

Abstract
Epidemiologic studies suggest that the incidence and severity of sepsis are ameliorated in patients on statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) for cholesterol lowering indications. We sought to understand the mechanism underlying such protection and hypothesized that simvastatin would be protective in mice against acute infection with Staphylococcus aureus, the primary etiologic agent in sepsis. Mice were treated with simvastatin or buffer for two weeks and were subsequently challenged with S. aureus intratracheally or intravenously. Relative to buffer-treated mice, bacterial killing was enhanced 4-fold (p=0.02), systemic dissemination was reduced, and lethality was decreased (hazard ratio 8.8, 95% CI 2.5 to 31.3, p=0.001) in mice that were pretreated with simvastatin for two weeks. Systemic inflammatory response was abrogated and the local elaboration of inflammatory mediators was diminished. Serum concentrations of pro-fibrinolytic protein C were elevated (p=0.034), while the concentration of pro-coagulant tissue factor in bronchoalveolar lavage fluids was attenuated (reduced 25%), p=0.001, in simvastatin-treated mice. Taken together, these data indicate that extended treatment with simvastatin is protective during infection with S. aureus through enhanced bacterial clearance, anti-inflammatory, and anti-coagulant activities. These studies provide insights into the mechanism by which statins confer protection in acute infection, support the notion that statins may be effective adjuncts in the treatment of sepsis, and provide a rationale for randomized control trials in patients that are at a high risk for infection and characterized by coagulopathy.
INTRODUCTION
Sepsis is an exaggerated, and often detrimental, response to infection characterized by systemic inflammation and coagulopathy [1]. Elucidation of the pathogenesis of sepsis has inspired interest in blocking important components of the pathways as novel therapeutic approaches. However, as multiple pro-coagulant and inflammatory pathways are triggered concurrently, blocking single components or mediators has been insufficient to favorably impact the course and outcome of sepsis in clinical trials.
Several reports indicate statins regulate systemic inflammation and coagulopathy associated with sepsis. Statins exert anti-inflammatory action independent of their cholesterol-lowering action [2, 3]. In in vitro studies, simvastatin inhibits cellular invasion by Staphylococcus aureus, the common etiologic agent in sepsis and healthcare-associated pneumonia [4]. In addition, S. aureus stimulates the expression of pro-coagulant factors during host cell invasion [5, 6]. Numerous epidemiologic studies and systematic reviews suggest that individuals on a statin regimen are at a decreased risk of death due to bacteremia, sepsis, or pneumonia [7-16]. However, most of the human reports are observational, with the inherent likelihood of selection or ascertainment bias, thereby casting doubt on the benefits of statins in infection [17-21]. Such observational reports provide relatively weak support for “off-label” use of statins as prophylaxis against infection. We hypothesize that pretreatment with simvastatin will protect mice against the deleterious effects of methicillin-resistant S. aureus (MRSA) pneumonia by modulating inflammatory response and ameliorating coagulopathy. The current study was undertaken to assess the cellular and molecular mechanisms of the beneficial effects of statins in host defense against acute infections. The objective of the current study was two-fold: 1) to assess the impact pretreatment with statin on sepsis-induced coagulopathy and 2) to examine the effects of statins on the outcome of experimental pneumonia due to S. aureus. We assessed the effect of extended low-dose simvastatin treatment on bacterial clearance, survival, coagulation factors, and inflammatory response in a mouse model of experimental S. aureus pneumonia. The findings in this study provide a rationale for randomized, controlled clinical studies of prophylaxis with statins in individuals at high risk for infection or that are in a prothrombotic state.
MATERIALS AND METHODS
Mice
All mice used in these studies were wild-type in the FVB/N genetic background. Mice were maintained in a barrier containment facility. Serologies were checked periodically for common murine pathogens in sentinel mice housed in the same room. Mice were handled according to the Institutional Animal Care and Use Committee guidelines at Cincinnati Children’s Hospital Medical Center. Mice were divided into three experimental conditions and treated as described below. Mice were age- and sex-matched between treatment groups for all experiments.
Experimental groups
Mice were treated as follows: 1) daily intraperitoneal (i.p.) injection of 200 μl of buffer (1% ethanol in normal saline); 2) daily i.p. injection of 6.3 μg (~0.25 mg/kg) of simvastatin suspended in buffer; and 3) daily i.p. injection of 200 μl of saline (for some studies). In preliminary experiments, no significant differences in lung bacterial burden were obtained between mice treated for two weeks and mice treated for three weeks. Subsequently, studies were performed on mice that had been pretreated with simvastatin for two weeks. Mice in the three groups were indistinct from one another at the beginning of the study. Mice were weighed every other day to monitor the change in weight gain.
Bacteria
A stock culture was maintained of luciferase-expressing Staphylococcus aureus Xen 29 (Xenogen Corporation, Alameda, CA; denoted as S. aureus Xen 29 in the text), a biotransformant of MRSA, which expresses luciferase from copies of the luxABCDE genes integrated as a chromosomal insert. To minimize variability in virulence, all bacteria were selected from aliquots of the same passage that had been frozen at −70°C in 20% glycerol/PBS. For each experiment, an aliquot of bacteria was thawed and plated on tryptic soy/5% sheep blood agar. A colony was inoculated in 4 ml of trypticase soy broth and grown to late logarithmic phase. Bacteria were pelleted from the media, washed in sterile PBS, and resuspended in 4 ml of Hank’s Balanced Salt Solution (HBSS) supplemented with 4.5 mM glucose. Bacteria were enumerated as colony-forming units (cfu) on blood agar plates from serial dilutions in PBS. For each experiment, the inoculum was confirmed by plating dilutions of the aliquot used.
Bacterial challenge
To assess bacterial killing, mice were challenged with 1 × 107 cfu S. aureus Xen 29 suspended in 100 μl PBS by intratracheal (IT) instillation. Mice were euthanized by i.p. injection of sodium pentobarbital 24 h post-infection, the lungs weighed, homogenized and dilutions plated for quantitative culture. The numbers of colonies were expressed as cfu/g lung tissue. To assess systemic dissemination of infection, splenic homogenates were plated and incubated overnight at 37°C and the number of colonies counted 24 or 48 h later. For each experiment, 8-10 mice from each group were infected. Studies were conducted twice and results pooled. Blood was aspirated via cardiac puncture. For each time point, sera were separated from blood specimens and stored at −20°C until analyzed for C-reactive protein or for protein C activity. For all other analyses, lung tissues were homogenized in sterile PBS, centrifuged at 250 × g for 10 minutes. Supernatants were stored at −20°C until analyses.
Survival studies
Six-week-old mice pretreated by daily i.p. injection with normal saline, buffer or simvastatin for two weeks were challenged with 5 × 108 cfu of S. aureus Xen 29 suspended in 100 μl for IT instillation or 1 × 108 for intravenous administration at the tail vein using a 30-gauge needle. Water and food were provided ad libitum during the period of observation. Mice were monitored every 12 h for overt disease states (inactivity, ruffled fur, cachexia). Moribund were euthanized.
Spatial distribution and longitudinal assessment of infection by luminescent imaging
For imaging, mice were anesthetized for bioluminescence measurements in an in vivo imaging system (IVIS, Caliper Life Science, Alameda, CA). The mice were anesthetized via inhalation of 2.5% isoflurane blended with oxygen and were imaged for 15 seconds to 5 min with a charge-coupled device camera which displays the intensity luminescence as computer-generated pseudocoloration superimposed on photographic images as previously described [22]. In preliminary experiments, groups of mice were challenged with 104 to 109 cfu of bioluminescent S. aureus Xen 29 IT to determine the limit of sensitivity and to assess correlation with cfu counts. Luminescent images acquired with the IVIS were quantified using the Living Image software (Caliper Life Sciences). The number of photons/s/cm2 was calculated from the region of the thoracic cavity encompassing the entire infected site.
Bronchoalveolar fluid (BALF) cell count
Five, 5 to 6-week-old mice were pretreated with simvastatin or with buffer for two weeks as described above. Mice were exsanguinated 24 h after challenge with S. aureus Xen 29 using a lethal dose of sodium pentobarbital i.p. The lungs were lavaged and BALF assessed for total and differential cell counts as described previously [23].
Lung morphology and immunohistochemistry
To assess lung histopathology, 24 h following challenge with S. aureus Xen 29, lungs from four, 5 to 6-week-old mice from each of the treatment groups were inflation-fixed with 4% paraformaldehyde for immunohistochemistry and light microscopy. Five-micron paraffin sections were stained with hematoxylin and eosin or immunostained for luciferase using a polyclonal antiserum directed against firefly luciferase (Abcam, Cambridge, MA). Parallel sections were incubated with pre-immune rabbit serum to verify the specificity of immunostaining.
Enzyme-linked immunosorbent assay (ELISA) for mouse C-reactive protein (CRP)
Sera obtained from mice challenged with S. aureus were assessed for the concentrations of C-reactive protein by enzyme-linked immunoassay using a mouse CRP ELISA kit as prescribed by the manufacturer of the kit (Life Diagnostics Inc, West Chester, PA).
Myeloperoxidase (MPO) assay
MPO activity was assessed in BALF or from lung homogenates obtained from uninfected mice or at 24 h post challenge by incubating 10 μl (in uninfected mice) or 10 μl of 1:100 dilution (infected mice) of BALF with 100 μl of 3, 3′, 5, 5′ tetramethylbenzidine in stabilized hydrogen peroxide (Sigma, St. Louis, MO) in a 96-well plate. The reaction was terminated after 30 min of incubation at room temperature with 50 μl of 0.5 M sulfuric acid. MPO activity was estimated from the optical absorbance at 450 nm relative to a standard curve generated with purified human neutrophil myeloperoxidase (Athens Research and Technology, Athens, GA). MPO content was expressed as relative MPO activity (Δ OD460 nm/mg protein). One unit was defined as the amount of enzyme required to decompose 1 μmole of hydrogen peroxide at 25°C, pH 6.0. Each sample was assayed in triplicate.
Cytokine/chemokine quantitative real-time RT-PCR
Total cellular RNA was extracted from lung tissues with RNeasy reagent according to manufacturer’s protocol (Qiagen Inc., Valencia, CA). Complementary DNA (cDNA) was synthesized from 1 μg of RNA using SuperScript III (Invitrogen, Carlsbad, CA). Quantification of transcripts of relevant cytokines/chemokines was performed by relative quantitative real-time RT-PCR (ABI PRISM 7700 SDS; PE Biosystems, Foster City, CA). Gene expression of the target sequence was normalized to the housekeeping gene β-actin or to 18S ribosomal RNA. All cDNA samples were tested in triplicate. All reagents for quantitative PCR were purchased from Applied Biosystems (Carlsbad, CA). Data are expressed as fold change relative to unchallenged normal saline-treated mice.
Assessment of protein C activity and tissue factor concentration
Protein C activity was determined in serum using a chromogenic assay (Biophen Protein C 2.5, Aniara, Mason, OH). Samples were diluted 1:2 in saline, followed by incubation with Protac (5 minutes, 37°C) to generate activated protein C, and incubation with the substrate SaPC-21 (5 minutes, 37°C) to form a colored product. After stopping the reaction with 20% acetic acid, the colored product, directly correlated to protein C activity, was measured at 405 nm using a Bio-Rad Model 680 Microplate Reader (Hercules, CA). Tissue factor concentration was measured in bronchoalveolar lavage fluid using the Actichrome Tissue Factor assay (American Diagnostica, Inc., Stamford, CT). Serially diluted lipidated tissue factor standard and undiluted samples were incubated with factors VIIa and X (15 minutes, 37°C) in the presence of Ca2+ to form tissue factor/FVIIa complexes and to convert factor X to Xa. Following incubation (20 minutes, 37°C) with Spectrozyme, a chromogenic Xa substrate, the reaction was stopped with glacial acetic acid and absorbance was measured at 405 nm. Tissue factor concentration was interpolated from a lipidated tissue factor standard curve.
Statistical analyses
Data are expressed as mean ± SEM. For bacterial clearance, data are reported as cfu/g lung tissue. Differences between groups were assessed by one-way ANOVA, and differences between means were assessed by contrast comparisons and the Student-Newman-Keuls test (GraphPad, La Jolla, CA). Non-parametric survival distributions were estimated to examine differences in survival among the three groups of mice. Differences between the groups were analyzed by Kaplan-Meier curve statistics.
RESULTS
Role of simvastatin in bacterial killing and survival following acute lung infection with S. aureus Xen 29
In preliminary experiments, mice were challenged with pathogens after 1, 2 or 3 weeks of daily i.p. injection of simvastatin or of buffer. Bacterial killing was not significantly different between buffer-treated mice and mice that were pretreated with simvastatin for 1 week. Similarly, there was no additional benefit, with respect to bacterial killing, to treating mice for 3 weeks compared to a two-week treatment with simvastatin. Subsequently, all bacterial challenges were performed after two weeks of treatment. This is a low-dosage regimen compared to doses used for controlling cholesterol in humans (see Discussion). Mean body weight at the beginning of experiments and weight gains during the pretreatment period were not significantly different between the three groups of mice (data not shown). Lung bacterial burden was reduced 65% at 24 h following acute infection with S. aureus Xen 29 in mice pretreated with simvastatin compared to control mice that received buffer, Figure 1, p=0.01. Bacterial clearance was not significantly different (p=0.87) between mice that received buffer (1% ethanol in normal saline) and mice that were treated with normal saline, p=0.65. Bacterial burden, as assessed by cfu counts on serial dilutions of lung homogenates, was corroborated with images obtained with IVIS at 24 h post infection (Figure 2), in which photon counts from bioluminescent bacteria were reduced 80% in simvastatin-treated mice compared to buffer-treated mice, p=0.02. Similarly, counts from lung homogenates assessed with a luminometer also were reduced 76%, p=0.001, in simvastatin-treated mice challenged with S. aureus Xen 29. Survival following challenge by IT instillation was enhanced in simvastatin-treated mice, p≤0.04 relative to mice that received normal saline or buffer, Figure 3A. Similarly, consistent with increased bacterial clearance, lethality was reduced in simvastatin-treated mice following intravenous challenge with S. aureus, p=0.03, Figure 3B. The median survival was 90 h versus 54 h in buffer-treated mice (hazard ratio 6.7, 95% confidence interval, 1.9 – 23.0). Similarly, the dose required to kill 50% of mice (LD50) by 72 h after challenge was increased by more that 1 log, p≤0.002, in simvastatin-treated mice. Lung histopathology was less severe in simvastatin-treated mice with respect to neutrophil infiltration of the alveolar space, alveolar septal thickening, and intra-alveolar debris, Figure 4A. Alveolar macrophages were heavily stained for luciferase (Figure 4B) and staining persisted for up to 72 h in buffer-treated mice (data not shown). Systemic dissemination of bacteria from the site of inoculation, as assessed from the amounts of bacteria recovered from splenic homogenates, was reduced >90% (15±8 vs. 159±42 cfu in buffer-treated mice) at 24 h post challenge in simvastatin-treated mice, p=0.01.
Figure 1. Pretreatment with simvastatin diminishes lung bacterial burden following challenge with S. aureus.
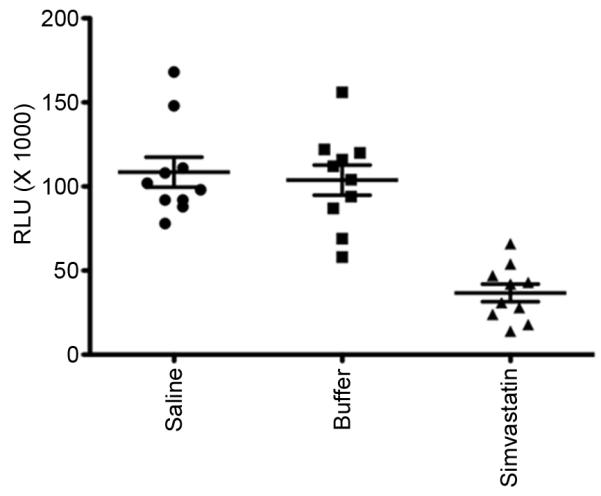
The luminescence emitted (RLU) from 100 μl of lung homogenates obtained 24 h post challenge was quantified with a microtiter plate luminometer reader and expressed as relative light units (RLU). *p≤0.05. n=8-10 mice per group.
Figure 2. Assessment of spatial distribution of bacteria following challenge.

Images were acquired using the in vivo imaging system (IVIS, Caliper Life Sciences) 24 h following challenge with bioluminescent S. aureus Xen 29.
Figure 3. Survival is enhanced in simvastatin-treated mice.
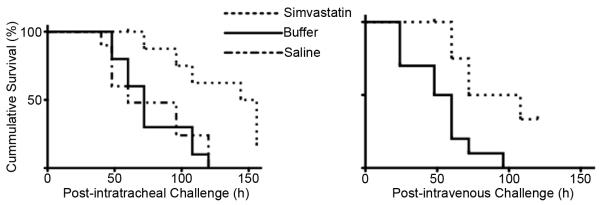
Lethality of S. aureus infection was assessed following intratracheal (Panel A) or intravenous challenge (Panel B). *p<0.04. n=10 mice per group.
Figure 4. Pulmonary histopathology is attenuated in simvastatin-treated mice.

Paraffin sections obtained 24 h post infection with S. aureus Xen 29 which expresses luciferase were stained with hematoxylin and eosin (Panel A) or immunostained for luciferase (Panel B).
Lung and systemic inflammatory response to lung infection with S. aureus
In uninfected mice, the differential cell counts and myeloperoxidase activity in BALF from saline-, buffer- or simvastatin-treated mice were not significantly different, p>0.55, Figure 5A. Following IT challenge with S. aureus Xen 29, lung neutrophil recruitment was significantly ameliorated in simvastatin-treated mice compared to saline- or buffer-treated mice, p≤0.02, Figure 5A. Similarly, MPO activity in BALF was reduced 51% in mice that were pretreated with simvastatin than control mice, Figure 5B, p=0.001. Expression of inflammatory mediators TNF-α, RANTES, MIP-2, IL-1β and IL-6 was reduced by 40-64% in S. aureus-challenged simvastatin-treated mice compared to buffer-treated mice, Figure 6, p≤0.05. Expression of IL-10 was not significantly altered by pretreatment with simvastatin. Systemic inflammation assessed by CRP concentration in the serum was reduced 20% in S. aureus-challenged simvastatin-treated compared to saline or buffer-treated mice, 821 vs. 641, p≤0.05, Figure 7.
Figure 5. Simvastatin pretreatment ameliorates lung cellular inflammatory response to infection.

A. Bronchoalveolar fluid (BALF) obtained 24 h post challenge was assessed for differential cell counts. B. Myeloperoxidase (MPO) activity was assessed in BALF as described in Methods. *p≤0.05. U=Uninfected mice, I=Infected mice. n= 5 mice per group.
Figure 6. Pretreatment with simvastatin attenuates expression of the genes for soluble inflammatory mediators following challenge with S. aureus.

Expression of the target sequences (TNF-α, IL-6, IL-1β, RANTES, MIP2 and IL-10) was assessed by qRT-PCR and normalized to β-actin or to 18S ribosomal RNA. Data are expressed as fold change relative to unchallenged mice pretreated with normal saline. *p≤0.05. U=Uninfected mice, I=Infected mice. n= 5 mice per group.
Figure 7. Simvastatin blunts systemic inflammatory response to infection.
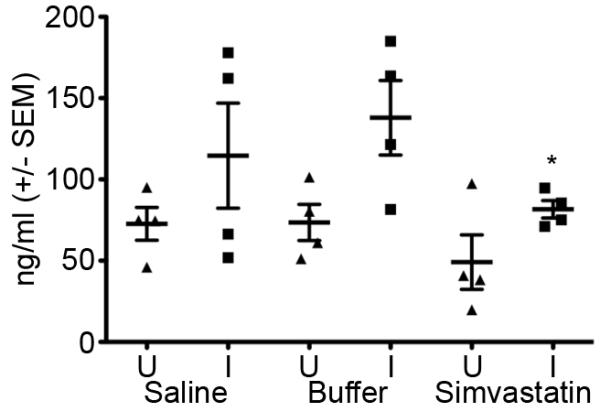
Sera from uninfected mice (U) or mice challenged with S. aureus Xen 29 (I) were assessed for the concentration of C reactive protein (CRP) by ELISA. *p≤0.05. U=Uninfected mice, I=Infected mice. n= 5 mice per group.
Assessment of coagulopathy in S. aureus-challenged mice
In simvastatin-treated mice, protein C activity in the serum was elevated prior to infection, p≤0.05, Figure 8A. Following infection, protein C activity remained elevated in the simvastatin-treated group and increased in the saline and buffer-treated groups, p≤0.05. Tissue factor levels were not significantly different between simvastatin and buffer-treated mice prior to infection, p>0.05. Following infection, tissue factor increased in the buffer-treated group, p≤0.05, but remained unchanged in simvastatin-treated mice, p>0.05, Figure 8B.
Figure 8. Simvastatin attenuates coagulopathy.

A. Sera from simvastatin- or buffer-treated mice challenged with S. aureus Xen 29 were analyzed for protein C activity using a chromogenic assay. B. Bronchoalveolar fluids from mice challenged with S. aureus Xen 29 were assessed for tissue factor activity as described in Methods. *p≤0.05. n=5 mice per group.
DISCUSSION
In the current study, the beneficial effects of simvastatin in experimental pneumonia were demonstrated in the absence of antibiotic treatment, suggesting that prophylaxis with simvastatin enhanced innate host defense, and that statins may be considered as a candidate for adjunctive therapy for bacterial infection. The enhanced bacterial clearance in this study was associated with diminished severity of lung histopathology, decreased systemic dissemination of bacteria from the site of inoculation, and with the modulation of inflammatory and procoagulant mediators. Taken together, these mechanisms may have contributed to the improved survival following challenge with MRSA in mice pretreated with simvastatin.
Although similar strategies targeting individual mediators of the cascade of inflammatory processes engendered in sepsis have been unsuccessful, the beneficial effects of simvastatin in the current study may be due to the fact that statins modulate multiple key components of sepsis. In addition to enhanced bacterial clearance, simvastatin affected coagulation factors and this may have provided additional protection at the level of the host during subsequent infection. In the clinical setting, one of the earliest signs of infection is an increase in pro-coagulant factors, detectable prior to changes in clinical or microbial markers [24]. Protein C and activated protein C (APC) possess potent fibrinolytic activity, capable of decreasing microvascular thrombosis commonly associated with infection [25]. The important role played by the protein C pathway in sepsis is underscored by the improved outcome of patients treated with recombinant APC [26]. APC enhances fibrinolysis by inactivating the coagulation factors Va and VIIIa and inhibiting expression of tissue factor [25]. In addition, protein C [27] and APC [25] possesses anti-inflammatory activities. S. aureus infection was associated with increased protein C levels. Decreased lethality of S. aureus in mice pretreated with simvastatin suggests that the protection by simvastatin may include augmenting endogenous levels of protein C prior to infection. In the current study, infection-induced increase in tissue factor concentration was also attenuated in the simvastatin-treated group. This may have been secondary to the inhibition of S. aureus invasion of endothelial cells and macrophages, as suggested by other reports [4, 5, 28, 29]. Finally, tissue factor levels could have been attenuated by the enhancement of the protein C pathway, as this pathway down-regulates the pro-coagulant tendency induced by sepsis [25]. Taken together, our data suggest that improved outcome following infection may have resulted from the pleiotropic actions of statins.
The underlying mechanisms for the enhanced bacterial clearance by pretreatment with simvastatin remain to be fully characterized. In vitro bactericidal activity in the absence of serum has been reported at a mean minimal inhibitory concentration far exceeding what is estimated for plasma levels for a 40 mg clinical dose (29.2 mg/l vs. 0.0209 mg/L) [30]. In vivo, cervastatin at a concentration 16-fold higher than the level employed in the current study administered 24 h and 1 h prior to infection enhanced the clearance of S. aureus in mice [31]. Although pulmonary and systemic bacterial burdens were significantly lower in simvastatin-treated mice in the current study, we could not demonstrate any direct bactericidal effect of simvastatin (data not shown), consistent with in vitro reports in which bactericidal activity was not detected when bacteria were incubated with simvastatin at therapeutic concentrations in the presence of serum, in an attempt to model in vivo bioavailability [4]. Our data are particularly exciting because, unlike previous studies, the dose of simvastatin used was to be 100 fold lower than concentrations equivalent to an 80 mg clinical dose [32]. Whether the mechanism for clearance is the same as the higher dosage regimens employed in other studies or it is mediated through an alternative mode of action remains to be determined. This could have significant clinical implications especially in the treatment of antibiotic resistant strains of bacteria such as the methicillin-resistant S. aureus used in this study.
Our study indicates that enhanced bacterial clearance, dampening of inflammatory response and modulation of coagulation may be principal mechanisms through which statins contribute to host defense. Our data suggest the potential usefulness of simvastatin in ameliorating the pro-coagulant response to bacterial infection/sepsis and support the conduct of a randomized, controlled clinical trial of the benefits of statins in sepsis using coagulation status, bacterial clearance, and clinical outcomes as endpoints.
Acknowledgments
This work was funded by the National Institutes of Health National Heart, Lung and Blood Institute Grants R37 HL056285 (HA) and R15HL092504 (SM).
References
- 1.Neyrinck AP, Liu KD, Howard JP, Matthay MA. Protective mechanisms of activated protein C in severe inflammatory disorders. Br J Pharmacol. 2009;158(4):1034–47. doi: 10.1111/j.1476-5381.2009.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miida T, Hirayama S, Nakamura Y. Cholesterol-independent effects of statins and new therapeutic targets: ischemic stroke and dementia. J Atheroscler Thromb. 2004;11(5):253–64. doi: 10.5551/jat.11.253. [DOI] [PubMed] [Google Scholar]
- 3.Liao JK. Clinical implications for statin pleiotropy. Curr Opin Lipidol. 2005;16(6):624–9. doi: 10.1097/01.mol.0000191913.16321.60. [DOI] [PubMed] [Google Scholar]
- 4.Horn MP, Knecht SM, Rushing FL, Birdsong J, Siddall CP, Johnson CM, Abraham TN, Brown A, Volk CB, Gammon K, Bishop DL, McKillip JL, McDowell SA. Simvastatin inhibits Staphylococcus aureus host cell invasion through modulation of isoprenoid intermediates. J Pharmacol Exp Ther. 2008;326(1):135–43. doi: 10.1124/jpet.108.137927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veltrop MH, Beekhuizen H, Thompson J. Bacterial species- and strain-dependent induction of tissue factor in human vascular endothelial cells. Infect Immun. 1999;67(11):6130–8. doi: 10.1128/iai.67.11.6130-6138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heying R, van de Gevel J, Que YA, Piroth L, Moreillon P, Beekhuizen H. Contribution of (sub)domains of Staphylococcus aureus fibronectin-binding protein to the proinflammatory and procoagulant response of human vascular endothelial cells. Thromb Haemost. 2009;101(3):495–504. [PubMed] [Google Scholar]
- 7.Liappis AP, Kan VL, Rochester CG, Simon GL. The effect of statins on mortality in patients with bacteremia. Clin Infect Dis. 2001;33(8):1352–7. doi: 10.1086/323334. [DOI] [PubMed] [Google Scholar]
- 8.Almog Y, Shefer A, Novack V, Maimon N, Barski L, Eizinger M, Friger M, Zeller L, Danon A. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004;110(7):880–5. doi: 10.1161/01.CIR.0000138932.17956.F1. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda H, Yuen PS, Hu X, Zhou H, Star RA. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int. 2006;69(9):1535–42. doi: 10.1038/sj.ki.5000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spitzer AL, Harris HW. Statins attenuate sepsis. Surgery. 2006;139(3):283–7. doi: 10.1016/j.surg.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 11.Kruger P, Fitzsimmons K, Cook D, Jones M, Nimmo G. Statin therapy is associated with fewer deaths in patients with bacteraemia. Intensive Care Med. 2006;32(1):75–9. doi: 10.1007/s00134-005-2859-y. [DOI] [PubMed] [Google Scholar]
- 12.Kruger S, Merx MW. Nonuse of statins--a new risk factor for infectious death in cardiovascular patients? Crit Care Med. 2007;35(2):631–2. doi: 10.1097/01.CCM.0000254069.48562.13. [DOI] [PubMed] [Google Scholar]
- 13.Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG. Statins and sepsis: multiple modifications at multiple levels. Lancet Infect Dis. 2007;7(5):358–68. doi: 10.1016/S1473-3099(07)70111-1. [DOI] [PubMed] [Google Scholar]
- 14.Donnino MW, Cocchi MN, Howell M, Clardy P, Talmor D, Cataldo L, Chase M, Al-Marshad A, Ngo L, Shapiro NI. Statin therapy is associated with decreased mortality in patients with infection. Acad Emerg Med. 2009;16(3):230–4. doi: 10.1111/j.1553-2712.2009.00350.x. [DOI] [PubMed] [Google Scholar]
- 15.Dobesh PP, Klepser DG, McGuire TR, Morgan CW, Olsen KM. Reduction in mortality associated with statin therapy in patients with severe sepsis. Pharmacotherapy. 2009;29(6):621–30. doi: 10.1592/phco.29.6.621. [DOI] [PubMed] [Google Scholar]
- 16.Viasus D, Garcia-Vidal C, Gudiol F, Carratala J. Statins for community-acquired pneumonia: current state of the science. Eur J Clin Microbiol Infect Dis. 2010;29(2):143–52. doi: 10.1007/s10096-009-0835-0. [DOI] [PubMed] [Google Scholar]
- 17.Mahboobi SK, Shohat EZ, Jellinek SP, Rose M. Systemic infections can decrease the threshold of statin-induced muscle injury. South Med J. 2006;99(4):403–4. doi: 10.1097/01.smj.0000209273.52754.86. [DOI] [PubMed] [Google Scholar]
- 18.Vincent A, Miller JA. Statins for sepsis: a cautionary note. Intensive Care Med. 2006;32(5):795. doi: 10.1007/s00134-006-0143-4. [DOI] [PubMed] [Google Scholar]
- 19.Drage SM, Barber VS, Young JD. Statins and sepsis: panacea or Pandora’s box? Lancet Infect Dis. 2007;7(2):80. doi: 10.1016/S1473-3099(07)70003-8. author reply 80-1. [DOI] [PubMed] [Google Scholar]
- 20.Golomb BA, Evans MA. Statin adverse effects : a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs. 2008;8(6):373–418. doi: 10.2165/0129784-200808060-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopterides P, Falagas ME. Statins for sepsis: a critical and updated review. Clin Microbiol Infect. 2009;15(4):325–34. doi: 10.1111/j.1469-0691.2009.02750.x. [DOI] [PubMed] [Google Scholar]
- 22.Kadurugamuwa JL, Sin LV, Yu J, Francis KP, Kimura R, Purchio T, Contag PR. Rapid direct method for monitoring antibiotics in a mouse model of bacterial biofilm infection. Antimicrob Agents Chemother. 2003;47(10):3130–7. doi: 10.1128/AAC.47.10.3130-3137.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akinbi HT, Epaud R, Bhatt H, Weaver TE. Bacterial killing is enhanced by expression of lysozyme in the lungs of transgenic mice. J Immunol. 2000;165(10):5760–6. doi: 10.4049/jimmunol.165.10.5760. [DOI] [PubMed] [Google Scholar]
- 24.van der Poll T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit Care. 2008;12(Suppl 6):S3. doi: 10.1186/cc7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esmon CT. The protein C pathway. Chest. 2003;124(3 Suppl):26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 26.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 27.Castellino FJ, Ploplis VA. The protein C pathway and pathologic processes. J Thromb Haemost. 2009;7(Suppl 1):140–5. doi: 10.1111/j.1538-7836.2009.03410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eto M, Kozai T, Cosentino F, Joch H, Luscher TF. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105(15):1756–9. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- 29.Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, Shiomi M, Schoen FJ, Libby P. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103(2):276–83. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 30.Jerwood S, Cohen J. Unexpected antimicrobial effect of statins. J Antimicrob Chemother. 2008;61(2):362–4. doi: 10.1093/jac/dkm496. [DOI] [PubMed] [Google Scholar]
- 31.Chaudhry MZ, Wang JH, Blankson S, Redmond HP. Statin (cerivastatin) protects mice against sepsis-related death via reduced proinflammatory cytokines and enhanced bacterial clearance. Surg Infect (Larchmt) 2008;9(2):183–94. doi: 10.1089/sur.2006.077. [DOI] [PubMed] [Google Scholar]
- 32.Newman TB, Hulley SB. Carcinogenicity of lipid-lowering drugs. Jama. 1996;275(1):55–60. [PubMed] [Google Scholar]