

Abstract
Cellular-Src (c-Src) encodes a plasma membrane-associated tyrosine protein kinase, which plays a vital role in signaling pathways related to cellular development and carcinogenesis (1,2). It was the first proto-oncogene to be described and is the cellular homologue in humans of the viral oncogene of Rous sarcoma virus, the chicken tumor virus discovered by Peyton Rous in 1911 (3). More recently, c-Src has been implicated in connexin43 (Cx43) remodeling in epicardial border zone myocytes following myocardial infarction (MI) (4).
Keywords: connexin43, gap junctions, myocardial infarction, Src, sudden death
Cx43 is the major ventricular gap junction protein in mammalian hearts, and the intercellular coupling mediated by these gap junctions allow for normal electrical conduction (5–7). Cx43 interacts with a scaffolding protein zonula-occludens-1 (ZO-1) to localize at intercalated disks (8,9). After a MI, phosphorylation of c-Src leads to its activation (p-Src), which then disrupts the ZO-1/Cx43 interaction (4,10). Displacement of the Z0-1/Cx43 interaction can cause Cx43 relocalization from the intercalated disks to myocyte lateral membranes (4,11). Cx43 lateralization in the infarct border zone is associated with altered gap junction coupling and is believed to be an important mechanism underpinning electrical conduction slowing and the development of reentrant arrhythmias (12).
Sovari et al. (11) have previously shown that 1-(1,1-dimethylethyl)-1-(4-methylphenyl)-1H-pyrazolo[3,4-d] pyrimidin-4-amine (PP1), a c-Src inhibitor, can increase Cx43 levels at gap junctions, improve gap junction function, and reduce inducibility of ventricular tachycardia (VT) in angiotensin-converting enzyme overexpression (ACE8/9) mice. Activation of the renin-angiotensin system in these mice led to c-Src up-regulation, loss of Cx43 and gap function coupling, and arrhythmogenesis; these processes were mitigated by inhibition of c-Src (11) (Fig. 1).
Figure 1. Scheme of c-Src Kinase Pathway Modulating Cx43.
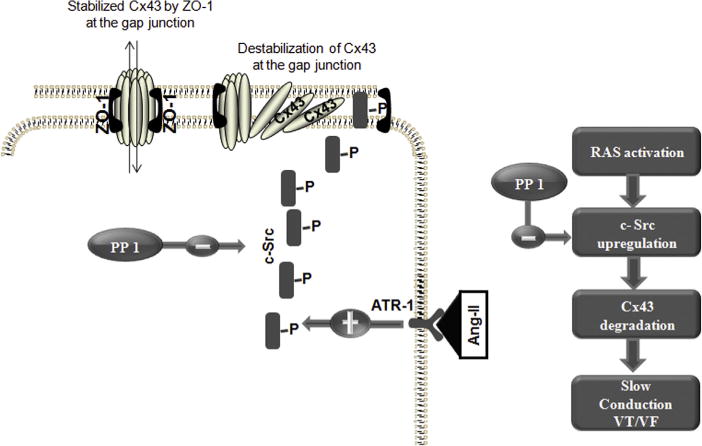
Angiotensin II (Ang II) activates and up-regulates c-Src, which in turn causes a dysregulation and degradation of connexin43 (Cx43) with impaired gap junction function. PP1 inhibits c-Src kinase and interrupts Ang II–mediated Cx43 reduction, myocyte uncoupling, and sudden arrhythmic death. ATR-1 = angiotensin II type 1 receptor; RAS = renin-angiotensin system; VF = ventricular fibrillation; VT = ventricular tachycardia; ZO = zonula-occludens. Reproduced with permission from Sovari et al. (11).
In this issue of the Journal, Rutledge et al. (13) expand on these prior findings by applying our current understanding of post-MI Cx43 pathophysiology and hypothesizing that inhibition of c-Src activation can improve Cx43 levels and thereby ameliorate arrhythmia inducibility following MI. Mice undergoing MI after coronary artery ligation were treated with c-Src inhibitors PP1 and AZD0530, which is currently in clinical development and has been used to treat human cancers (14,15). The authors showed that c-Src inhibition moderated Cx43 down-regulation after MI compared with infarcts in mice treated with an inactive analogue or with saline. In addition, the restoration in Cx43 levels in mice treated with c-Src inhibitors was correlated with observed findings of lower p-Src levels, enhanced conduction velocity, and reduced arrhythmia inducibility in these MI mice. Interestingly, the improved Cx43 levels did not appear to be due to redistribution of Cx43, but rather to PP1 effects on increased Cx43 transcription as well as decreased Cx43 degradation. Furthermore, the authors’ results suggest that the underlying post-MI mechanisms leading to reduced Cx43 levels and c-Src activation are similarly triggered throughout the myocardium, rather than being contained only to peri-infarct tissue.
Rutledge et al. (13) should be commended for continuing to elucidate a novel approach toward a potential new antiarrhythmic therapy via inhibition of c-Src. They have added to a growing literature of studies seeking to modulate gap junction remodeling in order to decrease post-infarct arrhythmias. These studies include targeted Cx43 gene transfer to a porcine infarct model (16), engraftment of Cx43-expressing embryonic cardiomyocytes post-infarct (17), use of growth hormone-releasing peptide to modulate Cx43 levels (18), and use of heavy ion radiation to up-regulate Cx43 levels (19,20).
However, further investigation will be necessary. Notably, post-translational phosphorylation is critical to almost all aspects of Cx43 regulation and gap junction function (21,22). The authors did find that c-Src inhibition increased slowly migrating forms of Cx43, which are felt to be related to phosphorylated Cx43 that are associated with enhanced gap junction function (23,24). Nevertheless, further studies are needed to understand the precise mechanisms and sites of Cx43 phosphorylation, and how c-Src inhibition relates to these different forms of Cx43. In addition, as the authors point out, although they found a beneficial outcome by using c-Src inhibitors after 2 weeks post-MI, the effects of c-Src inhibition immediately after an ischemic event has not be examined and, in fact, could be harmful. There is data to suggest that Cx43 down-regulation after an MI may be adaptive because restricting gap junction communication may prevent the spread of deleterious by-products of ischemia and thereby limit infarct size (25). Hence, the timing of c-Src inhibition is important; furthermore, duration of treatment will need to be determined because cardiac Cx43 has a relatively short half-life of about 1 to 2 h (26,27), and the substrate for arrhythmia may persist beyond the 2-week period after an MI. In addition, there are concerns about QT prolongation with this class of tyrosine kinase inhibitors (28), and it is not uncommon for a purportedly antiarrhythmic therapy to have pro-arrhythmic effects instead. Hence, more studies are needed to better understand these potential side effects due to tyrosine kinase inhibitors, including the risks of left ventricular dysfunction and hypertension (both systemic and pulmonary) (29,30). The long-term outcomes and possible cardiovascular side effects of this class of drugs are therefore still unknown for an already potentially vulnerable patient population. Finally, the clinical applicability of a mouse model has its limitations such that the role of c-Src inhibition in a larger animal infarct model is necessary.
In conclusion, c-Src and its importance in tumorigenesis have been widely studied. The findings of Rutledge et al. (13) have provided further insight into the role of c-Src inhibition and arrhythmogenesis. Furthermore, although PP1 is not used clinically, AZD0530 has been studied in several phase 1 and 2 human cancer trials and is therefore closer to clinical application (31–33). The analogy to tumorigenesis and cancer therapeutics may be appropriately fitting. Ventricular arrhythmias are believed to result from multiple factors and stochastic events; thus far, the mainstay treatments have involved relatively blunt tools including internal defibrillators, radiofrequency ablation of infarct border zones, and antiarrhythmic drug therapies nonspecifically targeting various ion channels. Similarly, until the advent of mechanistic-driven therapeutics, oncologic treatments have tended to involve surgical resection, radiation therapy, and relatively nondiscerning chemotherapy that can collaterally affect normal tissue while not fully targeting the underlying cause(s). However, over the last decades, advances in the understanding of the underlying molecular processes involved in tumorigenesis have contributed to more specific and precise therapeutics for a variety of cancers. Analogously, although it is not clear that c-Src inhibition or even modulating Cx43 after an infarct will be the key to reducing arrhythmias, studies such as this from Rutledge et al. (13) seeking mechanism-based therapies for ventricular arrhythmias provide further optimism that we are one step closer to an effective antiarrhythmic treatment.
Footnotes
Editorials published in the Journal of the American College of Cardiology reflect the views of the authors and do not necessarily represent the views of JACC or the American College of Cardiology.
Dr. Nguyen receives modest institutional research funding from Biosense Webster and Medtronic. Dr. Mestroni has reported that she has no relationships relevant to the contents of this paper to disclose.
References
- 1.Roskoski R., Jr Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun. 2004;324:1155–64. doi: 10.1016/j.bbrc.2004.09.171. [DOI] [PubMed] [Google Scholar]
- 2.Roskoski R., Jr Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol. 2001;2:467–75. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 4.Kieken F, Mutsaers N, Dolmatova E, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–12. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beyer EC, Paul DL, Goodenough DA. Connexin family of gap junction proteins. J Membr Biol. 1990;116:187–94. doi: 10.1007/BF01868459. [DOI] [PubMed] [Google Scholar]
- 6.Gutstein DE, Morley GE, Tamaddon H, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–9. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Severs NJ, Dupont E, Coppen SR, et al. Remodelling of gap junctions and connexin expression in heart disease. Biochim Biophys Acta. 2004;1662:138–48. doi: 10.1016/j.bbamem.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–4. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 9.Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–28. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giepmans BN, Hengeveld T, Postma FR, Moolenaar WH. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J Biol Chem. 2001;276:8544–9. doi: 10.1074/jbc.M005847200. [DOI] [PubMed] [Google Scholar]
- 11.Sovari AA, Iravanian S, Dolmatova E, et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–9. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–96. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 13.Rutledge CA, Ng FS, Sulkin MS, et al. c-Src kinase inhibition reduces arrhythmia inducibility and connexin43 dysregulation after myocardial infarction. J Am Coll Cardiol. 2014;63:928–34. doi: 10.1016/j.jacc.2013.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baselga J, Cervantes A, Martinelli E, et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clin Cancer Res. 2010;16:4876–83. doi: 10.1158/1078-0432.CCR-10-0748. [DOI] [PubMed] [Google Scholar]
- 15.Hennequin LF, Allen J, Breed J, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 16.Greener ID, Sasano T, Wan X, et al. Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction. J Am Coll Cardiol. 2012;60:1103–10. doi: 10.1016/j.jacc.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roell W, Lewalter T, Sasse P, et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450:819–24. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- 18.Yuan MJ, Huang H, Tang YH, et al. Effects of ghrelin on Cx43 regulation and electrical remodeling after myocardial infarction in rats. Peptides. 2011;32:2357–61. doi: 10.1016/j.peptides.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Amino M, Yoshioka K, Fujibayashi D, et al. Year-long upregulation of connexin43 in rabbit hearts by heavy ion irradiation. Am J Physiol Heart Circ Physiol. 2010;298:H1014–21. doi: 10.1152/ajpheart.00160.2009. [DOI] [PubMed] [Google Scholar]
- 20.Amino M, Yoshioka K, Tanabe T, et al. Heavy ion radiation up-regulates Cx43 and ameliorates arrhythmogenic substrates in hearts after myocardial infarction. Cardiovasc Res. 2006;72:412–21. doi: 10.1016/j.cardiores.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Marquez-Rosado L, Solan JL, Dunn CA, Norris RP, Lampe PD. Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim Biophys Acta. 2012;1818:1985–92. doi: 10.1016/j.bbamem.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno AP, Lau AF. Gap junction channel gating modulated through protein phosphorylation. Prog Biophys Mol Biol. 2007;94:107–19. doi: 10.1016/j.pbiomolbio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–86. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–72. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanno S, Kovacs A, Yamada KA, Saffitz JE. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J Am Coll Cardiol. 2003;41:681–6. doi: 10.1016/s0735-1097(02)02893-0. [DOI] [PubMed] [Google Scholar]
- 26.Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273:67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res. 2004;62:256–67. doi: 10.1016/j.cardiores.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 28.Lenihan DJ, Kowey PR. Overview and management of cardiac adverse events associated with tyrosine kinase inhibitors. Oncologist. 2013;18:900–8. doi: 10.1634/theoncologist.2012-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah DR, Shah RR, Morganroth J. Tyrosine kinase inhibitors: their on-target toxicities as potential indicators of efficacy. Drug Saf. 2013;36:413–26. doi: 10.1007/s40264-013-0050-x. [DOI] [PubMed] [Google Scholar]
- 30.Shah RR, Morganroth J, Shah DR. Cardiovascular safety of tyrosine kinase inhibitors: with a special focus on cardiac repolarisation (QT interval) Drug Saf. 2013;36:295–316. doi: 10.1007/s40264-013-0047-5. [DOI] [PubMed] [Google Scholar]
- 31.Renouf DJ, Moore MJ, Hedley D, et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2012;30:779–86. doi: 10.1007/s10637-010-9611-3. [DOI] [PubMed] [Google Scholar]
- 32.Mackay HJ, Au HJ, McWhirter E, et al. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adeno-carcinoma: a trial of the PMH phase II consortium. Invest New Drugs. 2012;30:1158–63. doi: 10.1007/s10637-011-9650-4. [DOI] [PubMed] [Google Scholar]
- 33.Fury MG, Baxi S, Shen R, et al. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) Anticancer Res. 2011;31:249–53. [PMC free article] [PubMed] [Google Scholar]