of filtered Pi is reabsorbed in the proximal renal tubule by a sodium (Na)-dependent Pi transport mechanism through the brush-boarder membrane (BBM), which acts as the rate-limiting step in overall Pi reabsorption [17]. Three families of NaPi cotransporter have been identified (types I, II, and III) [18–20]. The type II family of cotransporter consists of three highly homologous isoforms: type IIa and type IIc, which are expressed in the BBM of the renal proximal tubule [19, 20], and type IIb, which is not expressed in the kidney but is responsible for intestinal Pi absorption [21] (Fig. 1).
Fig 1.

Phosphorus homeostasis. ECF Extracellular fluid, Pi inorganic phosphorus, NaPi sodium cotransporter. Modification of Fig. 1 in Ghanekar et al. [10]
Renal tubular phosphate reabsorption is also mediated by multiple hormonal and non-hormonal factors. Parathyroid hormone (PTH) and dietary Pi intake are the principle regulators of this process [22]. High Pi intake and PTH inhibit NaPi cotransport across the BBM, influencing the endocytosis of NaPi types IIa and IIc [23]. Conversely, Pi deprivation stimulates the BBM NaPi IIa/c protein recruitment to the apical membrane [23]. Fibroblast growth factor-23 (FGF-23), a newly discovered regulator of renal Pi handling, has been shown to inhibit NaPi IIa/c-mediated cotransport [24, 25]. In addition, other hormones, including glucocorticoids [26] and thyroid hormone [27], have been shown to modulate renal tubular phosphate transport (Table 1).
Table 1. Physiologic regulation of renal phosphorus transport.
| Regulators of renal tubular phosphate transport |
|---|
| Dietary phosphorus intake |
| Hormonal |
| PTH |
| 1,25-Dihydroxyvitamin D |
| Glucocorticoids |
| Thyroid hormone |
| Growth hormone |
| Novel phosphaturic factors |
| FGF-23 |
| sFRP-4 |
| MEPE |
| PGE2 |
| Other |
| Acid–base status |
| Extracellular volume |
| Dietary potassium |
PTH, Parathyroid hormone; FGF-23, fibroblast growth factor- 23; sFRP-4, secreted frizzle related protein-4; MEPE, matrix extracellular phosphoglycoprotein; PGE2, prostaglandin E2
Natural history of renal phosphorus leak
Hypophosphatemia and renal phosphate wasting are common occurrences following kidney transplantation [28, 29]. These complications have been perceived as transient, occurring early after renal transplantation but rarely persisting for a long duration of time. However, under some circumstances, hypophosphatemia may be prolonged [30]. A normal serum Pi concentration does not preclude the persistence of renal Pi leak and persistent negative Pi balances, which may influence bone integrity. Most of the earlier studies did not take into account the normal cyclic variation of serum Pi concentrations [31]. Under ordinary circumstances, the serum Pi concentration rises post-prandially regardless of the total body Pi content [31] and, consequently, it is possible that low serum Pi is masked in a single specimen by this cyclic variation. Secondly, normal serum Pi concentrations may not exclude the inappropriately high fractional excretion of phosphate (FEpi) since skeletal Pi mobilization precludes the development of hypophosphatemia [32]. The determination of serum Pi in children has its own limitations since Pi kinetics vary according to age and skeletal phosphate displacement during this stage of rapid growth. Due to increased Pi deposition, serum Pi levels are usually lower in children ≥10 years of age [33].
Although the natural history of renal Pi leak and hypophosphatemia has not been extensively examined, one study in 12 successful PRT children and young adults aged 6–36 months following transplantation revealed hypophosphatemia in 58% of this population [34]. This result was detected despite similar demographic characteristics between the two groups, including age, primary kidney disease, duration of hemodialysis before renal transplantation, warm and cold ischemic times of allograft, and histocompatibility between donors and recipients. Moreover, the tubular Pi reabsorption was found to be 65.1%±5.9 (standard deviation, SD) in hypophosphatemic subjects, which was significantly lower than the 85.4%±1.8 in normophosphatemic children (p<0.01) [34]. However, the renal tubular Pi reabsorption value in the latter study was slightly lower than the approximate ≥90% renal tubular Pi reabsorption rate reported in most children [35]. This renal Pi leak was an isolated defect since the serum bicarbonate concentration was similar in both groups, and there was no evidence of amino-aciduria or glycosuria. In addition, no difference in serum PTH concentration was detected between normophosphatemic and hypophosphatemic children. One may conclude that the renal Pi leak possibly occurred independently of prevailing serum PTH concentrations in the hypophosphatemic group of children.
Role of parathyroid hormone
Hyperparathyroidism is known to play a significant role in the development of post-transplant hypophosphatemia [29]. Elevated PTH levels have been shown to decline shortly after renal transplantation [36, 37], with, in most cases, the decline rate reaching a plateau 1 year following renal transplantation. However, hyperparathyroidism may persist for many years, resulting in renal Pi wastage [36]. There is evidence inferring that hyperparathyroidism alone may not be sufficient in the development of a renal Pi leak [32, 38]. In one study in PRT subjects, serum Pi concentrations and FEpi failed to normalize following intravenous calcium infusions [38]. Another in vitro study using opossum cells (as a model for the proximal renal tubular cells) incubated with sera from end stage renal disease patients, chronic kidney disease subjects, and early PRT subjects demonstrated diminished Na-dependent Pi uptake in all three groups [32]. In addition, this diminished Na-dependent Pi uptake did not change following the addition of a bovine PTH inhibitor. These results suggest that PTH may not be the sole causal factor in PRT phosphorus wasting.
Secondary hyperparathyroidism is the most common bone disease in children undergoing renal dialysis [39, 40]. In one study performed in children and adolescents (mean age 12±2 years) following successful renal transplantation, quantitative bone histomorphometry showed an increase in osteoid perimeter, and eroded bone surfaces [7] changes were associated with a serum PTH above the upper range of normal in 22% of the patients. The results of this study suggest that persistent hyperparathyroidism may only occur in a minority of pediatric PRT subjects.
Role of phosphatonins
To date, several studies have suggested that persistent hyperparathyroidism may not be the only mechanism responsible for hypophosphatemia following successful renal transplantation [32, 38, 41, 42]. The phenotypic presentation of PRT hypophosphatemia and renal Pi wastage is similar to that of several inherited and acquired disorders of Pi metabolism [43–45]. Similar to PRT hypophosphatemia, calcitriol levels are low in all of these disorders despite normal kidney function, hypophosphatemia, and elevated PTH, all of which are stimuli known to enhance 1,25(OH)2D synthesis [10, 37, 46–48] (Fig. 2).
Fig 2.
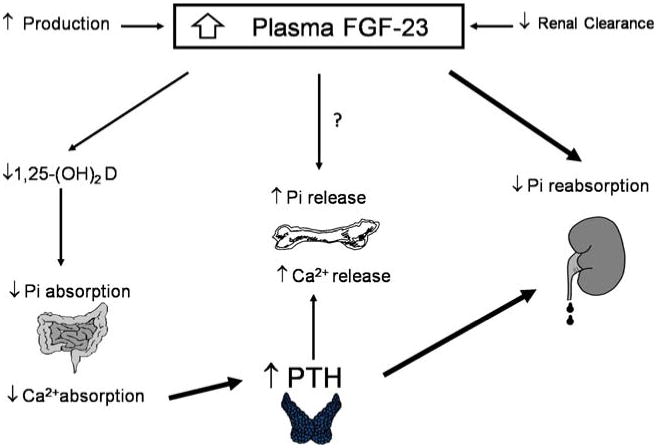
Potential physiologic role of fibroblast growth factor (FGF)-23 in post-renal transplant phosphorus wasting. PTH Parathyroid hormone
FGF-23 is an approximately 26-kDa protein that is not cleared by dialysis [49]. The cellular effects of the FGF family are mediated by FGF receptors (FGFRs) [50]. A specific receptor for FGF-23 has yet to be identified, and it is possible that its effects are mediated through more than a single receptor [51] since four genes code for the seven FGFRs, which are tyrosine kinase family members [52]. It has been shown that FGF-23 binds to and activates FGFRs in various tissues that co-express Klotho, a transmembrane protein co-factor that determines the tissue specificity of FGF-23 [53, 54]. It has also been postulated that a FGFR–Klotho complex, expressed in the distal renal tubule in paracrine fashion, regulates proximal renal tubular phosphorus reabsorption and 1,25(OH)2D production [55]. One such communicating factor may be prostaglandin, since the inhibition of prostaglandin synthesis with indomethacin has been shown to improve hypophosphatemia in Hyp mice [56]. The direct effect of FGF-23 on bone has not been fully elucidated. However, in one in vitro study, FGF-23 was shown to control osteoblastic cell differentiation [57].
Several clinical studies have determined that FGF-23 is a phosphatonin which plays a key role in the pathogenesis of inherited and acquired phosphorus wasting disorders [43–45]. FGF-23 has been shown to inhibit renal 1-α hydroxylase, lower calcitriol synthesis, and cause renal phosphorus wasting [58]. It has also been shown to have a physiologic role in normal phosphorus homeostasis [51, 59, 60]. Serum concentrations of FGF-23 are also known to be elevated in patients with chronic kidney disease (CKD) [49, 61–63]. Although the rise in serum FGF-23 may be due to its reduced renal clearance, the results from several studies have suggested that a progressive rise in FGF-23 may occur with the progression of CKD, possibly contributing to decreased 1,25(OH)2D synthesis [61, 62, 64]. In patients with kidney disease, serum Pi concentration may also play a role in the secondary increase of serum FGF-23, which by enhancing urinary Pi excretion in the remaining nephrons mitigates the rise in serum Pi.
A distinct relationship between plasma FGF-23 concentrations and renal failure was reported in a study of 49 children on continuous cycling peritoneal dialysis. In this population, FGF-23 levels were significantly elevated in subjects with secondary hyperparathyroidism [65]. In addition, the FGF-23 concentrations were shown to be much higher in anuric patients than in those with residual urine output, suggesting FGF-23 clearance by the kidney. The serum FGF-23 levels were also directly correlated with serum Pi concentrations, implying that Pi plays a regulatory role in FGF release [65]. A separate study reported a marked decrease in serum FGF-23 shortly after the improvement of renal function following successful renal transplantation, suggesting rapid FGF-23 clearance. However, plasma FGF-32 concentrations were significantly and inversely correlated with plasma Pi (r2=0.661, p<0.05), but not with creatinine clearance [66]. This result suggested that the remaining FGF-23 may have played a role in the development of hypophosphatemia in PRT recipients. In another longitudinal prospective study conducted by Bhan et al., 85% of the 27 living donor transplant recipients developed hypophosphatemia. In this study, the mean PRT FGF-23 concentration decreased distinctly within the first week following transplantation, although its level continuously remained above normal [48]. Moreover, a stronger correlation was shown with FGF-23 and hypophosphatemia than with PTH [48]. Based on these observations, Bhan et al. suggested that PRT hypophosphatemia may be in a state of tertiary ‘hyperphosphatoninism’ [48] (Fig. 3). The underlying pathophysiologic mechanism of persistently elevated FGF-23 in this population remains unknown. In an animal study using PTH-cyclin D1 transgenic mice, a model of primary hyperparathyroidism, FGF-23 concentrations were found to be directly correlated with serum PTH and inversely correlated with serum Pi concentration. Moreover, serum FGF-23 levels declined in this animal model following a parathyroidectomy, which is suggestive of the regulatory role of PTH in serum FGF-23 concentrations [67]. Therefore, it is plausible to suggest that tertiary hyperphosphatonemia may be, in part, due to the persistence of PTH secretion in PRT recipients. FGF-23 is highly expressed in osteocytes [68], and it is possible that osteocytes communicate with osteoblasts on the bone surface that express PTH receptors [69].
Fig 3.

Pre- and post-transplant FGF-23 (a) and PTH (b) concentrations in subjects with and without severe hypophosphatemia (<1.5 mg/dl). The relative risk of severe hypophosphatemia was stronger with FGF-23 (5.3, p=0.02) than with PTH (0.2, p=0.01) when compared to those falling below the corresponding median levels. Reprinted with permission by Bhan et al. [48]
The role of phosphatonins other than FGF-23 has recently been suggested in other genetic and acquired Pi wasting syndromes [70, 71]. However, the contribution of these phosphatonins in PRT phosphorus wasting has not been revealed. A recent study in PRT recipients demonstrated the relationship between secreted frizzle related protein-4 (sFRP-4) and serum Pi concentrations [66].
Role of 1,25-dihydroxyvitamin D
Serum 1,25-dihydroxyvitamin D [1,25(OH)2D] concentrations are commonly inappropriately low following successful renal transplantation despite hypophosphatemia and hyperparathyroidism, both of which are known to enhance calcitriol synthesis [37, 46, 47]. The lower 1,25(OH)2D concentration in these subjects may be due to high concentrations of FGF-23, which has been shown to have an inhibitory effect on 1,25(OH)2D production. Experimental evidence has demonstrated that 1,25(OH)2D increases renal proximal tubular Pi reabsorption [46, 72, 73]. The influence of 1,25(OH)2D in renal Pi reabsorption in PRT subjects independent of PTH has not yet been explored. Despite conclusive evidence on the role of 1,25(OH)2D on renal Pi wasting in PRT subjects, low 1,25(OH)2D may impair renal Pi reabsorption and intestinal Pi absorption, leading to a negative Pi balance.
Role of immunomodulatory agents
It has been suggested that immunosuppressive drugs may contribute to the development of hypophosphatemia. Both high doses of steroids and tacrolimus have been implicated as a cause of renal phosphorus wasting [41, 74, 75]. However, it has also been argued that a high dose of steroids in normal subjects [38] and/or other solid organ transplantations is not associated with hypophosphatemia and renal Pi wastage [14, 15]. In one study of PRT patients, despite all subjects receiving similar immunosuppressive drugs, not all developed hypophosphatemia. Furthermore, hypophosphatemia disappeared with a longitudinal follow-up although the same immunosuppressive regimen continued [48]. From these studies, one can conclude that although various immunosuppressive drugs may lead to renal phosphorus wastage, the confounding effect of other potential phosphaturic agents cannot be totally excluded.
Role of phosphorus wasting in post-renal transplantation bone disease
The pathophysiologic mechanisms of bone disease after solid organ transplantation are complex [76]. However, bone disease following renal transplantation is different from that following other solid organ transplants since bone density loss is persistent and, as a result, the cumulative fracture risk progressively increases with time. This is unlike the situation in other solid organ transplantations in which fracture rates plateau [9, 14, 76–78] (Fig. 4). In most instances, immunosuppressive drugs have been suggested as a leading cause in the development of post-transplantation bone disease [13]. Despite this hypothesis, several bone histomorphometric studies in PRT patients have shown features, such as defective bone mineralization, which are uncharacteristic of steroid-induced osteoporosis [79, 80]. The most recent study conducted by Monier-Faugere et al. demonstrated defective mineralization in the majority of PRT patients despite normal serum vitamin D and Pi concentrations [79]. However, this study ignored the pathogenic role of renal Pi wasting.
Fig 4.

The difference in cumulative risk of bone fracture in post-renal transplantation compared to other solid organ transplantation. Reprinted with permission by Ghanekar et al. [10]
At the present time, it is imperative to consider the measurement of FEpi in fasting urinary specimens. However, treatment with oral Pi may not negate renal Pi leak and may actually increase serum FGF-23 levels to further diminish renal tubular Pi reabsorption. A major misunderstanding among practicing physicians has been that the low bone mineral density (BMD) parameters established by the World Health Organization do not distinguish between osteoporosis and defective bone mineralization. Therefore, it is likely that most of the PRT patients labeled with the diagnosis of steroid-induced osteoporosis may actually have bone mineralization defects and osteomalacia. Thus, the lack of attention to correct this abnormality may result in increased cumulative risk of bone fracture in this population. Baseline and annual BMD analysis in PRT patients is logical as long as practicing physicians take into consideration that low BMD in this population may not solely be due to the effects of steroids on the bone.
To date, only a single study has shown a relationship between serum FGF-23 and bone histology on dialysis and/or after successful renal transplantation. This study, which was conducted in children on chronic peritoneal dialysis, reported an inverse relationship between serum FGF-23 concentrations and static/dynamic parameters of bone mineralization [65]. However, it is possible that the residual effect of previous treatment with vitamin D metabolites obscured the direct relationship between FGF-23 and bone mineralization parameters. Despite a lack of surveillance, persistent renal Pi wasting and negative Pi balance may be specifically important in PRT children and adolescents since peak bone density is attained as a result of normal bone growth [81]. Since bone strength is, in part, due to the material properties of the bone which, in turn, depend on bone mineral content, the lower bone Pi content can lead to decreased stiffness and, ultimately, diminished bone strength [82]. Both calcium (Ca) and Pi play an important role in bone formation, with a Ca:Pi ratio in human bone of 1.5–1.0.
Conclusion and future perspective
Since PRT hypophosphatemia is of notable clinical significance, it is necessary to further define the prevalence and natural history of PRT Pi wasting and negative Pi balance in both pediatric and adult PRT populations. Ideally, attempts should be made to develop novel therapeutic agents directly targeting FGF-23 production or inhibitory molecules targeting organ receptors. Until this has been realized, treatment with Ca sensor analogs appear to be logical since autonomous hyperphosphatonemia is, in part, correlated with excess PTH secretion. Long-term double blind control studies will be required to establish such a relationship.
Acknowledgments
The author would like to acknowledge Ms. Hadley Armstrong for her primary role in the preparation and editorial review of this manuscript.
Footnotes
Disclosures: The author has nothing to disclose.
References
- 1.Papalois VE, Najarian JS. Pediatric kidney transplantation: historic hallmarks and a personal perspective. Pediatr Transplant. 2001;5:239–245. doi: 10.1034/j.1399-3046.2001.005004239.x. [DOI] [PubMed] [Google Scholar]
- 2.Calne RY, White DJ, Thiru S, Evans DB, McMaster P, Dunn DC, Craddock GN, Pentlow BD, Rolles K. Cyclosporin A in patients receiving renal allografts from cadaver donors. Lancet. 1978;2:1323–1327. doi: 10.1016/s0140-6736(78)91970-0. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein G, Kremer AB, Barnes L, Hirsch RL. OKT3 monoclonal antibody reversal of renal and hepatic rejection in pediatric patients. J Pediatr. 1987;111:1046–1050. doi: 10.1016/s0022-3476(87)80054-9. [DOI] [PubMed] [Google Scholar]
- 4.Jensen CW, Jordan ML, Schneck FX, Shapiro R, Tzakis A, Hakala TR, Starzl TE. Pediatric renal transplantation under FK 506 immunosuppression. Transplant Proc. 1991;23:3075–3077. [PMC free article] [PubMed] [Google Scholar]
- 5.Vincenti F, Kirkman R, Light S, Bumgardner G, Pescovitz M, Halloran P, Neylan J, Wilkinson A, Ekberg H, Gaston R, Backman L, Burdick J. Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal transplantation. Daclizumab Triple Therapy Study Group. N Engl J Med. 1998;338:161–165. doi: 10.1056/NEJM199801153380304. [DOI] [PubMed] [Google Scholar]
- 6.Saland JM. Osseous complications of pediatric transplantation. Pediatr Transplant. 2004;8:400–415. doi: 10.1111/j.1399-3046.2004.00167.x. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez CP, Salusky IB, Kuizon BD, Ramirez JA, Gales B, Ettenger RB, Goodman WG. Bone disease in children and adolescents undergoing successful renal transplantation. Kidney Int. 1998;53:1358–1364. doi: 10.1046/j.1523-1755.1998.00866.x. [DOI] [PubMed] [Google Scholar]
- 8.Smets YF, van der Pijl JW, de Fijter JW, Ringers J, Lemkes HH, Hamdy NA. Low bone mass and high incidence of fractures after successful simultaneous pancreas-kidney transplantation. Nephrol Dial Transplant. 1998;13:1250–1255. doi: 10.1093/ndt/13.5.1250. [DOI] [PubMed] [Google Scholar]
- 9.Vautour LM, Melton LJ, 3rd, Clarke BL, Achenbach SJ, Oberg AL, McCarthy JT. Long-term fracture risk following renal transplantation: a population-based study. Osteoporos Int. 2004;15:160–167. doi: 10.1007/s00198-003-1532-y. [DOI] [PubMed] [Google Scholar]
- 10.Ghanekar H, Welch BJ, Moe OW, Sakhaee K. Post-renal transplantation hypophosphatemia: a review and novel insights. Curr Opin Nephrol Hypertens. 2006;15:97–104. doi: 10.1097/01.mnh.0000203187.49890.cc. [DOI] [PubMed] [Google Scholar]
- 11.Epstein S. Post-transplantation bone disease: the role of immunosuppressive agents and the skeleton. J Bone Miner Res. 1996;11:1–7. doi: 10.1002/jbmr.5650110102. [DOI] [PubMed] [Google Scholar]
- 12.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vega D, Sakhaee K. Osteoporosis following solid organ transplantation. Future Rheumatol. 2007;2:341–345. [Google Scholar]
- 14.Shane E, Rivas M, Staron RB, Silverberg SJ, Seibel MJ, Kuiper J, Mancini D, Addesso V, Michler RE, Factor-Litvak P. Fracture after cardiac transplantation: a prospective longitudinal study. J Clin Endocrinol Metab. 1996;81:1740–1746. doi: 10.1210/jcem.81.5.8626827. [DOI] [PubMed] [Google Scholar]
- 15.Ninkovic M, Skingle SJ, Bearcroft PW, Bishop N, Alexander GJ, Compston JE. Incidence of vertebral fractures in the first three months after orthotopic liver transplantation. Eur J Gastroenterol Hepatol. 2000;12:931–935. doi: 10.1097/00042737-200012080-00013. [DOI] [PubMed] [Google Scholar]
- 16.Takeda E, Taketani Y, Sawada N, Sato T, Yamamoto H. The regulation and function of phosphate in the human body. Biofactors. 2004;21:345–355. doi: 10.1002/biof.552210167. [DOI] [PubMed] [Google Scholar]
- 17.Levi M, Kempson SA, Lotscher M, Biber J, Murer H. Molecular regulation of renal phosphate transport. J Membr Biol. 1996;154:1–9. doi: 10.1007/s002329900127. [DOI] [PubMed] [Google Scholar]
- 18.Biber J, Custer M, Werner A, Kaissling B, Murer H. Localization of NaPi-1, a Na/Pi cotransporter, in rabbit kidney proximal tubules. II. Localization by immunohistochemistry. Pflugers Arch. 1993;424:210–215. doi: 10.1007/BF00384344. [DOI] [PubMed] [Google Scholar]
- 19.Custer M, Lotscher M, Biber J, Murer H, Kaissling B. Expression of Na-P(I) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Physiol. 1994;266:F767–F774. doi: 10.1152/ajprenal.1994.266.5.F767. [DOI] [PubMed] [Google Scholar]
- 20.Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K. Growth-related renal type II Na/Pi cotransporter. J Biol Chem. 2002;277:19665–19672. doi: 10.1074/jbc.M200943200. [DOI] [PubMed] [Google Scholar]
- 21.Hilfiker H, Hattenhauer O, Traebert M, Forster I, Murer H, Biber J. Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc Natl Acad Sci USA. 1998;95:14564–14569. doi: 10.1073/pnas.95.24.14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murer H, Hernando N, Forster I, Biber J. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev. 2000;80:1373–1409. doi: 10.1152/physrev.2000.80.4.1373. [DOI] [PubMed] [Google Scholar]
- 23.Tenenhouse HS. Phosphate transport: molecular basis, regulation and pathophysiology. J Steroid Biochem Mol Biol. 2007;103:572–577. doi: 10.1016/j.jsbmb.2006.12.090. [DOI] [PubMed] [Google Scholar]
- 24.Berndt TJ, Schiavi S, Kumar R. “Phosphatonins” and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol. 2005;289:F1170–F1182. doi: 10.1152/ajprenal.00072.2005. [DOI] [PubMed] [Google Scholar]
- 25.Yan X, Yokote H, Jing X, Yao L, Sawada T, Zhang Y, Liang S, Sakaguchi K. Fibroblast growth factor 23 reduces expression of type IIa Na+/Pi co-transporter by signaling through a receptor functionally distinct from the known FGFRs in opossum kidney cells. Genes Cells. 2005;10:489–502. doi: 10.1111/j.1365-2443.2005.00853.x. [DOI] [PubMed] [Google Scholar]
- 26.Levi M, Shayman JA, Abe A, Gross SK, McCluer RH, Biber J, Murer H, Lötscher M, Cronin RE. Dexamethasone modulates rat renal brush border membrane phosphate transporter mRNA and protein abundance and glycosphingolipid composition. J Clin Invest. 1995;96:207–216. doi: 10.1172/JCI118022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alcalde AI, Sarasa M, Raldua D, Aramayona J, Morales R, Biber J, Murer H, Levi M, Sorribas V. Role of thyroid hormone in regulation of renal phosphate transport in young and aged rats. Endocrinology. 1999;140:1544–1551. doi: 10.1210/endo.140.4.6658. [DOI] [PubMed] [Google Scholar]
- 28.Ambuhl PM, Meier D, Wolf B, Dydak U, Boesiger P, Binswanger U. Metabolic aspects of phosphate replacement therapy for hypophosphatemia after renal transplantation: impact on muscular phosphate content, mineral metabolism, and acid/base homeostasis. Am J Kidney Dis. 1999;34:875–883. doi: 10.1016/S0272-6386(99)70045-4. [DOI] [PubMed] [Google Scholar]
- 29.Levi M. Post-transplant hypophosphatemia. Kidney Int. 2001;59:2377–2387. doi: 10.1046/j.1523-1755.2001.00755.x. [DOI] [PubMed] [Google Scholar]
- 30.Felsenfeld AJ, Gutman RA, Drezner M, Llach F. Hypophosphatemia in long-term renal transplant recipients: effects on bone histology and 1, 25-dihydroxycholecalciferol. Miner Electrolyte Metab. 1986;12:333–341. [PubMed] [Google Scholar]
- 31.Portale AA, Halloran BP, Morris RC., Jr Physiologic regulation of the serum concentration of 1, 25-dihydroxyvitamin D by phosphorus in normal men. J Clin Invest. 1989;83:1494–1499. doi: 10.1172/JCI114043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green J, Debby H, Lederer E, Levi M, Zajicek HK, Bick T. Evidence for a PTH-independent humoral mechanism in post-transplant hypophosphatemia and phosphaturia. Kidney Int. 2001;60:1182–1196. doi: 10.1046/j.1523-1755.2001.0600031182.x. [DOI] [PubMed] [Google Scholar]
- 33.Heaney RP, Abrams S, Dawson-Hughes B, Looker A, Marcus R, Matkovic V, Weaver C. Peak bone mass. Osteoporos Int. 2000;11:985–1009. doi: 10.1007/s001980070020. [DOI] [PubMed] [Google Scholar]
- 34.Garabedian M, Silve C, Levy D, Bourdeau A, Ulmann A, Broyer M, Balsan S. Chronic hypophosphatemia in kidney transplanted children and young adults. Adv Exp Med Biol. 1980;128:249–254. doi: 10.1007/978-1-4615-9167-2_30. [DOI] [PubMed] [Google Scholar]
- 35.Brodehl J, Krause A, Hoyer PF. Assessment of maximal tubular phosphate reabsorption: comparison of direct measurement with the nomogram of Bijvoet. Pediatr Nephrol. 1988;2:183–189. doi: 10.1007/BF00862587. [DOI] [PubMed] [Google Scholar]
- 36.Bernheim J, Touraine JL, David L, Faivre JM, Traeger J. Evolution of secondary hyperparathyroidism after renal transplantation. Nephron. 1976;16:381–387. doi: 10.1159/000180623. [DOI] [PubMed] [Google Scholar]
- 37.Claesson K, Hellman P, Frodin L, Rastad J. Prospective study of calcium homeostasis after renal transplantation. World J Surg. 1998;22:635–641. doi: 10.1007/s002689900446. discussion 41-2. [DOI] [PubMed] [Google Scholar]
- 38.Rosenbaum RW, Hruska KA, Korkor A, Anderson C, Slatopolsky E. Decreased phosphate reabsorption after renal transplantation: Evidence for a mechanism independent of calcium and parathyroid hormone. Kidney Int. 1981;19:568–578. doi: 10.1038/ki.1981.54. [DOI] [PubMed] [Google Scholar]
- 39.Salusky IB, Coburn JW, Brill J, Foley J, Slatopolsky E, Fine RN, Goodman WG. Bone disease in pediatric patients undergoing dialysis with CAPD or CCPD. Kidney Int. 1988;33:975–982. doi: 10.1038/ki.1988.96. [DOI] [PubMed] [Google Scholar]
- 40.Salusky IB, Ramirez JA, Oppenheim W, Gales B, Segre GV, Goodman WG. Biochemical markers of renal osteodystrophy in pediatric patients undergoing CAPD/CCPD. Kidney Int. 1994;45:253–258. doi: 10.1038/ki.1994.31. [DOI] [PubMed] [Google Scholar]
- 41.Graf H, Kovarik J, Stummvoll HK, Wolf A, Pinggera WF. Handling of phosphate by the transplanted kidney. Proc Eur Dial Transplant Assoc. 1979;16:624–629. [PubMed] [Google Scholar]
- 42.Parfitt AM, Kleerekoper M, Cruz C. Reduced phosphate reabsorption unrelated to parathyroid hormone after renal transplantation: implications for the pathogenesis of hyperparathyroidism in chronic renal failure. Miner Electrolyte Metab. 1986;12:356–362. [PubMed] [Google Scholar]
- 43.White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Jüppner H, Econs MJ. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86:497–500. doi: 10.1210/jcem.86.2.7408. [DOI] [PubMed] [Google Scholar]
- 44.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Jüppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 45.White KE, Evans WE, O'Riordan J, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 46.Riancho JA, de Francisco AL, del Arco C, Amado JA, Cotorruelo JG, Arias M, Gonzalez-Macias J. Serum levels of 1, 25-dihydroxyvitamin D after renal transplantation. Miner Electrolyte Metab. 1988;14:332–337. [PubMed] [Google Scholar]
- 47.Steiner RW, Ziegler M, Halasz NA, Catherwood BD, Manolagas S, Deftos LJ. Effect of daily oral vitamin D and calcium therapy, hypophosphatemia, and endogenous 1–25 dihydroxycholecalciferol on parathyroid hormone and phosphate wasting in renal transplant recipients. Transplantation. 1993;56:843–846. doi: 10.1097/00007890-199310000-00013. [DOI] [PubMed] [Google Scholar]
- 48.Bhan I, Shah A, Holmes J, Isakova T, Gutierrez O, Burnett SM, Jüppner H, Wolf M. Post-transplant hypophosphatemia: tertiary ‘hyperphosphatoninism’? Kidney Int. 2006;70:1486–1494. doi: 10.1038/sj.ki.5001788. [DOI] [PubMed] [Google Scholar]
- 49.Imanishi Y, Inaba M, Nakatsuka K, Nagasue K, Okuno S, Yoshihara A, Miura M, Miyauchi A, Kobayashi K, Miki T, Shoji T, Ishimura E, Nishizawa Y. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int. 2004;65:1943–1946. doi: 10.1111/j.1523-1755.2004.00604.x. [DOI] [PubMed] [Google Scholar]
- 50.Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, Linhardt RJ, Ornitz DM, Mohammadi M, White KE. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology. 2005;146:4647–4656. doi: 10.1210/en.2005-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imel EA, Econs MJ. Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol. 2005;16:2565–2575. doi: 10.1681/ASN.2005050573. [DOI] [PubMed] [Google Scholar]
- 52.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 53.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. [Google Scholar]
- 54.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD. FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol. 2008;19:2342–2350. doi: 10.1681/ASN.2007121301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baum M, Syal A, Quigley R, Seikaly M. Role of prostaglandins in the pathogenesis of X-linked hypophosphatemia. Pediatr Nephrol. 2006;21:1067–1074. doi: 10.1007/s00467-006-0126-2. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res. 2008;23:939–948. doi: 10.1359/jbmr.080220. [DOI] [PubMed] [Google Scholar]
- 58.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 59.Gupta A, Winer K, Econs MJ, Marx SJ, Collins MT. FGF-23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492. doi: 10.1210/jc.2004-0724. [DOI] [PubMed] [Google Scholar]
- 60.Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519–1524. doi: 10.1210/jc.2004-1039. [DOI] [PubMed] [Google Scholar]
- 61.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 62.Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, Fukagawa M. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44:250–256. doi: 10.1053/j.ajkd.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 63.Sato T, Tominaga Y, Ueki T, Goto N, Matsuoka S, Katayama A, Haba T, Uchida K, Nakanishi S, Kazama JJ, Gejyo F, Yamashita T, Fukagawa M. Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis. 2004;44:481–487. [PubMed] [Google Scholar]
- 64.Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Jüppner H, Wolf M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–2215. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 65.Wesseling-Perry K, Pereira RC, Wang H, Elashoff RM, Sahney S, Gales B, Jüppner H, Salusky IB. Relationship between plasma fibroblast growth factor-23 concentration and bone mineralization in children with renal failure on peritoneal dialysis. J Clin Endocrinol Metab. 2009;94:511–517. doi: 10.1210/jc.2008-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pande S, Ritter CS, Rothstein M, Wiesen K, Vassiliadis J, Kumar R, Schiavi SC, Slatapolsky E, Brown AJ. FGF-23 and sFRP-4 in chronic kidney disease and post-renal transplantation. Nephron Physiol. 2006;104:23–32. doi: 10.1159/000093277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–2688. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 68.Liu S, Tang W, Zhou J, Vierthaler L, Quarles LD. Distinct roles for intrinsic osteocyte abnormalities and systemic factors in regulation of FGF23 and bone mineralization in Hyp mice. Am J Physiol Endocrinol Metab. 2007;293:E1636–E1644. doi: 10.1152/ajpendo.00396.2007. [DOI] [PubMed] [Google Scholar]
- 69.Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, Jan De Beur SM, Schiavi SC, Kumar R. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67:54–68. doi: 10.1006/geno.2000.6235. [DOI] [PubMed] [Google Scholar]
- 72.Liang CT, Barnes J, Balakir R, Cheng L, Sacktor B. In vitro stimulation of phosphate uptake in isolated chick renal cells by 1, 25-dihydroxycholecalciferol. Proc Natl Acad Sci USA. 1982;79:3532–3536. doi: 10.1073/pnas.79.11.3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kurnik BR, Hruska KA. Effects of 1, 25-dihydroxycholecalciferol on phosphate transport in vitamin D-deprived rats. Am J Physiol. 1984;247:F177–F184. doi: 10.1152/ajprenal.1984.247.1.F177. [DOI] [PubMed] [Google Scholar]
- 74.Loffing J, Lotscher M, Kaissling B, Biber J, Murer H, Seikaly M, Alpern RJ, Levi M, Baum M, Moe OW. Renal Na/H exchanger NHE-3 and Na-PO4 cotransporter NaPi-2 protein expression in glucocorticoid excess and deficient states. J Am Soc Nephrol. 1998;9:1560–1567. doi: 10.1681/asn.v991560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Falkiewicz K, Nahaczewska W, Boratynska M, Owczarek H, Klinger M, Kaminska D, Wozniak M, Szepietowski T, Patrzalek D. Tacrolimus decreases tubular phosphate wasting in renal allograft recipients. Transplant Proc. 2003;35:2213–2215. doi: 10.1016/s0041-1345(03)00765-6. [DOI] [PubMed] [Google Scholar]
- 76.Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab. 2005;90:2456–2465. doi: 10.1210/jc.2004-1978. [DOI] [PubMed] [Google Scholar]
- 77.Feller RB, McDonald JA, Sherbon KJ, McCaughan GW. Evidence of continuing bone recovery at a mean of 7 years after liver transplantation. Liver Transpl Surg. 1999;5:407–413. doi: 10.1002/lt.500050507. [DOI] [PubMed] [Google Scholar]
- 78.Pichette V, Bonnardeaux A, Prudhomme L, Gagne M, Cardinal J, Ouimet D. Long-term bone loss in kidney transplant recipients: a cross-sectional and longitudinal study. Am J Kidney Dis. 1996;28:105–114. doi: 10.1016/s0272-6386(96)90138-9. [DOI] [PubMed] [Google Scholar]
- 79.Monier-Faugere MC, Mawad H, Qi Q, Friedler RM, Malluche HH. High prevalence of low bone turnover and occurrence of osteomalacia after kidney transplantation. J Am Soc Nephrol. 2000;11:1093–1099. doi: 10.1681/ASN.V1161093. [DOI] [PubMed] [Google Scholar]
- 80.Carlini RG, Rojas E, Weisinger JR, Lopez M, Martinis R, Arminio A, Bellorin-Font E. Bone disease in patients with long-term renal transplantation and normal renal function. Am J Kidney Dis. 2000;36:160–166. doi: 10.1053/ajkd.2000.8289. [DOI] [PubMed] [Google Scholar]
- 81.Burckhardt P, Michel C. The peak bone mass concept. Clin Rheumatol. 1989;8(Suppl 2):16–21. doi: 10.1007/BF02207228. [DOI] [PubMed] [Google Scholar]
- 82.Seeman E. Bone quality. Osteoporos Int. 2003;14(Suppl 5):S3–7. doi: 10.1007/s00198-003-1465-5. [DOI] [PubMed] [Google Scholar]