

Abstract
The brain is an ever-changing organ that encodes memories and directs behavior. Neuroanatomical studies have revealed structural plasticity of neural architecture, and advances in gene expression technology and epigenetics have demonstrated new mechanisms underlying the brain’s dynamic nature. Stressful experiences challenge the plasticity of the brain, and prolonged exposure to environmental stress redefines the normative transcriptional profile of both neurons and glia, and can lead to the onset of mental illness. A more thorough understanding of normal and abnormal gene expression is needed to define the diseased brain and improve current treatments for psychiatric disorders. The efforts to describe gene expression networks have been bolstered by microarray and RNA-sequencing technologies. The heterogeneity of neural cell populations and their unique microenvironments, coupled with broad ranging interconnectivity, makes resolving this complexity exceedingly challenging and requires the combined efforts of single cell and systems level expression profiling to identify targets for therapeutic intervention.
Keywords: epigenetics, gene expression, microarray, RNA-seq, stress
Introduction
Each cell in the body exhibits a typical pattern of gene expression that characterizes its phenotype. Gene expression networks (GEN) define these expression patterns within a cell and reflect the molecular dynamics underlying its adaptation to the environment. The brain is a plastic structure that responds rapidly to various inputs from the environment, as was first recognized by “enriched environment” studies [1]. The concept of neuroplasticity has come to include synaptic turnover, shrinkage and elongation of dendrites, and potentially neurogenesis in the adult brain, despite the recognition that neurogenesis may be limited [2]. This ability to respond and adapt is crucial for survival, and serves as the molecular basis of higher order processing such as learning and memory. In some cases, excess exposure to environmental cues can become detrimental, manifesting in psychiatric disorders such as bipolar disorder (BPD), major depressive disorder (MDD) and post-traumatic stress disorder (PTSD) [3, 4]. Understanding the components of GENs and how they interact with each other to facilitate adaptation will aid in discovering better treatments for the diseased brain and in deciphering the causes of their onset.
Traditionally, examination of GENs was a slow and laborious process because researchers were limited to looking at the reaction of individual genes in response to a manipulation. Techniques such as in situ-hybridization and northern blotting give semi-quantitative measurements of RNA levels, but suffer from a lack of scalability, making whole transcriptome analysis prohibitive in terms of time and cost. The search for methods to speed up transcript expression analysis has evolved from differential display [5], to sequential analysis of gene expression (SAGE)[6], to gene microarrays [7], and to RNA sequencing [8], all of which have contributed to the identification and categorization of GENs. Microarray and RNA-sequencing technologies have recently allowed examination of the whole transcriptome in a single experiment [9, 10]. This increase in target throughput has allowed more complex networks to be elucidated in a shorter amount of time. These technologies are not without their limitations, but nonetheless offer the ability to rapidly examine increasingly complex regulatory interactions within GENs. High throughput sequencing also provides the capacity to correlate changes in the transcriptome with the unique underlying genotype of the individual.
Through the use of these technologies, much has been learned about how RNAs, proteins, and DNA each contribute to the regulation of GENs. Modulation of just one component can drastically affect the expression of numerous others. For example, a modest increase in a transcription factor such as c-myc, which has been estimated to regulate as much as 15% of all human genes, can have dramatic effects on multiple pathways and lead to the development of cancers [11]. GEN’s are often auto-regulated, with transcriptional outputs coding for enhancers or repressors of further transcription [12]. Depending on the stimulus, this can create an amplification where tens to hundreds of genes are transcribed or silenced.
Changes in gene expression can also occur through epigenetic modifications. Epigenetics embodies the quest for understanding how modifications to the proteins and functional groups that bind and sequester our DNA regulate gene expression without fundamentally changing the DNA sequence [13, 14]. Modifications to the tails of histones associated with DNA can have a great impact on the regulation of the underlying gene through diverse mechanisms such as sequestering a transcription start site or opening up an enhancer sequence to allow protein binding [14]. Direct methylation of cytosine bases in DNA is another epigenetic mechanism that has a variety of implications for gene expression in a context dependent manner [15]. The addition of a methyl group near a transcription start site can serve to prevent initiation of transcription [16], whereas methylation within the gene body is associated with transcribed genes, particularly in the brain [17]. Finally, a stimulus that produces a change in activity of epigenetic modifying enzymes, such as histone deacetylases (HDACs) and histone acetyltransferases (HATs), can have drastic effects on the expression of numerous genes by increasing or decreasing the rate at which the histone marks can be modified. Importantly, the use of drugs that can manipulate the levels of these modifying enzymes are already being explored for the treatment of mental illnesses [18].
Further complicating the picture, multiple stimuli can induce diverse changes in GENs and different individuals can exhibit unique levels of reactivity to the same stimuli. The type of stress, intensity, duration, and the developmental stage of an animal have important implications for the transcriptional output, such as the differences in transcription observed after either acute or chronic stress[19]. Differences in reactivity may be the result of alterations to the GEN landscape by environmental factors that occur during development, such as stress and exposure to pollutants, or as a result of aging. For example, the same stressor produces a different pattern of hippocampal gene expression in a young unstressed mouse than in an older mouse with a history chronic stress [19]. Understanding how external factors cause semi-permanent and permanent changes in GENs will be key to formulating better methods for disease prevention, as well as possible treatments for disease. Here we examine the present state of mapping GENs in the brain in response to stress, and speculate about where new advances might lie.
Glucocorticoids drive changes to GENs after stress
Glucocorticoids (GCs) are the main signaling molecules through which stress initiates gene expression changes in the brain [20]. GCs bind glucocorticoid receptors (GR), which become activated transcription factors once bound [21]. Newly stimulated GRs are translocated to the nucleus and bind to a specific DNA sequence, referred to as glucocorticoid response elements (GRE), to activate new transcriptional events (Fig 1A). Depending on strength and frequency of activation, GR can act to enhance or inhibit new transcription [22]. While GCs are administered clinically to suppress inflammation, they can also stimulate the immune system and modulate cellular and humoral immune balance[23]. GC actions in the brain remain an area of intense study (see Box 1). Besides genomic effects (Fig 1A), GRs translocate to the mitochondria where they biphasically regulate calcium balance and free radical levels [24]. They can also act at the synapse to stimulate the release of endocannabinoids and inhibit presynaptic glutamate and GABA release (Fig. 1B)[25]. Finally, they can stimulate glutamate release via a membrane associated form of the mineralocorticoid receptor (MR) [26] (Box 1).
Figure 1.
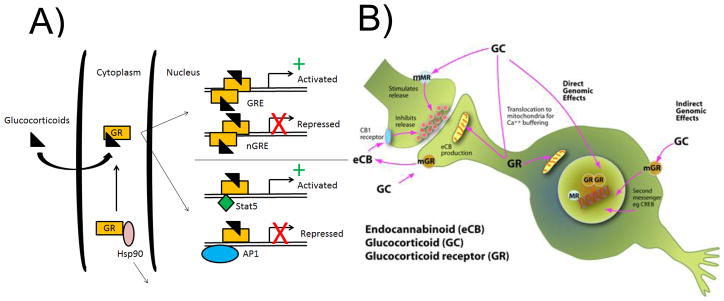
Diverse mechanisms of glucocorticoid receptor signaling. GR was initially identified as a transcription factor that is translocated to the nucleus upon ligand binding to alter gene expression (A). Activated GR can bind the glucocorticoid response element (GRE) to activate transcription (+) or to the negative GRE to suppress transcription (X). Further, GR can also act together with other transcription factors, such as Stat5 and AP1, to activate or suppress transcription at other genomic sites [104]. More recently, several studies have demonstrated that GR can function as a signaling molecule throughout the cell. In some neurons, it is localized at post-synaptic dendritic spines that are located far from the soma (mGR; membrane-bound GR) and can also be translocated to the mitochondria to alter Ca+ release (B). (A is adapted from Reichardt and Schutz 1997 and B was drawn by Susan Strider)
Box 1. Glucocorticoid Signaling.
Since the identification of adrenal steroid receptors in the hippocampus [105], its role in the actions of stress on the brain has been a major focus of many laboratories [106]. Decades of research have yielded a more thorough understanding of its subcellular localization, its binding affinity, its associated proteins, and its cellular effects when activated. Here we briefly examine what is known about GR and fellow steroid receptor the mineralocorticoid receptor (MR).
GR and MR are activated when they bind GCs released from the adrenal glands. GR is expressed in most cell types throughout the body, whereas MR is expressed more specifically in brain regions such as the hippocampus and at lower levels or not at all in other brain regions [107, 108]. Interestingly, the nuclear form of MR has a higher affinity for GCs than does GR: MR is almost fully occupied under basal conditions [106] and thus nuclear GR responds to fluctuations of GC levels [33]. However, there is a non-genomic, membrane-associated form of MR that responds to stress levels of CORT and stimulates glutamate release [26] (Fig. 1).
Under resting conditions, GR and MR that are destined for the cell nucleus are located in the cytosol and are bound by various chaperone proteins to stabilize their 3-D conformation. Hsp-70 and Hsp-90 are two chaperones that have a great impact on GR assembly and activity. Importantly, Hsp-70 and Hsp-90 are ATP-dependent and thus rely on co-chaperone proteins such as Hsp-40 and BAG-1 for proper function. The chaperones and co-chaperones complexed with GR help to determine its affinity for CORT. Changes in expression of GR, chaperones, or co-chaperones can all affect cellular outputs of stress, and many are under investigation for their roles in stress-related disease [109]. When GCs are released, they bind to the ligand-binding domain of GR and cause Hsp-70 to dissociate from the complex. New chaperone proteins such as FKBP51 and p23 produce a conformation that has high affinity for GCs. Upon binding, FKBP51 is displaced by FKBP52 and allows for translocation to the nucleus to affect gene transcription [110].
Once GR is inside the nucleus it can act as either a transcriptional activator or repressor [111]. When the DNA binding domain recognizes the palindromic glucocorticoid response element (GRE) in the DNA, GR acts as an enhancer of transcription and in concert with other transcription factors recruits the basal transcriptional machinery. GR has also been shown to bind negative glucocorticoid response elements (nGRE) to block new transcriptional events, although consensus sequences for nGREs have not been thoroughly described. In addition to directly acting as a transcription factor, GR has been shown to affect transcription through non-genomic protein-protein interactions [112]. This protein cross-talk enables GR to produce transcriptional changes without binding DNA.
While cortisol in humans and corticosterone (both referred to hereafter as CORT) in rodents are the main GCs released during stressful events, pharmacological administration of these substances give differing molecular responses from those produced in standard models of stress such as Forced Swim Test (FST) and Chronic Restraint Stress (CRS) [19, 27]. In addition to acting as a transcription factor, GR can also alter the activity of other proteins that may or may not act as transcription factors themselves, such as AP-1 [28](Fig. 1B). Complicating the matter further is the finding that activation of alternate pathways alters the transcriptional output of activated GR [29]. Rao et al., (2011) examined the gene expression response of GC-induced transcriptional activation with and without activation of the NF-κB pathway using TNFα. Concurrent activation of both pathways in vitro produced a unique transcriptional pattern that was distinct from the expression patterns observed with activation of either of the pathways alone. This is not entirely surprising, given the strong regulatory interconnectedness of the two pathways [30], but nonetheless raises new questions about individual responses to stress. How does the presence or absence of other factors influence an organism’s capacity to deal with stress? What are these other factors, and how many are there?
Different stressors have unique effects on GENs
There are four factors known to determine the differential response of GENs to stress. First, the type of stressor plays a key role in the transcriptional output of stress, as different modalities may activate competing pathways. In many cases, a single ligand is capable of binding multiple receptors, though usually with differing affinity. For instance, CORT binds both GR and MR, though it binds nuclear MR with greater affinity [31, 32], and the nuclear GR is more responsive to fluctuating ultradian levels of CORT [33]. Depending on the type of stressor, the amount of CORT released can differ, leading to differences in GR activation and transcriptional output. For example, acute swim stress and acute restraint both significantly elevate CORT in rodents, but not to the same level [34].
The intensity and duration of stressors must also be considered because they can determine whether the response is adaptive and protective, or maladaptive and detrimental [35]. Efficiently turning on and off a response to an acute stressor leads to adaptation by a process referred to as “allostasis” (the active process of maintaining homeostasis)[36, 37], which includes such positive effects as enhanced memory of danger and increased adaptive immunity [38, 39]. Conceptually, the shift from the adaptive to the negative effects of stress has been described as an inverted U-shape, where increasing the intensity as well as duration of the stressor and the stress response leads to impairment of memory and immune function as well as promoting cardiovascular disease and metabolic syndrome [35] that is termed “allostatic load/overload” [37, 40]. In the hippocampus, chronic restraint stress (CRS) that promotes allostatic load leads to altered reactivity in other pathways than those activated by acute stress and leads to a unique gene expression profile compared with acute stress or CORT administration alone [19](Fig. 2A).
Figure 2.

Microarray profiles illustrate the dynamic nature of in vivo gene expression networks. A: Venn diagrams illustrating genes that are significantly increased (up arrow) or decreased (down arrow) by an acute forced swim stress (FST; yellow), chronic restraint stress (CRS; blue), or corticosterone injection (Cort; purple) from whole hippocampal microarrays. The lack of overlap between conditions demonstrates that acute and chronic stress produce unique gene expression profiles and that neither response is equivalent to corticosterone administration alone. B: Venn diagrams illustrate genes that are significantly changed by an acute forced swim stress (FST; yellow), a forced swim stress administered immediately after CRS (CRS+FST; orange), or a forced swim stress administered after the mice have been given three weeks in their home cage to recover from a three week CRS (Rec+FST; green). While the same acute stress was used in each condition, mice with a history of chronic stress (orange and green) showed a unique gene expression profile from naive animals (FST; yellow). Further, mice that were allowed to recover from a chronic stress (green) still exhibited a distinct response from the naive animals (yellow), suggesting that recovery from stress does not necessarily result in a return to the stress-naive baseline. C: Scatter plot of all genes from the microarray in which genes changed by CRS (x-axis) are plotted against genes changed after recovery from CRS (y-axis). The majority of genes that were increased by CRS were found to be decreased in recovery (10, 682; lower right quadrant), suggesting that many genes do return to baseline. However, 2,905 genes (upper right) were elevated by CRS and remained elevated after recovery or were decreased by CRS and remained decreased after recovery (3,608; lower right). Adapted with permission from Gray et al., 2013 [19].
Gray et al. (2013) showed CRS can change gene expression in the hippocampus in a number of pathways including NF-κB, cytoskeletal remodeling pathways, and insulin signaling pathways. In addition, CRS produced an altered landscape of reactivity in the hippocampus. When an animal was exposed to a novel, acute stressor following CRS the transcriptional output was dramatically different from that of a naïve animal that received the same acute stress (Fig. 2B). The mechanisms that lead to this altered reactivity are currently under investigation. Further, mice were allowed to recover after CRS to investigate how some changes in reactivity persist while others revert back to baseline. While the majority of genes return to basal levels after a 3 week recovery period after CRS, many genes establish a new baseline that reflects the stress history of the animal (Fig. 2C). Finally, despite recovery from CRS the transcriptome’s reactivity to a novel acute stressor was distinct from the naïve acute stress response (Fig. 2B; green circle), suggesting that recovery from CRS is not a return to the stress-naïve baseline but reflects a new setpoint and reactivity profile for the GEN of the hippocampus.
Lastly, the developmental stage at which an animal is exposed to a stressor plays a fundamental role in the response and future outcome. In general, resilient individuals can adapt and recover when given a period of rest following a stressful event. However, stressors that occur prenatally [41, 42] and during a critical post-natal period [43–45] can have lasting effects that impede development, shape adult behavior, and even lead to disease [46]. Adolescent and pubertal animals have a unique stress response as well, which leads to an increased incidence in physical and mental dysfunction [47]. Finally, studies of aged rodents have revealed a diminished capacity to recover from stress compared with younger peers [48]. Old rats subjected to chronic restraint exhibited the characteristic dendritic retraction in the prefrontal cortex compared to unstressed controls, yet unlike the young and middle age rats that restored their dendritic length after the stress was ended, aged rats did not show any recovery of dendritic length [49]. Yet, when recovery of dendritic shrinkage occurs in prefrontal cortex, the dendrites that grow out are more proximal to the cell body than those that were shortened by CRS, indicating that even though total dendritic length had recovered the neurons do not return to pre-stress morphology [50]. Together, these studies illustrate how both neuronal morphology and the transcriptome can recover after stress, but are not restored to a stress-naïve state.
The interaction between genes and environment shape the GEN landscape
How the brain responds to influences from the environment is a fundamental question that underlies all of neuroscience. The genome itself plays a central role in shaping and determining patterns of gene expression, as well as how experience-dependent changes create individual differences that determine behavior and in some cases onset of disease. Identification of key factors that respond to external cues has been a focus of keen interest for understanding general brain function. Recently, many of the “risk genes” associated with mental illness have been hypothesized to survive evolutionarily in the human genome because they can confer many adaptable benefits in a supportive, non-stressful environment [51].
Genetic predispositions can alter GEN reactivity
One gene that has been widely studied because its levels in the brain are associated with risk for mental disorders is brain derived neurotrophic factor (BDNF). BDNF plays a crucial role in the growth and maintenance of neurons and has been extensively investigated in response to stress [52, 53]. Studies of patients with MDD have shown decreased serum and plasma levels of BDNF [54, 55] and post-mortem studies have shown decreased protein levels in the hippocampus [56–58]. People with a specific single nucleotide polymorphism (SNP) that results in a methionine substitution for valine (Val66Met) in their BDNF gene are more prone to some psychiatric disorders [59, 60]. BDNF has also been implicated in the structural and behavioral changes seen after stress, and its expression has been linked to changes in the GEN of numerous brain regions [61]. Further, the endurance of BDNF polymorphisms in the genome suggests that it may confer aspects of enhanced adaptability in the right environment despite its correlation with increased incidence of mood disorders.
BDNF expression has proven to be highly dynamic with stress, as time course studies have shown immediate increases in BDNF in acutely stressed rats peaking at 60 minutes post-stress, followed by a significant decrease at 180 minutes [62]. The levels of BDNF expression following chronic stress remain highly dynamic, as the time-point at which BDNF levels are evaluated appears to determine whether BDNF is increased or decreased. Chronic stress was originally thought to decrease levels of BDNF in the hippocampus [63]. However, Naert et al. showed that chronic restraint stress shifts the baseline expression level of BDNF higher than control, and that the acute response from each stress is a decrease in BDNF levels, not an elevation of BDNF levels [64]. This is the opposite of what is seen in acutely stressed animals [62], and is a response that lasts beyond the cessation of stress and into recovery [19]. This example is illustrative of one of the major limitations of these technologies, in that longitudinal studies from the same animal are not possible. Examining multiple time points rather than following how expression changes over time leads to speculations about regulation that can only be answered by increasing the number of data points sampled. However, even snapshots give useful indications about patterns of expression, and their relevance should not be discounted.
Environmental factors can alter GEN reactivity
One area of research that has shown great promise in elucidating the mechanisms underlying altered reactivity has focused on the epigenome (see Box 2). The revelation that a modification of the histones through methylation, acetylation, or various other functional groups leads to lasting and profound changes in gene expression has brought us one step closer to understanding many major diseases [65, 66]. Some epigenetic changes can be extremely long lasting, as in the case of the Dutch Winter Famine: adults screened six decades after being prenatally exposed to Nazi-imposed famine were shown to have less DNA methylation on specific genes than siblings who were not exposed [67]. The prenatal environment appears to be vitally important for setting up epigenetic marks for proper development, and as a result can be sensitive to external exposures that may have a great impact on future outcomes [68]. Moreover, Michael Meaney and others have shown that this critical period of epigenetic programming lasts beyond the womb and into early life. In addition to exposures from the external environment, parental behavior can play a role in the offspring’s adult behavior and predisposition to disease [69, 70]. For instance, the licking and grooming behavior of female rats when rearing their pups has been shown to have consistent epigenetic consequences that last into adulthood [71]. Similar epigenetic changes are seen in human studies of childhood abuse, suggesting that there may be an evolutionarily conserved mechanism that imprints early life experiences on the epigenome, if not directly in the germ line, at least via behavioral transmission [72, 73].
Box 2. The changing meaning of epigenetics.
The term epigenetics has taken on a number of different meanings since C.H. Waddington coined it in 1942. The structure of DNA was still a mystery at the time, but the idea that genes produced a phenotype based on interactions with their environment holds a great deal of truth. At the conception of the term, however, the mechanism was thought to depend on a tissues position with the organism, leading to differentiation into a variety of cell types and the emergence of characteristics not present or predictable from the prior developmental state.
The discovery of the structure of DNA and the association of histone proteins has opened up a new field of study, yielding valuable insights into gene regulation. A number of post-translational modifications to tails of histone proteins have been shown to reliably correlate with enhancement or repression of transcription of associated genes. The ability to identify the typical modifications produced by a stimulus gives an idea of the mechanism through which changes in gene expression occur. Specifically, addition or removal of functional groups in the form of methylation, acetylation, phosphorylation, sumolyation, and ubiquitination of lysine residues on histone tails produce unique consequences for gene transcription. In addition to modifications to histone tails, direct modification of DNA via the addition of methyl groups locally represses gene expression. These modifications to the histones or the DNA itself are commonly referred to as epigenetic modifications [14]. Non-coding RNAs also play a role in epigenetic regulation of gene expression [113, 114]
In its current form, epigenetics seeks to understand the interactions between our genes and the environment. How does experience shape the patterns of gene expression within each of our cells? Epigenetic modifications provide a plausible explanation for how different experiences can lead to altered patterns of gene expression even between two genetically identical individuals, such as monozygotic twins. These findings have provided a large boost to the concept of “nuture” in the ever-present apposition with “nature,” because experience seems to play a significant role in regulating gene expression in both healthy and diseased cells.
One striking revelation about epigenetic modifications is that they may be heritable across generations [46]. Some epigenetic marks acquired during the lifetime of an animal have been seen in their gametes and subsequently in the following generation [74]. For instance, male mice exposed to chronic stress have altered expression of micro RNAs (miRNA) in their sperm, a phenomenon that has been linked to epigenetic marks [75]. The offspring of these mice have reduced stress responsivity and altered gene expression in various brain regions associated with the stress response. This raises the critical question of how much our experience affects the gene expression of our offspring.
Following critical periods of development when many permanent epigenetic marks are established, modifications to histone proteins continue throughout adulthood to affect gene expression. They can often be very rapid in response to acute stress and result in both transient and lasting changes [76, 77]. The fact that some modifications are not maintained may be the result of the function that they serve, such as forming new memories. The hippocampus is an important brain structure for the formation of declarative, episodic, and spatial learning and memory [78]. Transient modifications to histones during a stressful event may play a role in activating the machinery to craft a new memory. When the event is over, the mark is removed to prevent aberrant memory formation [79]. Other possibilities include adaptive mechanisms used to deal with the stress that are no longer needed following its cessation, such as activation of neurotrophic factors and anti-apoptotic pathways that protect neurons. However, if these pathways were left active it could become detrimental, as epigenetic marks have been recently linked to certain cancers [80].
Modification of a histone protein tail in proximity to a gene may have great consequence for the expression of that gene, but what happens when modifications occur to histones outside of coding regions? For many years non-coding DNA was thought to be useless, and was termed “junk DNA”. New insights have revealed that this is likely not the case, as many non-coding regions have been shown to be highly relevant in regulation of expression of other genes [81, 82]. Chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) has shown both coding and non-coding regions that may be sensitive to modification. When ChIP-seq reveals that specific genes are being regulated by a particular histone mark, the particular histone can then become a target of possible treatment to correct the imbalance. However, when ChIP-seq reveals that a non-coding region is being regulated, its relevance should not be dismissed. For example, ChIP-seq on H3K9me3 in the hippocampus of acutely stressed rats revealed an increase in regulation of retrotransposable elements [83]. H3K9me3 has been shown to be a silencing mark, indicating that an increase in expression may be preventing stress induced genome instability through blocking movement of retrotransposable elements. However, in chronic stress the same increase in the H3K9me3 mark is not seen, indicating there may be a loss of this compensatory mechanism. Deregulated retrotransposable elements allowed to freely move throughout the genome could have innumerable effects on GENs depending on where they ultimately land. Similar changes in H3K9me3 have been observed in other brain regions in response to environmental manipulations [84]. A more thorough understanding of how and where retrotransposons move will determine whether they contribute to disease, and whether they might be a viable target for treatment.
Alternative mechanisms of GEN regulation that can be driven by environmental factors
Other mechanisms that effect GEN dynamics are a result of P bodies and stress granules. P bodies are ribonucleoprotein complexes that are found in germ cells and somatic cells that serve to regulate RNA metabolism [85, 86]. They are composed of RNA decapping enzyme, exonucleases, helicases, RNA interference effectors, Argonaute proteins, microRNAs, and mRNAs [87]. P bodies have been linked to degradation and repression of mRNA translation, as well as sequestration of mRNAs that are later released for temporal regulation of translation [88]. Interestingly, BDNF has been shown to rapidly induce the appearance of P bodies in neurons. Expression of BDNF causes micro RNA (miRNA) biogenesis mediated through the protein Dicer, leading to increased P body abundance [89]. This pathway is involved in regulating the translational specificity produced by BDNF [90, 91], indicating that when BDNF levels are altered, as in stress, different sets of proteins are translated.
Stress granules are similar to P bodies in composition, but have a few distinguishing characteristics and are induced by environmental stress and aging. Unlike P bodies, stress granules contain translational initiation machinery such as eIF3 and ribosomal proteins [92]. The appearance of stress granules might signify abnormal transcriptional regulation, and could become a biomarker for identifying stress-induced damage in cells. Both P bodies and stress granules appear to play a role in regulation of GENs and warrant further investigation.
Examining GENs from regional to single cell specificity
Initial studies using microarray and RNA-seq in the brain have relied upon RNA extraction from manually dissected tissue, which results in the collection of a wide diversity of cell types. Depending on the size of the animal, wet dissection can be accomplished at the time of sacrifice in a reliable manner to separate distinct brain regions and test for changes in GENs. Yet, even within brain regions there are distinct sub-regions with different populations of cells that make them functionally unique, such as the hippocampus, which when analyzed as a whole could mask important changes. One technique that has been developed to deal with this difficulty is laser capture microdissection (LCM)[93]. LCM allows a researcher to isolate subpopulations of cells under microscopic visualization. This offers specificity through direct visualization, in which RNA can be isolated from as little as a single cell. However, LCM requires that tissue be fixed prior to RNA extraction, which may have drastic effects on gene expression [94].
The effect of transcriptional changes identified in a single cell can be difficult to decipher in the larger context of its brain region, let alone at the systems level. It is also unclear whether the expression of this single cell is representative of other similar cells in close proximity, or if it is an abnormal representation due to a transient change in its microenvironment [95]. Different techniques have been developed to look at a homogeneous population of cells within a small sub-region of the brain. For many applications, fluorescence activated cell sorting (FACS) can provide a pure population of cells that then can be analyzed for expression patterns. However, this technique also has drawbacks, as it relies on cell specific membrane bound markers or transgenic reporter lines in order to sort a homogeneous population. Not all cell types have been identified based on surface marker expression, and many require numerous markers to achieve specificity; even then it is not always clear whether a uniquely functional population has been isolated. Another technique, immunopanning, also suffers from this limitation [96] and has been shown to alter gene expression in ways that are believed to reflect the stress of the method itself on the cells [94].
To achieve increased cell type specificity for GEN analysis, one method relies on genetically unique tags. bacterial artificial chromosomes (BACs) have been used to create transgenic mice that contain an EGFP attached to the L10a subunit of the ribosome. BACs allow for the insertion of large regions of DNA surrounding a gene of interest, and therefore will include more endogenous regulatory regions. The BAC is also designed so that the EGFP tag is under the control of a cell type-specific promoter, ensuring that it is only expressed in the cell type of interest. Then, utilizing translating ribosomal affinity purification (TRAP), mRNAs bound to the ribosomes can be isolated and analyzed from a single cell sub-type, hence “BAC-TRAP [97]”. The Heintz lab has generated nearly 100 BAC-TRAP mice for studying a wide variety of cell populations (Personal communication).
Another technology has emerged to address one of the limitations present in all of the RNA isolation techniques previously mentioned. None of them allow exploration of the transcriptome from live tissue while it is still present in its natural microenvironment. Lovatt et al. developed a transciptome in vivo analysis (TIVA) tag that allows for noninvasive and spatially precise transcriptome profiling of single cells [98]. This method relies on loading cells with an mRNA capture molecule that can be light-activated by FRET before tissue homogenization. The capture molecule can then be used for purification of RNA expressed from only the FRET-activated cell prior to homogenization. This avoids isolation of RNA from surrounding cells and those induced by mechanical disruption of the tissue. TIVA is of particular interest for study of the brain, where seemingly identical neurons can have markedly different gene expression profiles stemming from responses to stimuli with spatial specificity, as in sensory neurons [99, 100].
Selecting the appropriate RNA isolation technique for the biological question is essential. Subsequently, microarray and RNA-seq technologies provide high throughput avenues for exploring changes in GENs in either whole brain regions or single cells. However, microarray and RNA-seq are not without their drawbacks. One of the major limitations of these technologies is the inability to do longitudinal studies. Examining transcript levels as a snapshot at the time of sacrifice gives an expression pattern that may not accurately reflect transcriptional fluctuations of a manipulation. Increasing the number of time points examined can help to alleviate this problem, but this requires large numbers of animals and is not always feasible. Also, changes in expression patterns do not necessarily imply functional consequence. An increase in transcription of a gene can be met with compensatory mechanisms that reduce its translational output through changes in mRNA stability, RNA interference (RNAi), or sequestration in p-bodies or stress granules [101]. Characterization of post-transcriptional regulation has been aided by techniques such as individual nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP), which allows genome-wide analysis of RNA binding proteins and their interaction sites [102]. Equating changes in expression with differences in protein levels is an important aspect of demonstrating functional relevance. Still, microarray and RNA-seq technology offer the ability to examine increasingly complex GENs in relatively little time.
Conclusions
The widespread adoption of high throughput technologies has brought with it great new insights into gene expression patterns and gene interactions. Yet, linking expression maps to regulatory mechanisms remains a challenge. How do the changes observed in the expression of 100 genes alter the activity of 100 more? This level of complexity requires equally complex data sets for which the analysis tools are still being developed. Microarray and RNA-seq have been combined with other technologies such as ChIP-seq to locate positions in the epigenome where regulation is occurring and correlate those findings with changes in mRNA levels. These patterns will facilitate the examination of how site-specific epigenetic modifications can impact gene expression and their effect on behavior and disease [103].
The overall complexity of the stress response makes it a difficult problem to address at a molecular level. Finding the relevant targets and underlying mechanisms behind differences in individual susceptibility are still elusive goals. The range of antidepressant treatments currently prescribed to treat psychiatric disorders remains unsatisfactory because of the delayed onset of therapeutic effects, and a lack of overall efficacy in treating the disease. Further, a wide range of side effects that are often worse than the disease itself stem from a lack of directed specificity. Using microarray and RNA-seq to look at expression patterns in combination with various RNA isolation techniques will bring us closer to understanding normative gene expression patterns on both a micro and systems level, while concurrently investigating how a stimulus such as stress leads to aberrant expression, function, and ultimately disease.
Acknowledgments
This project was supported by NIH grants MH41256 and the Hope for Depression Research Foundation grant RGA#13-004 to BSM and the Gary R. Helman Fellowship to JDG.
Abbreviations
- BDNF
brain derived neurotrophic factor
- CRS
chronic restraint stress
- GC
glucocorticoid
- GEN
gene expression network
- GR
glucocorticoid receptor
- MDD
major depressive disorder
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Bennett E, Diamond M, Krech D, Rosenzweig M. Chemical and anatomical plasticity of brain. Science. 1964;146:610–9. doi: 10.1126/science.146.3644.610. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS. Stress, sex, and neural adaptation to a changing environment: mechanisms of neuronal remodeling. Ann N Y Acad Sci. 2010;1204(Suppl):E38–59. doi: 10.1111/j.1749-6632.2010.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sapolsky RM. Why stress is bad for your brain. Science. 1996;273:749–50. doi: 10.1126/science.273.5276.749. [DOI] [PubMed] [Google Scholar]
- 4.McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci. 2004;1032:1–7. doi: 10.1196/annals.1314.001. [DOI] [PubMed] [Google Scholar]
- 5.Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–71. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 6.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–7. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 7.Evans SJ, Datson NA, Kabbaj M, Thompson RC, et al. Evaluation of Affymetrix Gene Chip sensitivity in rat hippocampal tissue using SAGE analysis. Serial Analysis of Gene Expression. Eur J Neurosci. 2002;16:409–13. doi: 10.1046/j.1460-9568.2002.02097.x. [DOI] [PubMed] [Google Scholar]
- 8.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gearhart J, Pashos EE, Prasad MK. Pluripotency redux--advances in stem-cell research. N Engl J Med. 2007;357:1469–72. doi: 10.1056/NEJMp078126. [DOI] [PubMed] [Google Scholar]
- 12.Calhoun DH. Autoregulation of gene expression. Annu Rev Microbiol. 1975;29:275–99. doi: 10.1146/annurev.mi.29.100175.001423. [DOI] [PubMed] [Google Scholar]
- 13.Rozek LS, Dolinoy DC, Sartor MA, Omenn GS. Epigenetics: relevance and implications for public health. Annu Rev Public Health. 2014;35:105–22. doi: 10.1146/annurev-publhealth-032013-182513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 15.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 16.Venolia L, Gartler SM. Comparison of transformation efficiency of human active and inactive X-chromosomal DNA. Nature. 1983;302:82–3. doi: 10.1038/302082a0. [DOI] [PubMed] [Google Scholar]
- 17.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77:126–35. doi: 10.1124/mol.109.061333. [DOI] [PubMed] [Google Scholar]
- 19.Gray JD, Rubin TG, Hunter RG, McEwen BS. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol Psychiatry. 2013 doi: 10.1038/mp.2013.175. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baxter JD, Rousseau GG. Glucocorticoid hormone action: an overview. Monogr Endocrinol. 1979;12:1–24. doi: 10.1007/978-3-642-81265-1_1. [DOI] [PubMed] [Google Scholar]
- 21.Rousseau GG, Baxter JD, Tomkins GM. Glucocorticoid receptors: relations between steroid binding and biological effects. J Mol Biol. 1972;67:99–115. doi: 10.1016/0022-2836(72)90389-0. [DOI] [PubMed] [Google Scholar]
- 22.De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol. 2009;23:281–91. doi: 10.1210/me.2008-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 24.Du J, McEwen BS, Manji HK. Glucocorticoid receptors modulate mitochondrial function. Commun Integr Biol. 2009;2:1–3. doi: 10.4161/cib.2.4.8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:791–7. doi: 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karst H, Berger S, Turiault M, Tronche F, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA. 2005;102:19204–7. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacDougall MJ, Howland JG. Acute stress, but not corticosterone, disrupts short- and long-term synaptic plasticity in rat dorsal subiculum via glucocorticoid receptor activation. Cereb Cortex. 2013;23:2611–9. doi: 10.1093/cercor/bhs247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–41. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 29.Rao NA, McCalman MT, Moulos P, Francoijs KJ, et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011;21:1404–16. doi: 10.1101/gr.118042.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 31.Veldhuis HD, Van Koppen C, Van Ittersum M, De Kloet ER. Specificity of the adrenal steroid receptor system in rat hippocampus. Endocrinology. 1982;110:2044–51. doi: 10.1210/endo-110-6-2044. [DOI] [PubMed] [Google Scholar]
- 32.Krozowski ZS, Funder JW. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci USA. 1983;80:6056–60. doi: 10.1073/pnas.80.19.6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stavreva DA, Wiench M, John S, Conway-Campbell BL, et al. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol. 2009;11:1093–102. doi: 10.1038/ncb1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart LQ, Roper JA, Young WS, 3rd, O’Carroll AM, et al. Pituitary-adrenal response to acute and repeated mild restraint, forced swim and change in environment stress in arginine vasopressin receptor 1b knockout mice. J Neuroendocrinol. 2008;20:597–605. doi: 10.1111/j.1365-2826.2008.01704.x. [DOI] [PubMed] [Google Scholar]
- 35.McEwen BS. The physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 36.Sterling P, Eyer J. Allostasis: A New Paradigm to Explain Arousal Pathology. In: Fisher S, Reason J, editors. Handbook of Life Stress, Cognition and Health. New York: John Wiley & Sons; 1988. pp. 629–49. [Google Scholar]
- 37.McEwen BS. Protective and damaging effects of stress mediators. New England J Med. 1998;338:171–9. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- 38.Roozendaal B, Cahill L, McGaugh JL. Interaction of emotionally activated neuromodulatory systems in regulating memory storage. In: Ishikawa K, McGaugh JL, Sakata H, editors. Brain process and memory. Amsterdam: Elsevier; 1996. pp. 39–54. [Google Scholar]
- 39.Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune cells--from barracks to boulevards to battlefields: a tale of three hormones--Curt Richter Award winner. Psychoneuroendocrinology. 2012;37:1345–68. doi: 10.1016/j.psyneuen.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43:2–15. doi: 10.1016/s0018-506x(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 41.Rodrigues AJ, Leao P, Pego JM, Cardona D, et al. Mechanisms of initiation and reversal of drug-seeking behavior induced by prenatal exposure to glucocorticoids. Mol Psychiatry. 2012;17:1295–305. doi: 10.1038/mp.2011.126. [DOI] [PubMed] [Google Scholar]
- 42.Morgan CP, Bale TL. Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci. 2011;31:11748–55. doi: 10.1523/JNEUROSCI.1887-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eiland L, McEwen BS. Early life stress followed by subsequent adult chronic stress potentiates anxiety and blunts hippocampal structural remodeling. Hippocampus. 2012;22:82–91. doi: 10.1002/hipo.20862. [DOI] [PubMed] [Google Scholar]
- 44.Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology. 2008;149:4892–900. doi: 10.1210/en.2008-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei Q, Fentress HM, Hoversten MT, Zhang L, et al. Early-life forebrain glucocorticoid receptor overexpression increases anxiety behavior and cocaine sensitization. Biol Psychiatry. 2012;71:224–31. doi: 10.1016/j.biopsych.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gapp K, von Ziegler L, Tweedie-Cullen RY, Mansuy IM. Early life epigenetic programming and transmission of stress-induced traits in mammals: how and when can environmental factors influence traits and their transgenerational inheritance? BioEssays. 2014;36:491–502. doi: 10.1002/bies.201300116. [DOI] [PubMed] [Google Scholar]
- 47.Romeo RD, Minhas S, Svirsky SE, Hall BS, et al. Pubertal shifts in adrenal responsiveness to stress and adrenocorticotropic hormone in male rats. Psychoneuroendocrinology. 2014;42:146–52. doi: 10.1016/j.psyneuen.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bloss EB, Janssen WG, McEwen BS, Morrison JH. Interactive effects of stress and aging on structural plasticity in the prefrontal cortex. J Neurosci. 2010;30:6726–31. doi: 10.1523/JNEUROSCI.0759-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldwater DS, Pavlides C, Hunter RG, Bloss EB, et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyce WT, Ellis BJ. Biological sensitivity to context: I. An evolutionary-developmental theory of the origins and functions of stress reactivity. Dev Psychopathol. 2005;17:271–301. doi: 10.1017/s0954579405050145. [DOI] [PubMed] [Google Scholar]
- 52.Lindsay RM, Thoenen H, Barde YA. Placode and neural crest-derived sensory neurons are responsive at early developmental stages to brain-derived neurotrophic factor. Dev Biol. 1985;112:319–28. doi: 10.1016/0012-1606(85)90402-6. [DOI] [PubMed] [Google Scholar]
- 53.Hofer MM, Barde YA. Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature. 1988;331:261–2. doi: 10.1038/331261a0. [DOI] [PubMed] [Google Scholar]
- 54.Lee BH, Kim YK. The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investig. 2010;7:231–5. doi: 10.4306/pi.2010.7.4.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong W, Fan J, Yuan C, Zhang C, et al. Significantly decreased mRNA levels of BDNF and MEK1 genes in treatment-resistant depression. Neuroreport. 2014 doi: 10.1097/WNR.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, et al. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–15. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- 57.Karege F, Vaudan G, Schwald M, Perroud N, et al. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res. 2005;136:29–37. doi: 10.1016/j.molbrainres.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 58.Dunham JS, Deakin JF, Miyajima F, Payton A, et al. Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. J Psychiatr Res. 2009;43:1175–84. doi: 10.1016/j.jpsychires.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 59.Sen S, Nesse RM, Stoltenberg SF, Li S, et al. A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology. 2003;28:397–401. doi: 10.1038/sj.npp.1300053. [DOI] [PubMed] [Google Scholar]
- 60.Verhagen M, van der Meij A, van Deurzen PA, Janzing JG, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry. 2010;15:260–71. doi: 10.1038/mp.2008.109. [DOI] [PubMed] [Google Scholar]
- 61.Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214–27. doi: 10.1016/j.neuroscience.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marmigere F, Givalois L, Rage F, Arancibia S, et al. Rapid induction of BDNF expression in the hippocampus during immobilization stress challenge in adult rats. Hippocampus. 2003;13:646–55. doi: 10.1002/hipo.10109. [DOI] [PubMed] [Google Scholar]
- 63.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–77. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naert G, Ixart G, Maurice T, Tapia-Arancibia L, et al. Brain-derived neurotrophic factor and hypothalamic-pituitary-adrenal axis adaptation processes in a depressive-like state induced by chronic restraint stress. Mol Cell Neurosci. 2011;46:55–66. doi: 10.1016/j.mcn.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang Y, Langley B, Lubin FD, Renthal W, et al. Epigenetics in the nervous system. J Neurosci. 2008;28:11753–9. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heijmans BT, Tobi EW, Stein AD, Putter H, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 69.Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–8. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- 70.Gleason G, Liu B, Bruening S, Zupan B, et al. The serotonin1A receptor gene as a genetic and prenatal maternal environmental factor in anxiety. Proc Natl Acad Sci USA. 2010;107:7592–7. doi: 10.1073/pnas.0914805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci USA. 2006;103:3480–5. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gilbert R, Widom CS, Browne K, Fergusson D, et al. Burden and consequences of child maltreatment in high-income countries. Lancet. 2009;373:68–81. doi: 10.1016/S0140-6736(08)61706-7. [DOI] [PubMed] [Google Scholar]
- 73.Borghol N, Suderman M, McArdle W, Racine A, et al. Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol. 2012;41:62–74. doi: 10.1093/ije/dyr147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soubry A, Hoyo C, Jirtle RL, Murphy SK. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. BioEssays. 2014;36:359–71. doi: 10.1002/bies.201300113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodgers AB, Morgan CP, Bronson SL, Revello S, et al. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. 2013;33:9003–12. doi: 10.1523/JNEUROSCI.0914-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szyf M. Therapeutic implications of DNA methylation. Future Oncol. 2005;1:125–35. doi: 10.1517/14796694.1.1.125. [DOI] [PubMed] [Google Scholar]
- 77.Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, et al. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci USA. 2009;106:20912–7. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eichenbaum H, Otto T, Cohen NJ. The hippocampus--what does it do? Behav Neural Biol. 1992;57:2–36. doi: 10.1016/0163-1047(92)90724-i. [DOI] [PubMed] [Google Scholar]
- 79.Reul JM, Hesketh SA, Collins A, Mecinas MG. Epigenetic mechanisms in the dentate gyrus act as a molecular switch in hippocampus-associated memory formation. Epigenetics. 2009;4:434–9. doi: 10.4161/epi.4.7.9806. [DOI] [PubMed] [Google Scholar]
- 80.Smith AA, Huang YT, Eliot M, Houseman EA, et al. A novel approach to the discovery of survival biomarkers in glioblastoma using a joint analysis of DNA methylation and gene expression. Epigenetics. 2014;9 doi: 10.4161/epi.28571. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taft RJ, Pheasant M, Mattick JS. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays. 2007;29:288–99. doi: 10.1002/bies.20544. [DOI] [PubMed] [Google Scholar]
- 82.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 83.Hunter RG, Murakami G, Dewell S, Seligsohn M, et al. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc Natl Acad Sci USA. 2012;109:17657–62. doi: 10.1073/pnas.1215810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maze I, Feng J, Wilkinson MB, Sun H, et al. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci USA. 2011;108:3035–40. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kotaja N, Bhattacharyya SN, Jaskiewicz L, Kimmins S, et al. The chromatoid body of male germ cells: similarity with processing bodies and presence of Dicer and microRNA pathway components. Proc Natl Acad Sci USA. 2006;103:2647–52. doi: 10.1073/pnas.0509333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Teixeira D, Sheth U, Valencia-Sanchez MA, Brengues M, et al. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA. 2005;11:371–82. doi: 10.1261/rna.7258505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–46. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 88.Brengues M, Teixeira D, Parker R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 2005;310:486–9. doi: 10.1126/science.1115791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang YW, Ruiz CR, Eyler EC, Lin K, et al. Dual regulation of miRNA biogenesis generates target specificity in neurotrophin-induced protein synthesis. Cell. 2012;148:933–46. doi: 10.1016/j.cell.2012.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schratt GM, Nigh EA, Chen WG, Hu L, et al. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–77. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yin Y, Edelman GM, Vanderklish PW. The brain-derived neurotrophic factor enhances synthesis of Arc in synaptoneurosomes. Proc Natl Acad Sci USA. 2002;99:2368–73. doi: 10.1073/pnas.042693699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–6. doi: 10.1038/nrm2694. [DOI] [PubMed] [Google Scholar]
- 93.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, et al. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 94.Okaty BW, Sugino K, Nelson SB. A quantitative comparison of cell-type-specific microarray gene expression profiling methods in the mouse brain. PLoS One. 2011;6:e16493. doi: 10.1371/journal.pone.0016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–73. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cahoy JD, Emery B, Kaushal A, Foo LC, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heiman M, Schaefer A, Gong S, Peterson JD, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–48. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lovatt D, Ruble BK, Lee J, Dueck H, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11:190–6. doi: 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eberwine J, Lovatt D, Buckley P, Dueck H, et al. Quantitative biology of single neurons. J R Soc Interface. 2012;9:3165–83. doi: 10.1098/rsif.2012.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lumpkin EA, Caterina MJ. Mechanisms of sensory transduction in the skin. Nature. 2007;445:858–65. doi: 10.1038/nature05662. [DOI] [PubMed] [Google Scholar]
- 101.Wachter A. Gene regulation by structured mRNA elements. Trends Genet. 2014;30:172–81. doi: 10.1016/j.tig.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 102.Konig J, Zarnack K, Rot G, Curk T, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–15. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rusk N. CRISPRs and epigenome editing. Nat Methods. 2014;11:28. doi: 10.1038/nmeth.2775. [DOI] [PubMed] [Google Scholar]
- 104.Reichardt HM, Schutz G. Glucocorticoid signalling--multiple variations of a common theme. Mol Cell Endocrinol. 1998;146:1–6. doi: 10.1016/s0303-7207(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 105.McEwen BS, Weiss J, Schwartz L. Selective retention of corticosterone by limbic structures in rat brain. Nature. 1968;220:911–2. doi: 10.1038/220911a0. [DOI] [PubMed] [Google Scholar]
- 106.DeKloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocrine Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 107.McEwen BS, Gerlach J, Micco D. Putative glucocorticoid receptors in hippocampus and other regions of the rat brain. In: Isaacson R, Pribram K, editors. Hippocampus, Structure and Development. New York: Plenum Press; 1975. pp. 285–322. [Google Scholar]
- 108.Ahima R, Krozowski Z, Harlan R. Type I corticosteroid receptor-like immunoreactivity in the rat CNS: distribution and regulation by corticosteroids. JCompNeurol. 1991;313:522–38. doi: 10.1002/cne.903130312. [DOI] [PubMed] [Google Scholar]
- 109.Pratt WB, Morishima Y, Murphy M, Harrell M. Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol. 2006:111–38. doi: 10.1007/3-540-29717-0_5. [DOI] [PubMed] [Google Scholar]
- 110.Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34(Suppl 1):S186–95. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 111.Reichardt HM, Schutz G. Glucocorticoid signalling - multiple variations of a common theme. Mol Cell Endocrinol. 1998;146:1–6. doi: 10.1016/s0303-7207(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 112.Groeneweg FL, Karst H, de Kloet ER, Joels M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J Endocrinol. 2011;209:153–67. doi: 10.1530/JOE-10-0472. [DOI] [PubMed] [Google Scholar]
- 113.Mehler MF. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog Neurobiol. 2008;86:305–41. doi: 10.1016/j.pneurobio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hunter RG, McEwen BS, Pfaff DW. Environmental stress and transposon transcription in the mammalian brain. Mob Genet Elements. 2013;3:e24555. doi: 10.4161/mge.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]