

Abstract
The ever-increasing threat of multi-drug resistant bacterial infections has spurred renewed interest in alternative approaches to classical antibiotic therapy. In contrast to other mammals, humans do not express the galactose-α-1,3-galactosyl-β-1,4-N-acetyl-glucosamine (α-Gal) epitope. As a result of exposure of humans to α-Gal in the environment, a large proportion of circulating antibodies are specific for the trisaccharide. In this study, we examine whether these anti-Gal antibodies can be recruited and redirected to exert anti-bacterial activity. We show that a specific DNA aptamer conjugated to an α-Gal epitope at its 5′ end, herein termed an alphamer, can bind to group A Streptococcus (GAS) bacteria by recognition of a conserved region of the surface-anchored M protein. The anti-GAS alphamer was shown to recruit anti-Gal antibodies to the streptococcal surface in an α-Gal-specific manner, elicit uptake and killing of the bacteria by human phagocytes, and slow growth of invasive GAS in human whole blood. These studies provide a first in vitro proof of concept that alphamers have the potential to redirect pre-existing antibodies to bacteria in a specific manner and trigger an immediate antibacterial immune response. Further validation of this novel therapeutic approach of applying α-Gal technology in in vivo models of bacterial infection is warranted.
Keywords: Aptamer, Galactose-α-1, 3-galactosyl-β-1, 4-N-acetyl-glucosamine, α-Gal, Anti-Gal, Antibiotic therapy, Drug development, Antibodies, Group A Streptococcus, M protein, Phagocytosis
Introduction
The emergence and rapid spread of multi-drug resistant (MDR) bacteria represent a severe threat to the global health and economy. In 2012, the Director-General of the World Health Organization, Dr. Margaret Chan, warned that we risk entering a “post-antibiotic era”, which “means, in effect, an end to modern medicine as we know it” [1]. The incidence of MDR, associated morbidity and mortality, clinical and economic impact, and unmet need for novel drug development were highlighted in a recent US Centers for Disease Control and Prevention (CDC) report [2]. In the USA alone, an estimated two million people develop serious MDR bacterial infections annually, resulting in at least 23,000 deaths [2].
The present MDR crisis underscores an urgent need for the development of novel antibacterial therapeutics. Current clinical development efforts are largely focused on modifying existing antibiotic classes (e.g., beta-lactams, fluoroquinolones, aminoglycosides) or the screening or design of small molecule inhibitors to a few as yet unexploited bacterial enzyme targets [3, 4]. While some progress has been made, the development pipeline remains precariously thin. The effectiveness of such “classical” antibiotics involves targeting of core processes such as bacterial nucleic acid, protein, or cell wall synthesis, frequently providing broad-spectrum activity. However, broad-spectrum activities may adversely impact the human commensal microflora homeostasis, which can provoke serious medical complications, e.g., Clostridium difficile colitis, while exerting a continual selective pressure for MDR evolution.
Additional approaches to prevent or treat bacterial infections involve active vaccination to prevent bacterial diseases or passive immunization with therapeutic monoclonal antibodies (mAbs) consisting of immunoglobulin G (IgG) or M (IgM) [5]. While no vaccine has been approved against the current most important MDR bacteria, the ESKAPE organisms, significant progress in vaccine development has been made with some having entered into clinical trials as reviewed in [6–9]. Therapeutic antibodies provide antibacterial activity via mechanisms including bacterial opsonization for recognition by phagocytic Fc receptors leading to bacterial uptake and destruction, or complement activation leading to C3b deposition, which allows recognition by phagocyte complement receptors, or initiates direct lysis of susceptible bacteria. However, despite significant development efforts, no mAb is yet approved for therapeutic use in humans against acute or chronic bacterial infections [10].
Here, we explore an entirely different approach to immunotherapy of bacterial infection—a strategy to redirect pre-existing high-titer immunoglobulins called natural anti-Gal antibodies to target a specific pathogen. Unlike other mammals such as swine, humans do not express the galactose-α-1, 3-galactosyl-β-1,4-N-acetyl-glucosamine (α-Gal) epitope on cell surface glycolipids and glycoproteins. As a result of α-Gal exposure as antigen of the gut microflora, humans naturally produce anti-Gal antibodies [11]. Remarkably, ~1 % of circulating antibodies and human B cells are α-Gal specific [12, 13]. These immunoglobulins are responsible for hyper-acute rejection in xenotransplantations in which organs from α-Gal-positive donor mammals such as pigs have been implanted into humans [14, 15].
We theorized that if a molecular adaptor could be provided that (a) bound effectively to a target antigen on bacteria and (b) simultaneously displayed the α-Gal epitope, pre-existing anti-Gal antibodies could be recruited to provide antibacterial activity. In essence, the goal is to instantly recruit therapeutic antibodies from the patient’s own serum, wherein the novel drug is a modular molecular adaptor rather than a mAb. For the adaptor function, we chose nucleic acid aptamers, which are single-stranded RNA or DNA oligonucleotides exhibiting high target affinity and specificity [16]. Aptamer specificity has been exploited for development of diagnostics for detecting microbial infections [17]. Moreover, aptamers are emerging as novel therapeutic agents [18] with currently one aptamer, pegaptanib (Macugen), approved by the US Food and Drug Administration for use against age-related macular degeneration.
In this study, we seek a first proof of principal of an α-Gal-based antibacterial immunotherapy, utilizing as a model the major human pathogen, group A Streptococcus (GAS). GAS is estimated to cause ~700 million cases of localized infection (pharyngitis, impetigo) and more than ~660,000 cases of invasive infection leading annually to ~160,000 deaths worldwide [19]. We identify the molecular target of a DNA aptamer (20A24P) proposed for diagnostic use [20] to be the conserved domain of the surface-expressed GAS M protein and derive an α-Gal conjugated “alphamer” version of 20A24P as therapeutic tool. We then evaluate the ability of this alphamer to redirect pre-existing anti-Gal antibodies to the GAS surface and promote opsonophagocytic clearance in vitro.
Material and methods
Aptamers
HPLC-purified 5′- or 3′-6-carboxyfluorescein (FAM)-labeled and 3′-biotin-TEG aptamers were purchased from Integrated DNA Technologies (Coralville, IA), HPLC-purified 5′-α-Gal aptamers ±3′-FAM label, herein referred to as alphamers, were provided by Biosearch Technologies (Novato, CA). See Supplementary Methods online for details on aptamer preparation for assays. Secondary aptamer structures were modeled with the Mfold web server for nucleic acid folding prediction [21]; QGRS-Mapper was used to predict G-quartet formation [22].
Bacteria
Several GAS strains representing clinically relevant M serotypes were used for the study. These are listed in Supplementary Methods online along with bacterial mutants and culture conditions.
Aptamer/alphamer and M protein antiserum IgG binding to bacteria
FAM-labeled aptamer or alphamer and M protein antiserum IgG binding to streptococci was assayed by flow cytometry (see Supplementary Methods online).
ELISA with recombinant M1 protein to determine aptamer target
The binding of 3′-biotinylated GAS and control aptamers to recombinant his6-tagged full-length or truncated M1 proteins and control proteins was examined by ELISA (see Supplementary Methods online).
Purification of IgG and IgM mouse antibodies and measurement of mouse and human anti-α-Gal titers
Transgenic GT−/− mice expressing the variable region of the anti-α-Gal mAb M86 heavy chain produce α-Gal-reactive antibodies [23]. IgG and IgM from these animals were separated as described in [24]; the anti-Gal titers in mouse IgG and IgM and human IgG (human IVIG (hIVIG); Gamunex-C, Talecris Biotherapeutics) were determined by ELISA (see Supplementary Methods online for details).
Recognition of alphamers bound to GAS by anti-Gal antibodies
Streptococci were incubated with 5′-α-Gal, 3′-FAM GAS or control alphamers, or 3′-FAM aptamers with no α-Gal, or vehicle. Subsequently, the bacteria were exposed to purified mouse antibodies or vehicle. Antibodies bound to the bacteria were detected with Alexa Fluor®647 secondary antibodies. Aptamer/alphamer binding (green fluorescence) and antibody binding (red fluorescence) were simultaneously analyzed by flow cytometry. See Supplementary Methods online for details.
Phagocytosis studies
Green fluorescent GAS was incubated with alphamers, aptamers, or vehicle. Then, polyclonal mouse IgG or hIVIG as anti-Gal antibody sources were added. Subsequently, the pre-opsonized bacteria were co-incubated with human blood neutrophils. The percentage of neutrophils with ingested bacteria was determined by fluorescence microscopy as described [25]. Neutrophil purification and experimental conditions are described in Supplementary Methods online.
Opsonophagocytic and human whole blood killing experiments
GAS cells were pre-incubated with GAS or control alphamers. In opsonophagocytosis experiments, a mixture of human neutrophils and hIVIG was added. Samples with hIVIG and no phagocytes served as controls. In blood killing assays, heparinized human whole blood was added to the alphamer-coated streptococci. At various time points, the bacterial concentrations in the samples were determined. The procedures are detailed in Supplementary Methods online.
Statistical analyses
Phagocytosis data were analyzed using unpaired, two-tailed t test. Opsonophagocytic and whole blood killing data were analyzed by two-way ANOVA with Bonferroni post tests. Statistical tests were performed using GraphPad Prism (GraphPad Software) or Microsoft® Excel® software. P values <0.05 were considered statistically significant.
Ethics permissions
Permission to collect human blood under informed consent was approved by the UCSD Human Research Protection Program. Mouse blood was collected as approved by the University of Massachusetts Medical School Worcester Institutional Animal Care and Use Committee.
Results
Prioritization of group A Streptococcus (GAS) aptamers
Hamula and colleagues reported a panel of 39- to 80-nucleotide (nt) DNA aptamers that bind to clinically prevalent GAS serotypes for diagnostic purposes [20]. Green fluorescent FAM-labeled versions of nine of these (listed in Table 1) and two negative control aptamers (RAND-39 and RAND-80) were synthesized and tested for binding to live GAS SF370 cells by flow cytometry. In the binding buffer described by Hamula et al. [20], the 80-nt GAS aptamer 5′-FAM-20A24P bound significantly to stationary phase GAS, whereas 5′-FAM-RAND-80-treated bacteria showed background fluorescence equivalent to vehicle-treated cells (Fig. 1a). In contrast to 5′-FAM-20A24P, absent or only weak binding was detected for the other eight 5′-FAM GAS aptamers and 5′-FAM-RAND-39 (data not shown). 5′-FAM-20A24P also bound to GAS in Hank’s balanced salt solution with calcium and magnesium (HBSS+/+), Dulbecco’s phosphate-buffered saline (DPBS), and RPMI-1640 (data not shown). 5′-FAM-20A24P bound to representative strains of the serotypes M1, M1T1, M2, M3, M4, M12, and M77, confirming the reported broad-spectrum aptamer reactivity [20] but did not bind to the M49 strain NZ131 (data not shown). Of note, 5′-FAM-20A24P also bound to stationary phase cells of a highly encapsulated animal-passaged GAS 5448 strain [26] (data not shown), an invasive isolate representative of the hyper-virulent M1T1 clone that has emerged as the leading cause of invasive GAS infections worldwide [27]. 3′-FAM-20A24P bound to GAS 5448 (Fig. 1a) indicating that FAM placement on the 5′ end was not required for aptamer binding. In a dose titration experiment with 5′-FAM-20A24P and strain 5448, the aptamer exhibited dose-dependent reactivity (Fig. 1b). 20A24P was prioritized for further experimentation.
Table 1.
GAS and control DNA aptamers used in initial prioritization experiments
| GAS aptamersa | Size | Aptamer sequencesb |
|---|---|---|
| 15A3P | 80 nt | 5′-TTCACGGTAGCACGCATAGGGACAGCAAGCCCAAGCTGGG TGTGCAAGGTGAGGAGTGGGCATCTGACCTCTGTGCTGCT-3′ |
| 20A1P | 80 nt | 5′-TTCACGGTAGCACGCATAGGCAGAACGCACCCGCACACCT CCATCACTCGCATGCACCCCCATCTGACCTCTGTGCTGCT-3′ |
| 20A8P | 80 nt | 5′-AGCAGCACAGAGGTCAGATGCCCCACGAATCGTTACTCTGG TCCTCTATTTCTCCTCCCCCCTATGCGTGCTACCGTGAA-3′ |
| 20A8 | 40 nt | 5′-CCCCACGAATCGTTACTCTGGTCCTCTATTTCTCCTCCCC-3′ |
| 20A9P | 79 nt | 5′-AGCAGCACAGAGGTCAGATGCACACGCTGAAGAAACTGAG GTCGTAGGTTTTCTTCGGGCCTATGCGTGCTACCGTGAA-3′ |
| 20A9 | 39 nt | 5′-CACACGCTGAAGAAACTGAGGTCGTAGGTTTTCTTCGGG-3′ |
| 20A12P | 78 nt | 5′-TTCACGGTAGCACGCATAGGGCCCGACACTCGTCCACCCGA TACCTCTCATGTGTCCCCATCTGACCTCTGTGCTGCT-3′ |
| 20A14P | 80 nt | 5′-AGCAGCACAGAGGTCAGATGGGCATGGGGAAGAGAAAGC GGGATAACTTCGTTACCGGGCCCTATGCGTGCTACCGTGAA-3′ |
| 20A24P | 80 nt | 5′-AGCAGCACAGAGGTCAGATGGGGGGAAGACACAGAGAAA GGCCGGGGTGAAGTGTAGAGGCCTATGCGTGCTACCGTGAA-3′ |
| Negative control aptamers | Size | Aptamer sequencesb |
| RAND-80 | 80 nt | 5′-TTCACGGTAGCACGCATAGGGTGCAAGCTGAGGCAAGCAA CAGCGGAGGTGCGTTGAGGGCATCTGACCTCTGTGCTGCT-3′ |
| RAND-39 | 39 nt | 5′-AGTAGAGACTGGCGTGCTGGTTCTCACATCGAATCGTAG-3′ |
The aptamer are named as in the original publication [20]
All GAS aptamers and negative control aptamers were labeled with a 6-carboxyfluorescein (FAM) on the 5′-end or 3′-end
Fig. 1.

Initial binding tests with DNA aptamer 20A24P and live GAS cells. a Stationary phase GAS SF370 or GAS 5448 cells were incubated for 45 min at 37 °C with the indicated end concentrations of 5′- or 3′-FAM labeled 80-nucleotide (nt) GAS aptamer 20A24P (solid black lines), the 80-nt negative control aptamer 5′-FAM-RAND-80 (dashed lines), or aptamer vehicle (gray solid). Subsequently, the bacteria were washed and subjected to flow cytometry to measure the fluorescence intensities of 20,000 bacterial particles per sample. b Performed as above, GAS 5448 incubated with 5′-FAM labeled GAS aptamer 20A24P over a range of concentrations. The histograms show FL-1 channel overlays for representative samples of several experiments performed. Each sample was run in duplicate with similar results
Truncation of 20A24P leads to increased affinity of GAS aptamer
Seven truncated 5′-FAM-20A24P variants ranging from 22 to 69 nt in length were synthesized (see Supplementary Table 1 for sequences and Supplemental Fig. 1A for secondary structures). These were assayed for binding to GAS 5448 to help pinpoint the minimal functional 20A24P sequence. Aptamers 20A24P.A2, 20A24P.A3, 20A24P.A4, 20A24P.A8, and 20A24P.A9 bound significantly to GAS, with 20A24P.A3 showing the strongest reactivity (Fig. 2a). In dose titration experiments, 20A24P.A3 showed increased GAS binding compared to the parental aptamer at 10–1000 nM concentrations (Fig. 2b). Given the guanosine residue prevalence in 20A24P and that 20A24P.A3 and 20A24P.A8 displayed binding to GAS whereas 20A24P.A6 did not, it is likely that the parental aptamer forms a G-quartet that is required for aptamer binding starting with G20 and ending with G60 as predicted by the QGRS-Mapper tool (Supplemental Fig. 1B). Subsequent experiments were done with 20A24P and/or 20A24P.A3.
Fig. 2.

Truncation of GAS aptamer 20A24P from 80 to 52 nucleotides leads to enhanced binding to live GAS 5448 cells. a Binding of the indicated 5′-FAM labeled truncated versions of the GAS aptamer 20A24P (solid black lines) and 39-nt negative control aptamer RAND-39 (dashed lines) at 500 nM end concentration or aptamer vehicle (gray solid). For the data shown for 20A24P.A9, 3′-FAM labeled GAS aptamer (solid line) and 52-nt negative control aptamer RAND-52 (dashed line) were used. The length of the respective GAS aptamers is indicated in parentheses. b Binding comparison of parental 5′-FAM 80-nt GAS aptamer 20A24P (black solid lines) and 5′-FAM truncated 52-nt aptamer 20A24P.A3 (dashed lines) at the indicated aptamer end concentrations vs. aptamer vehicle (gray solid). For all samples, the bacteria were incubated with the aptamers or vehicle for 37 °C at 45 min, then washed and subjected to flow cytometry to measure the fluorescence intensities of 20,000 bacterial particles per sample. Each sample was run in duplicate with similar results
20A24P and 20A24P.A3 binding to GAS is growth-phase dependent
5′-FAM-20A24P.A3 was assayed for binding to live GAS 5448 at different growth stages. 5′-FAM-20A24P.A3 binding was absent in early exponential phase bacteria, weak binding was seen with mid-exponential phase GAS, and strong reactivity was seen during the late exponential and stationary growth phases (Fig. 3). The results were confirmed with 5′-FAM-20A24P (data not shown). Reduced aptamer reactivity with exponential phase streptococci could not be explained by antigen masking by the hyaluronan capsule or aptamer degradation by DNase SdaI, since the aptamers lacked binding to exponential phase 5448 capsule (ΔhasA) and DNase (Δsda1) negative mutants, respectively, whereas 20A24P bound well to the mutants in the stationary phase (data not shown).
Fig. 3.
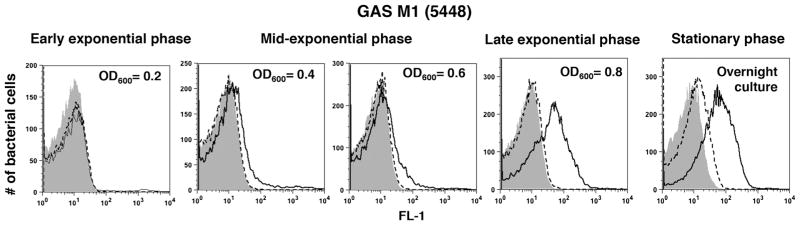
Growth phase dependence of 20A24P.A3 binding to live GAS cells. GAS 5448 cells were grown in THB media to the indicated optical densities at 600 nm (OD600), then washed and photometrically adjusted to the same bacterial concentrations. Then, the binding of 5′-FAM versions of the 52-nt GAS aptamer 20A24P.A3 (black solid lines) to the bacteria was tested as described in Figs. 1 and 2. 5′-FAM 52-nt aptamer RAND-52 (dashed lines) and aptamer vehicle (gray solid) served as negative controls. FL-1 histogram overlays for the indicated stages of growth for a representative experiment of several performed are shown. For each experiment, the samples were run in duplicate yielding similar results
Identification of the conserved region of M protein as the 20A24P target
To identify the molecular target of 20A24P and 20A24P.A3, aptamer binding to several isogenic GAS 5448 mutants was tested in the stationary growth phase. Both aptamers showed equivalent binding to wild-type GAS and a capsule-deficient ΔhasA mutant, indicating that the capsule was not the aptamer target (data not shown). Similarly, the aptamers bound well to a mutant lacking the cysteine protease SpeB (ΔspeB) (data not shown), proving that the aptamer target did not require proteolytic SpeB processing, which is expressed strongly in stationary phase at the peak aptamer reactivity. Importantly, both 20A24P and 20A24P.A3 did not bind to a M1 protein-deficient (Δemm1) mutant, and reactivity was restored in a complemented mutant strain expressing emm1 on a plasmid pM1 (Fig. 4a shows 20A24P data). This indicated that the 20A24P and 20A24P.A3 target was the M1 protein or that aptamer binding to another target required M1 presence. To determine if the aptamer target was the M protein, 3′-biotinylated 20A24P was synthesized and its binding to recombinant full-length M1 protein and several truncated M protein versions tested by ELISA (Fig. 4b illustrates the M1 protein variants). Biotinylated 20A24P reproducibly bound strongly to full-length M1 protein and a C-terminal region containing the conserved S domain, C repeats and D repeats (SCD), whereas minimal binding was observed to the variants HC, AB, BC (see photographs of ELISA wells in Fig. 4b) or negative control proteins. These results pinpointed the M protein as the 20A24P target; the aptamer specifically reacted with the conserved protein SCD region.
Fig. 4.

M protein is the target of aptamer 20A24P on GAS cells. a The binding of 5′-FAM GAS aptamer 20A24P (black solid lines) at a concentration of 500 nM to stationary phase cells of live GAS 5448 wild type, an isogenic M1-deficient emm1 mutant, and a complemented emm1 mutant was determined by flow cytometry. Five hundred nanomolar 5′-FAM 80-nt aptamer RAND-80 (dashed lines) and aptamer vehicle (gray solid) served as negative controls. The histogram overlays show the FL-1 fluorescence intensities of 20,000 bacterial particles per sample. b By ELISA, the binding of 3′-biotinylated 20A24P to immobilized recombinant full-length mature M1 protein and truncated versions, which are schematically depicted, was tested. Biotinylated aptamer RAND-80 and vehicle served as negative controls. The photographs show representative results of several experiments performed. Yellow color indicates binding of an aptamer to the respective protein, whereas colorless wells indicate lack of binding of a given test reagent to the immobilized proteins
After the M protein was identified as the 20A24P aptamer target, we re-examined why lower aptamer binding was observed in the exponential phase of GAS growth. M protein-specific rabbit and mouse hyper-immune antisera were used to determine whether detectable M protein expression correlated with 20A24P binding. First, we demonstrated that antiserum IgG binding to GAS wild-type bacteria was M protein specific, since both mouse and rabbit IgG bound more strongly to GAS wild-type 5448 bacteria compared to an isogenic M protein deficient emm1 mutant (Supplemental Fig. 2A). As shown in Supplemental Figure 2B, antiserum IgG binding to GAS was readily detectable in early exponential phase and increased in later growth phases. The increased antiserum IgG reactivity correlated with enhanced 20A24P binding in late exponential phase. Therefore, one potential reason for lower aptamer binding until late exponential phase might be that the M protein has to accumulate to a critical density before strong aptamer binding can be detected.
Preparation of 5′-α-Gal conjugated 20A24P and 20A24P.A3
5′-α-Gal, 3′-FAM versions of the GAS aptamers 20A24P and 20A24P.A3, herein designated as alphamers α20A24P and α20A24P.A3, were prepared and tested for reactivity with live GAS 5448. The 5′-α-Gal, 3′-FAM alphamer αRAND-80 served as negative control. α20A24P bound well to GAS 5448 and SF370, whereas the fluorescence shift of GAS exposed to αRAND-80 was only slightly greater compared to vehicle-treated cells (Fig. 5a). In contrast, the truncated alphamer α20A24P.A3 bound only weakly to GAS (data not shown). The lack of binding of α20A24P.A3 was attributable to the positioning of the 3′-FAM label, since non-α-Gal conjugated aptamer 20A24P.A3 with a 3′-FAM tag also lost GAS binding (data not shown).
Fig. 5.

Recognition of 5′-α-Gal GAS alphamers on bacteria by anti-α-Gal antibodies. 5′-α-Gal conjugated versions of the GAS aptamers 20A24P (α20A24P) and 20A24P.A3 (α20A24P.A3) as well as negative control RAND-80 (αRAND-80), herein referred to as alphamers, were prepared. a Binding of 5′-α-Gal, 3′-FAM α20A24P (black solid lines) and αRAND-80 (dashed lines) to live GAS 5448 and SF370 cells at an end concentration of 1 μM. The binding of transgenic mouse IgM (b, d) and IgG (c) antibodies to alphamer coated, live GAS 5448 cells was tested by flow cytometry. Bacteria were pre-incubated with 5′-α-Gal, 3′-FAM-labeled GAS alphamer α20A24P (Black solid lines in b, c), non-FAM-labeled 20A24P.A3 (black solid lines in d), 3′-FAM-labeled control alphamer αRAND-80 (dashed lines) or alphamer vehicle (gray solid). The binding of the primary antibodies was detected with secondary Alexa Fluor® 647 anti-mouse IgM (b, d) or anti-mouse IgG (c) antibodies. This allowed for simultaneous visualization of alphamer and mouse antibody binding in the FL-1 and FL-4 channels, respectively. Samples incubated with alphamers but no mouse IgM or IgG (–mouse IgM; –mouse IgG) served as negative controls to rule out unspecific secondary antibody binding to the bacteria. Representative results of several experiments performed are shown in the histogram overlays. e To determine if the α-Gal portion was critical for mouse antibody binding to 5′-FAM-α20A24P-coated GAS 5448, the ability of the alphamer and non-α-Gal-conjugated aptamer 5′-FAM-20A24P at 1 μM endconcentration to redirect mouse IgM antibodies to the bacteria was compared as above. Vehicle and 5′-FAM-αRAND-80 were used as controls. FL-1 (= Aptamer/alphamer binding) vs. FL-4 (= IgM binding) dot blots for one representative experiment of several performed are shown
Recognition of alphamers bound to GAS cells by mouse anti-Gal antibodies
We next determined if the GAS alphamers could serve as a molecular bridge to promote the binding of anti-Gal antibodies to the bacterial surface. As a consistent high-titer anti-Gal source, mouse antibodies purified from transgenic GT−/− mice expressing the anti-Gal antibody M86 [23] were utilized. Supplemental Fig. 2 shows the anti-Gal titers of these preparations. To determine mouse antibody binding, live GAS was pre-incubated with α20A24P, control alphamer αRAND-80, or vehicle. Then, bacteria were mixed with transgenic mouse IgM, or IgG, or buffer only, and surface-bound IgM and IgG were detected with Alexa Fluor® 647-labeled anti-mouse antibodies. By flow cytometry, both alphamer binding (FL-1 channel) and mouse antibody binding (FL-4 channel) were simultaneously measured. As seen in the leftmost FL-1 histogram overlay in Fig. 5b, 3′-FAM α20A24P bound well to GAS, whereas the αRAND-80-treated cells showed background fluorescence comparable to vehicle-treated bacteria. Strongly enhanced mouse IgM binding was detected on α20A24P-treated GAS (second to the left FL-4 histogram in Fig. 5b) indicating that IgM recognized the GAS alphamer on the streptococci. As negative controls, bacteria treated with α20A24P, αRAND-80, or vehicle were probed with secondary antibodies in the absence of mouse IgM. Here, α20A24P bound well to GAS (Fig. 5b, FL-1 histogram on the right), but no secondary antibody signal was observed (Fig. 5b, FL-4 histogram on the right), ruling out non-specific secondary antibody binding to α20A24P-treated GAS. Similar results were obtained with mouse IgG (Fig. 5c), which bound only to 3′-FAM-α20A24P-treated GAS.
As mentioned above, the 5′-α-Gal, 3′-FAM truncated GAS alphamer α20A24P.A3 only bound weakly to streptococci due to 3′ manipulation with FAM. Binding of non-FAM-labeled 5′-α-Gal alphamer α20A24P.A3 was confirmed functionally, since mouse IgM bound to GAS treated with 5′-α-Gal α20A24P.A3 but not to negative control alphamer-treated bacteria (Fig. 5d).
To demonstrate that the α-Gal modification was required for aptamer recognition by mouse antibodies, binding studies with 5′-α-Gal, 3′-FAM-α20A24P were performed in parallel with non-α-Gal conjugated 3′-FAM-20A24P. The α-Gal portion was necessary for mouse IgM binding to GAS suggesting that these antibodies were α-Gal specific (Fig. 5e). In the dot plots, 86 % of GAS cells exposed to 1 μM α20A24P and mouse IgM were FL-1 and FL-4 positive corresponding to alphamer and IgM binding, respectively, whereas 10 % of FL-1 negative cells were also FL-4 negative demonstrating that alphamer binding was required for IgM binding. In contrast, no IgM binding was seen for GAS cells treated with 3′-FAM-20A24P, despite 43 % of the cells being FL-1 positive (Fig. 5e). As expected, vehicle or negative control alphamer-treated cells were FL-1 and FL-4 negative (Fig. 5e).
Combined, the results demonstrated that the α-Gal portion of α20A24P mediated anti-Gal antibody recruitment to GAS.
Alphamers promote GAS phagocytosis by human neutrophils
A critical goal of alphamer-based therapeutics is promotion of opsonophagocytic bacterial clearance. We tested if α20A24P and α20A24P.A3 could stimulate GAS phagocytosis by human neutrophils in the presence of mouse and human anti-Gal IgG. Human neutrophils were added to green fluorescent GAS 5448 pre-incubated with anti-GAS alphamers, negative control alphamer, or vehicle control, all in the presence of mouse or human polyclonal anti-Gal IgG or vehicle control. The GAS alphamers did not confer phagocytosis over the negative controls in the absence of IgG (see vehicle control panel in Fig. 6a). In contrast, both GAS alphamers significantly increased phagocytosis in the presence of either transgenic mouse IgG or hIVIG (Fig. 6a). The hIVIG used was verified to possess a significant anti-Gal IgG titer (Supplemental Fig. 3).
Fig. 6.

GAS alphamers confer uptake of GAS M1T1 cells by human phagocytes in the presence of mouse and human polyclonal IgG in an alpha-Gal-specific manner. Green fluorescent GAS cells were pre-incubated with the indicated 5′-α-Gal conjugated GAS alphamers, non-alpha-Gal GAS aptamer, the respective control alphamer/aptamer, or vehicle only. Then, polyclonal mouse IgG or human IgG (hIVIG) and, finally, purified human blood neutrophils were added to the bacteria. After 20 min of co-incubation, the phagocytes were washed and examined for uptake of GAS by fluorescence microscopy. Extracellular bacteria were distinguishable from intracellular bacteria by counterstaining with ethidium bromide. Samples were run in quintuplicate or hexuplicate, and average percentages of neutrophils with phagocytosed bacteria ± SD are shown in the graphs. ***p<0.00005, unpaired t test, GAS alphamers vs. respective control alphamers and vehicle controls. Representative data of one experiment for each data set of at least two performed are shown
We next demonstrated that anti-Gal antibodies in both mouse and human IgG were responsible for stimulating phagocytosis. Specifically, 5′-α-Gal, 3′-FAM α20A24P but not parental 3′-FAM aptamer 20A24P (no α-Gal) promoted GAS phagocytosis in the presence of mouse or human IgG (Figs. 6b–c).
α20A24P confers killing of GAS by human phagocytes and impairs GAS growth in human blood
After finding that GAS alphamers promote phagocytosis, we determined whether this translates to bactericidal activity in opsonophagocytic killing and human whole blood experiments. Bacteria were opsonized with alphamers and then co-incubated with purified human neutrophils or whole blood. In the presence of 1 mg/mL hIVIG as anti-Gal source, α20A24P promoted GAS SF370 killing by human neutrophils compared to control αRAND-80 (Fig. 7a). Control samples without neutrophils demonstrated that the α20A24P-anti-bacterial effect was phagocyte-dependent (Fig. 7a). α20A24P impaired the growth of invasive isolate GAS 5448 in human whole blood when compared to control αRAND-80 (Fig. 7b).
Fig. 7.

Activity of GAS alphamer in opsonophagocytic killing assays and whole blood killing assay. a Late exponential phase GAS SF370 was pre-incubated with GAS alphamer α20A24P or control alphamer αRAND-80. After 20 min, neutrophils and human IgG (hIVIG) were added. Samples to which vehicle (HBSS+/+) and hIVIG instead of neutrophils were added served as no phagocyte controls. At various time points, the percentage of live colony forming units as compared to time point 0 min was determined. Quadruplicate to hexuplicate samples were run for each condition, and average bacterial percentage values as compared to the initial bacterial concentrations±SD plotted. *p<0.05; ***p<0.001, α20A24P vs. αRAND-80 in presence of neutrophils; two-way ANOVA with Bonferroni post test. b 1.1×105 CFU/mL of GAS M1T1 5448 were pre-incubated for 15 min with 1 μM of the GAS alphamer α20A24P (closed bars) or the control alphamer αRAND-80 (open bars) in a total volume of 30 μL. Then, 120 μL of human blood was added. Thus, the alphamer end concentrations were 200 nM. After 60, 120, 180, and 240 min of co-incubation, the bacterial concentrations were determined. Each sample was run in quadruplicate and average values± SD are plotted. *p<0.05, α20A24P vs. αRAND-80 two-way ANOVA with Bonferroni post tests
Discussion
The goal of this study was to provide first in vitro proof of principle of an immunotherapeutic approach using α-Gal-modified aptamers to recruit pre-existing antibodies for an antibacterial function. We demonstrated that M protein-specific 5′-α-Gal DNA alphamers bound a broad range of GAS serotypes, recruited anti-Gal IgG and IgM antibodies to the streptococcal surface in an α-Gal-dependent manner, and mediated bacterial uptake and killing by human neutrophils. Moreover, the GAS alphamer α20A24P was shown to reduce growth of invasive GAS in human blood.
Hamula et al. reported 20A24P as a high affinity aptamer reacting with several different M serotypes in a GAS species-specific manner [20]. In our study, we identified the abundant cell surface antigen M protein as the 20A24P target. 20A24P specifically bound to the highly conserved SCD C-terminal M protein region [28], which explains the broad-spectrum reactivity of the aptamer to multiple M serotype strains despite the high structural variability of the M protein N-terminal regions [29]. Of note, 20A24P may form a G-quartet, which appears to be required for binding to the bacteria. Truncation of 20A24P further increased the binding strength of the aptamer.
M protein is a major GAS virulence determinant, which, among other functions, is implicated in having roles in adhesion to and internalization into host cells and in inhibition of phagocytosis and complement deposition [29, 30]. It will be interesting to explore if α20A24P can inhibit M protein function and thereby alter GAS virulence traits. Importantly, the activity of α20A24P in phagocytosis assays was fully dependent on the α-Gal portion of the alphamer, indicating that the antibacterial effect was not due to M protein inhibition.
The M protein-specific aptamer 20A24P bound in a growth phase-dependent manner to GAS with highest reactivity observed in the late exponential and stationary growth phases. We hypothesized that the reason for low 20A24P binding in the early and mid-exponential growth phases was that the hyaluronic acid capsule may mask the aptamer target epitope, as expression of the capsule synthesis genes is highest in the exponential phase and diminishes in later growth stages [31, 32] coinciding with higher aptamer reactivity. However, the aptamer failed to bind to capsule-negative GAS cells in early and mid-exponential phase. Consistent with this, the conserved target region of 20A24P in the M protein is believed to extrude the bacterial cell wall [29]. Moreover, DNase SdaI expression did not influence aptamer binding. The M protein-encoding emm gene is highly expressed in the exponential growth phase [32]. Consistent with this, we showed detection of M protein by mouse and rabbit M protein-specific antisera on exponential phase GAS cells. Antiserum IgG reactivity with the streptococci increased in later stages of growth correlating with a higher degree of 20A24P aptamer binding. Therefore, one potential reason for the low aptamer binding until late exponential phase might be that the M protein has to accumulate to a critical density before strong 20A24P binding can be detected.
Mouse IgG and IgM from transgenic GT−/− mice producing anti-Gal antibodies [23] bound specifically to α20A24P-and α20A24P.A3-coated streptococci. Since the α20A24P α-Gal modification was essential for mouse IgM binding, we concluded that the immunoglobulins directed to the bacteria were anti-Gal antibodies. Uptake of α20A24P-covered GAS by human neutrophils in the presence of mouse or human IgG was also dependent on the α-Gal modification, further suggesting that anti-Gal antibodies recognized the alphamer on the bacteria and mediated their phagocytosis. Since the phagocytosis experiments were conducted without complement, it is likely that the bacterial engulfment via α20A24P and α20A24P.A3 and mouse or human IVIG was dependent on neutrophil Fc receptors.
The opsonophagocytosis experiments showed that α20A24P could not only mediate GAS uptake by neutrophils but also bacterial killing. In our model system, purified pooled normal human serum IgG (hIVIG) at 1 mg/kg could not mediate killing of GAS SF370 by human phagocytes during a 60-min incubation period. In contrast, ~50 % killing of the streptococci was achieved the presence of α20A24P in a phagocyte-dependent manner. These experiments establish proof of concept that a model alphamer can mediate antibacterial activity. Salvadori et al. showed previously that human sera with anti-GAS capsule titers could mediate various degrees of opsonophagocytic killing of GAS [33]. Moreover, the levels of M protein-specific antibodies in human serum correlate with protection against infection with the homologous serotype of GAS [34]. It would be interesting to see how α20A24P activity would compare side by side with human sera with high anti-GAS capsule or M protein titers or hyper-immune sera from humans treated with a GAS vaccine. However, such reagents are not readily available and comparative activity experiments were not in the scope of the current proof-of-principle study. Overall, there is no clear correlate available between the activity of GAS polyclonal or monoclonal antibodies (or alphamers) in opsonophagocytic killing assays and their in vivo efficacy in animal models of infection or humans. Therefore, future in vivo studies with alphamers that are specific for GAS or other bacteria will be critical to address the therapeutic alphamer potential.
α20A24P impaired the growth of invasive GAS M1T1 in human whole blood. Nevertheless, the alphamer was not able to fully inhibit bacterial growth. One possible explanation could be degradation of the DNA alphamer by blood nucleases. This limitation could theoretically be overcome by chemical modifications rendering the alphamer more nuclease-resistant [35] or reselection of a nuclease-resistant GAS-specific aptamer. In contrast to neutrophil phagocytosis and killing assays, active complement was present in whole blood assays. It remains to be evaluated if the anti-Gal antibodies bound to bacteria via α-Gal bridging can activate the classical complement pathway, which is an important component of the immune defense against bacteria [36].
Previous evidence for the potential of antibody-recruiting molecules for treating bacterial infections in humans was recently reviewed by McEnaney et al. [37]. Li et al. induced binding of endogenous anti-Gal antibodies to Escherichia coli tagged with α-Gal-mannose conjugates, which bound to the bacteria via a mannose-specific lectin [38]; anti-Gal-mediated antibacterial activity was not tested [38]. Kaewsapsak and colleagues [39] labeled Helicobacter pylori with dinitrophenyl for recognition by an anti-dinitrophenyl mAb and killing by human blood cells. Other studies with hapten-labeled bacteria demonstrated that monoclonal anti-avidin and anti-fluorescein antibodies can induce complement-mediated lysis and phagocytosis of E. coli [40] and Staphylococcus aureus and Streptococcus pneumoniae uptake by mouse macrophages [41]. Unlike ours, those studies testing antibacterial activity did not use endogenous human antibodies, but mAbs. Nevertheless, it can be contemplated that humans could be vaccinated with haptens to passively induce antibodies that would recognize hapten-tagged bacteria.
In summary, we showed that α-Gal linked to a GAS-specific DNA aptamer could label bacteria for recognition by pre-existing anti-Gal antibodies and can mediate host phagocytic uptake and killing in vitro. This proof-of-concept study raises the possibility that any other bacterial pathogen for which a specific alphamer could be designed could be targeted for recruitment of pre-existing human antibodies (summarized schematically in Fig. 8). Of note, since aptamers can be generated in a highly specific manner and would likely be administered for systemic infections, minimal side effects of a carefully designed alphamer to the beneficial host micro-flora would be expected. As for any other antiinfective drug, animal models of infection will be critical to examine the therapeutic potential of an alphamer (Fig. 8). Furthermore, human anti-Gal levels will have to be correlated with antibacterial alphamer activity and pharmacodynamics. Further exploration of this novel therapeutic approach of applying α-Gal technology to nuclease-resistant aptamers specific for additional pathogens of critical importance to human medicine such as multi-drug resistant species is warranted.
Fig. 8.

Summary scheme for characterizing aptamer-based drugs. In a process termed Systematic Evolution of Ligands by Exponential Enrichment (SELEX), bacterial whole cells or bacterial components, aptamer libraries, PCR, and sequencing are used to identify aptamer candidates for further characterization. Fluorescently tagged aptamer candidates are synthesized and prioritized based on their binding to several strains of the target bacterial species by flow cytometry. Then, alphamer versions of prioritized aptamers are prepared. After confirmation that the alphamers bind to bacteria and attract anti-Gal antibodies to the microbial surface, alphamer activity is tested in correlate of protection assays in vitro (complement deposition and lysis, phagocytosis assays) that may predict the in vivo activity of candidate molecules. Alphamers with strong in vitro activity could then be tested in mouse infection models and later in humans
Supplementary Material
Key Messages.
α-Gal-tagged aptamers lead to GAS opsonization with anti-Gal antibodies.
α-Gal-tagged aptamers confer phagocytosis and killing of GAS cells by human phagocytes.
α-Gal-tagged aptamers reduces replication of GAS in human blood.
α-Gal-tagged aptamers may have the potential to be used as novel passive immunization drugs.
Acknowledgments
The authors would like to thank Drs. Igor Kojic (Loxbridge Research, London, UK), Dimitri van Simaeys, and Paul Hatala (both Altermune Technologies) for helpful discussions. We are grateful to Chelsea Stewart and Dr. Partho Ghosh (Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA) for providing recombinant M protein constructs. The authors are thankful to Dr. Malak Kotb (University of North Dakota School of Medicine and Health Sciences, Grand Forks, ND) and Dr. Bernard W. Beall (CDC, Atlanta, GA) for providing GAS strains for the study and Dr. Mark Walker (University of Queensland) for providing anti-M1 rabbit antiserum. The work of Dr. Iacomini was supported by NIH grant 1R01AI083459-06.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00109-015-1280-4) contains supplementary material, which is available to authorized users.
Author contributions SAK, JHH, BH, and EL designed and conducted the experiments; SAK and VN wrote the manuscript; SAK, BH, RO, UG, CR, KBM, MW, and VN contributed to the study design. BH designed truncated aptamer variants. UG and JI provided critical research materials. SAK, MW, and VN directed the study.
Conflict of interest The aptamer work was funded in full by Altermune Technologies, LLC (Irvine, CA). Several of the authors are employees, have executive roles and/or are consultants to Altermune Technologies, LLC with respect to their efforts to develop aptamers as therapeutic drugs.
Contributor Information
Mike Westby, Email: mike.westby@altermune.com.
Victor Nizet, Email: vnizet@ucsd.edu.
References
- 1.Chan M. Combating antimicrobial resistance: time for action. Copenhagen, Denmark: 2012. Antimicrobial resistance in the European Union and the world. [Google Scholar]
- 2.Prevention CfDCa. Antibiotic resistance threats in the United States. 2013;2013 [Google Scholar]
- 3.Boucher HW, Talbot GH, Benjamin DK, Jr, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D Infectious Diseases Society of America. 10 × ‘20 Progress–development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis. 2013;56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pucci MJ, Bush K. Investigational antimicrobial agents of 2013. Clin Microbiol Rev. 2013;26:792–821. doi: 10.1128/CMR.00033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oleksiewicz MB, Nagy G, Nagy E. Anti-bacterial monoclonal antibodies: back to the future? Arch Biochem Biophys. 2012;526:124–131. doi: 10.1016/j.abb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Scully IL, Liberator PA, Jansen KU, Anderson AS. Covering all the bases: preclinical development of an effective Staphylococcus aureus vaccine. Front Immunol. 2014;5:109. doi: 10.3389/fimmu.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmad TA, El-Sayed LH, Haroun M, Hussein AA, El Ashry el SH. Development of immunization trials against Klebsiella pneumoniae. Vaccine. 2012;30:2411–2420. doi: 10.1016/j.vaccine.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Quintanilla M, Pulido MR, McConnell MJ. First steps towards a vaccine against Acinetobacter baumannii. Curr Pharm Biotechnol. 2013;14:897–902. doi: 10.2174/1389201014666131226123511. [DOI] [PubMed] [Google Scholar]
- 9.Priebe GP, Goldberg JB. Vaccines for Pseudomonas aeruginosa: a long and winding road. Expert Rev Vaccines. 2014;13:507–519. doi: 10.1586/14760584.2014.890053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ter Meulen J. Monoclonal antibodies in infectious diseases: clinical pipeline in 2011. Infect Dis Clin North Amer. 2011;25:789–802. doi: 10.1016/j.idc.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun. 1988;56:1730–1737. doi: 10.1128/iai.56.7.1730-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galili U, Rachmilewitz EA, Peleg A, Flechner I. A unique natural human IgG antibody with anti-alpha-galactosyl specificity. J Exp Med. 1984;160:1519–1531. doi: 10.1084/jem.160.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galili U, Anaraki F, Thall A, Hill-Black C, Radic M. One percent of human circulating B lymphocytes are capable of producing the natural anti-Gal antibody. Blood. 1993;82:2485–2493. [PubMed] [Google Scholar]
- 14.Galili U. Interaction of the natural anti-Gal antibody with alpha-galactosyl epitopes: a major obstacle for xenotransplantation in humans. Immunol Today. 1993;14:480–482. doi: 10.1016/0167-5699(93)90261-i. [DOI] [PubMed] [Google Scholar]
- 15.Macher BA, Galili U. The Galalpha1,3Galbeta1,4GlcNAc-R (alpha-Gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim Biophys Acta. 2008;1780:75–88. doi: 10.1016/j.bbagen.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banerjee J, Nilsen-Hamilton M. Aptamers: multifunctional molecules for biomedical research. J Mol Med. 2013;91:1333–1342. doi: 10.1007/s00109-013-1085-2. [DOI] [PubMed] [Google Scholar]
- 17.Zimbres FM, Tarnok A, Ulrich H, Wrenger C. Aptamers: novel molecules as diagnostic markers in bacterial and viral infections? BioMed Res Intl. 2013;2013:731516. doi: 10.1155/2013/731516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sundaram P, Kurniawan H, Byrne ME, Wower J. Therapeutic RNA aptamers in clinical trials. Eur J Pharm Sci. 2013;48:259–271. doi: 10.1016/j.ejps.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 19.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 20.Hamula CL, Le XC, Li XF. DNA aptamers binding to multiple prevalent M-types of Streptococcus pyogenes. Analyt Chem. 2011;83:3640–3647. doi: 10.1021/ac200575e. [DOI] [PubMed] [Google Scholar]
- 21.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acid Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kikin O, D’Antonio L, Bagga PS. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34:W676–W682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cretin N, Iacomini J. Immunoglobulin heavy chain transgenic mice expressing Galalpha(1,3)Gal-reactive antibodies. Transplantation. 2002;73:1558–1564. doi: 10.1097/00007890-200205270-00007. [DOI] [PubMed] [Google Scholar]
- 24.Galili U, Anaraki F. alpha-galactosyl (Galalpha1-3Galbeta1-4GlcNAc-R) epitopes on human cells: synthesis of the epitope on human red cells by recombinant primate α1,3galactosyltransferase expressed in E.coli. Glycobiology. 1995;5:775–782. doi: 10.1093/glycob/5.8.775. [DOI] [PubMed] [Google Scholar]
- 25.Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, Gallo RL, Nizet V. D-alanylation of teichoic acids promotes group a streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol. 2005;187:6719–6725. doi: 10.1128/JB.187.19.6719-6725.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 27.Aziz RK, Kotb M. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis. 2008;14:1511–1517. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollingshead SK, Fischetti VA, Scott JR. A highly conserved region present in transcripts encoding heterologous M proteins of group A streptococci. Infect Immun. 1987;55:3237–3239. doi: 10.1128/iai.55.12.3237-3239.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oehmcke S, Shannon O, Morgelin M, Herwald H. Streptococcal M proteins and their role as virulence determinants. Clin Chim Acta. 2010;411:1172–1180. doi: 10.1016/j.cca.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 30.Smeesters PR, McMillan DJ, Sriprakash KS. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 2010;18:275–282. doi: 10.1016/j.tim.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 31.Crater DL, van de Rijn I. Hyaluronic acid synthesis operon (has) expression in group A streptococci. J Biol Chem. 1995;270:18452–18458. doi: 10.1074/jbc.270.31.18452. [DOI] [PubMed] [Google Scholar]
- 32.Unnikrishnan M, Cohen J, Sriskandan S. Growth-phase-dependent expression of virulence factors in an M1T1 clinical isolate of Streptococcus pyogenes. Infect Immun. 1999;67:5495–5499. doi: 10.1128/iai.67.10.5495-5499.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salvadori LG, Blake MS, McCarty M, Tai JY, Zabriskie JB. Group A streptococcus-liposome ELISA antibody titers to group A polysaccharide and opsonophagocytic capabilities of the antibodies. J Infect Dis. 1995;171:593–600. doi: 10.1093/infdis/171.3.593. [DOI] [PubMed] [Google Scholar]
- 34.Dale JB. Current status of group A streptococcal vaccine development. Adv Exper Med Biol. 2008;609:53–63. doi: 10.1007/978-0-387-73960-1_5. [DOI] [PubMed] [Google Scholar]
- 35.Pestourie C, Tavitian B, Duconge F. Aptamers against extra-cellular targets for in vivo applications. Biochimie. 2005;87:921–930. doi: 10.1016/j.biochi.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Zipfel PF, Hallstrom T, Riesbeck K. Human complement control and complement evasion by pathogenic microbes—tipping the balance. Mol Immunol. 2013;56:152–160. doi: 10.1016/j.molimm.2013.05.222. [DOI] [PubMed] [Google Scholar]
- 37.McEnaney PJ, Parker CG, Zhang AX, Spiegel DA. Antibody-recruiting molecules: an emerging paradigm for engaging immune function in treating human disease. ACS Chem Biol. 2012;7:1139–1151. doi: 10.1021/cb300119g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Zacharek S, Chen X, Wang J, Zhang W, Janczuk A, Wang PG. Bacteria targeted by human natural antibodies using α-Gal conjugated receptor-specific glycopolymers. Bioorg Med Chem. 1999;7:1549–1558. doi: 10.1016/s0968-0896(99)00099-1. [DOI] [PubMed] [Google Scholar]
- 39.Kaewsapsak P, Esonu O, Dube DH. Recruiting the host’s immune system to target Helicobacter pylori’s surface glycans. ChemBioChem. 2013;14:721–726. doi: 10.1002/cbic.201300006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertozzi CB, Bednarski MD. A receptor-mediated immune response using synthetic glycoconjugates. J Am Chem Soc. 1992;114:5543–5546. [Google Scholar]
- 41.Krishnamurthy VM, Quinton LJ, Estroff LA, Metallo SJ, Isaacs JM, Mizgerd JP, Whitesides GM. Promotion of opsonization by antibodies and phagocytosis of Gram-positive bacteria by a bi-functional polyacrylamide. Biomaterials. 2006;27:3663–3674. doi: 10.1016/j.biomaterials.2006.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.