

Abstract
According to the ‘amyloid cascade hypothesis of Alzheimer’s disease’ first proposed about 16 years ago, the accumulation of Aβ peptides in the human central nervous system (CNS) is the primary influence driving Alzheimer’s disease (AD) pathogenesis, and Aβ peptide accretion is the result of an imbalance between Aβ peptide production and clearance. In the last 18 months multiple laboratories have reported two particularly important observations: (i) that because the microbes of the human microbiome naturally secrete large amounts of amyloid, lipopolysaccharides (LPS) and other related pro-inflammatory pathogenic signals, these may contribute to both the systemic and CNS amyloid burden in aging humans; and (ii) that the clearance of Aβ peptides appears to be intrinsically impaired by deficits in the microglial plasma-membrane enriched triggering receptor expressed in microglial/myeloid-2 cells (TREM2). This brief general commentary-perspective paper: (i) will highlight some of these very recent findings on microbiome-secreted amyloids and LPS and the potential contribution of these microbial-derived pro-inflammatory and neurotoxic exudates to age-related inflammatory and AD-type neurodegeneration in the host; and (ii) will discuss the contribution of a defective microglial-based TREM2 transmembrane sensor-receptor system to amyloidogenesis in AD that is in contrast to the normal, homeostatic clearance of Aβ peptides from the human CNS.
Keywords: Aβ42 peptides, Alzheimer’s disease (AD), amyloidogenesis, beta amyloid precursor protein (βAPP), hologenome, inflammation, innate-immunity, senile (amyloid) plaques (SP), triggering receptor expressed in microglial/myeloid cells-2 (TREM2)
AD amyloids and the amyloid cascade hypothesis
The ‘amyloid cascade hypothesis of Alzheimer’s disease’ proposes that the accumulation of amyloid-beta (Aβ) peptides in the inflammatory degeneration of neurons in the human central nervous system (CNS) is the primary influence driving Alzheimer’s disease (AD) pathogenesis [1]. These Aβ peptides of AD are originally derived from a polytopic, membrane-spanning, ~770 amino acid β-amyloid precursor protein (βAPP) though tandem beta- and gamma-secretase cleavage events [1–4]. Trafficking of the βAPP transmembrane holoprotein appears to be regulated by a large βAPP interactome that includes membrane integral and membrane peripheral adaptor proteins such as tetraspanin (TSPAN), secretase and sortilin proteins, and also by interactions with membrane-associated glycolipids and phospholipids [4–7] (Figure 1). Aβ peptide monomers are soluble, highly flexible, and have high aggregation propensity. While Aβ40 peptides prefer to associate with highly specialized microvessel endothelial cells that line the cerebral vasculature, the more neurotoxic, albeit less abundant and more hydrophobic Aβ42 peptides, form the central core of the senile plaque (SP). SPs are highly insoluble, pro-inflammatory parenchymal lesions that are progressively deposited during the course of AD [7–9]. The extra two hydrophobic amino acids in the Aβ42 peptide appear to convey many of the neurotoxic biophysical properties and self-aggregation of this slightly larger (42 amino acid) molecule [9–10]. Aβ42 peptides are not only highly immunogenic and pro-inflammatory but they may self-organize into ‘annular ring’ structures that allow hydrophobic side chains to face and interact with the plasma membrane, permitting charged/polar residues to face solvated channel pores. This allows uncontrolled leakage of ions into and/or out of the cell, thus destabilizing ionic homeostasis [9,10]. For example, excessively produced Aβ42 peptides may not only induce cellular toxicity directly through altered Aβ42 peptide-plasma membrane interactions and channel-mediated destabilization of ionic homeostasis, but also through direct interaction with cell adhesion molecules such as neuroligins and neurexins located in the post-synaptic cleft [9–12].
Figure 1. Highly schematicized depiction of the potential contribution of gastrointestinal (GI) tract microbiome-derived amyloids and lipopolysaccharide (LPS) to systemic and/or CNS amyloid burden – as.
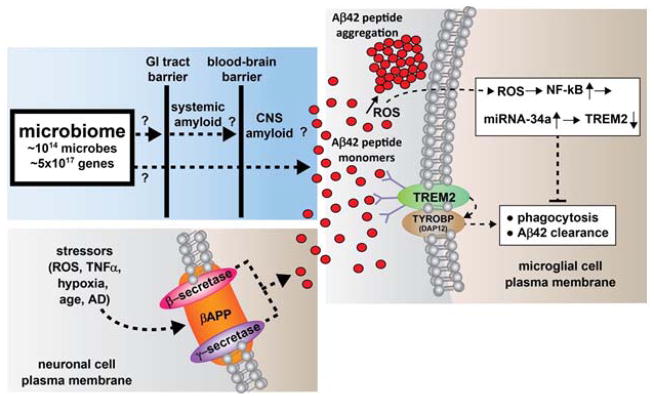
the major component if the human microbiome, gastrointestinal (GI) tract microbial sources of amyloid, LPS and/or other microbial-derived signaling molecules have potential to contribute to both systemic amyloid and CNS amyloid burden in their respective CNS compartments. The other major source of CNS amyloid – Aβ40 and Aβ42 peptide monomers (small red circles) – is generated from the tandem beta- and gamma-secretase (β- and γ-secretase; red and purple ovals, respectively) mediated cleavage of the neuronal cell plasma membrane-resident beta-amyloid precursor protein (βAPP, orange oval, lower left panel). The amyloid contribution from the microbiome may be increasingly important during the course of aging when both the GI tract barrier and blood-brain barrier become significantly more permeable to small molecules. Amyloidogenesis is further promoted, and phagocytosis and Aβ42 peptide clearance impaired, by insufficient TREM2 (green oval, right panel), a microglial cell plasma membrane receptor-sensor whose down-regulation has been shown to be mediated by increases in reactive oxygen species (ROS), NF-kB and miRNA-34a signaling. TREM2 function is linked to the TYROBP (DAP12) transmembrane protein (brown oval, right panel) whose abundance in unchanged in AD [42–54]. These or related mechanisms may operate (i) directly, via LPS/amyloid leakage through compromised GI tract or blood-brain barriers; (ii) directly, through deficits in the sensor/receptor TREM2; and/or (iii) indirectly, through LPS/amyloid-triggered cytokines or other small pro-inflammatory molecules which transit normally protective physiological barriers. Interestingly, microbes and their secretory exudates are extremely powerful pro-inflammatory and innate-immune system activators – gaining free access to the CNS would further induce these complement proteins and inflammatory cytokines which subsequently enhance vascular permeability, trigger host immunogenicity, and further induce the generation of ROS and NF-kB signaling. These neuropathogenic signals further promote amyloid aggregation and inflammatory degeneration characteristic of age-related neurological diseases including AD and other neurological disorders that exhibit defective Aβ42 peptide clearance mechanisms and progressive amyloidogenesis [4–11, 61–63]. See text for further details.
Interestingly, (i) Aβ42 peptide monomers, dimers, oligomers and fibrils each induce patterns of pro-inflammatory gene expression typical of the classical microglial-mediated innate-immune and inflammatory response induced by infectious agents such as bacterial LPS, a common lipopolysaccharide endotoxin secreted by the outer membrane of gram-negative bacteria [13,14]; (ii) the presence of bacterial LPS or endotoxin-mediated inflammation strongly contributes to amyloid neurotoxicity [15–17]; and (iii) AD amyloids, like prion amyloids, once formed, may induce a self-perpetuating process leading to amplification, aggregation and spreading of pathological protein assemblies [17–19]. Serial propagation of distinct strains of Aβ prion-like amyloids from AD patients has been recently observed [18–20]. Further, a number of recent studies support the evolving ideas: (i) that certain self-propagating amyloid-containing protein conformations feature in the pathogenesis of several common neurodegenerative diseases including AD; (ii) that pro-inflammatory and immunogenic aggregates of Aβ peptides may become self-propagating in AD brain; and (iii) that certain forms of Aβ peptides may be serially transmissible and hence important in the propagation of neurological diseases expressing pathological amyloids, such as in prion disease [18–24]. The contribution of microbial amyloids and LPS to the serial transmissibility of amyloidogenic Aβ peptide monomers and their capability to aggregate is currently not well understood. However, it has recently been shown that Aβ peptide fibrillogenesis is strongly potentiated by soluble bacterial endotoxins and viruses such as HSV-1, suggesting the contribution of infectious events and/or microbial-sourced factors to AD pathogenesis [16–23; see below].
The human microbiome and microbiome-derived amyloid
As for most mammals, Homo sapiens contain highly complex and remarkably dynamic communities of microbes collectively termed ‘the microbiome’ that forms a ‘metaorganism’ with commensal or symbiotic benefit to the human host [23–31]. Interestingly, the ~1014 microbial cells that comprise the human microbiome outnumber human host cells by approximately one hundred-to-one, the microbial genes of the microbiome outnumber human host genes by about one hundred-and-fifty to one, and together these microbes constitute the largest ‘diffuse organ system’ in the body, more metabolically active than the liver [24–27]. Interestingly, only two bacterial divisions (of the 52 divisions currently identified by metagenomics analysis) are prominent in GI tract microbiota, and these include the anaerobic Gram-negative Bacteroidetes (~48%) and the Gram-positive Firmicutes (~51%). The remaining 1% of phylotypes are distributed amongst the Cyanobacteria, Fusobacteria, Proteobacteria, Spirochaetes and Verrucomicrobia, along with various species of fungi, protozoa, viruses and other commensal microorganisms [23–36]. That the Bacteroidetes and Firmicutes were preferentially selected from the 52 bacterial divisions available in the biosphere is of evolutionary interest with implications for the ‘hologenome’ theory. This theory postulates: (i) that all plants and animals establish commensal or symbiotic relationships with microorganisms; and (ii) that it is not the individual organism, but rather the organism together with its associated microbial communities that should be considered as the basic unit of natural selection and eukaryotic evolution [23–27]. Gastrointestinal (GI) tract microbes that make up 99% of the human microbiome in part define a GI tract-CNS axis that provides two-way homeostatic communication, through cytokine, immunological, hormonal and neuronal signals [25–30]. What is of interest is that a remarkably wide variety of microbiome-resident species, including bacteria and fungi, generate significant quantities of functional lipopolysaccharides (LPS), amyloids and related microbial exudates [24,27,32]. While early scientific interpretations of the nature of the microbiome suggested that these secreted amyloids and other shed molecules served some immune-evasion and microbial survival strategy within the host, more current ideas support a significant microbiotic and symbiotic role of benefit to both microbiome and host [26–30,32,34,35]. Considering the 1014 microbiota of the human microbiome (chiefly bacteria, but also including protozoa, viruses and other commensal microorganisms) it is apparent that humans tolerate a substantial life-long exposure to LPSs and microbial-generated amyloid and related microbial secretory products, which could potentially contribute to the pathology of progressive neurological disorders with an amyloidogenic component [36–38]. Indeed, the extremely large number and variety of microbiome inhabitants and their capability to produce relatively enormous quantities of LPS, amyloid and LPS/amyloid related signaling molecules indicates that human physiology may be chronically exposed to a tremendous systemic burden of wide varieties of microbial amyloid. This exposure may be especially important during the course of aging when both the GI tract epithelium and blood-brain barriers become significantly more restructured and permeable [15,22–25,35–41].
TREM2 and amyloid clearance
Aβ40 and Aβ42 peptide monomers, continually generated primarily by neurons are notoriously ‘sticky’, flexible hydrophobic peptides that rapidly self-aggregate into higher order, pro-inflammatory dimers, oligomers and fibrils [3–6]. The removal and clearance of excessive Aβ40 and Aβ42 peptide monomers thus represents a constant ‘homeostatic task’ for neurons. The clearance of free Aβ peptide monomers would limit the supply of Aβ species that would otherwise be progressively aggregated into highly insoluble, neurotoxic, pathogenic SP or cerebrovascular lesions [5–10] (see Figure 1). Indeed highly efficient systems have evolved for Aβ40 and Aβ42 peptide monomer removal in the CNS and one microglial-based receptor-sensor-phagocytosis system for Aβ amyloid peptide monomer clearance appears to be the triggering receptor expressed in myeloid/microglial cells-2 (TREM2) [42–54].
TREM2 is a variably glycosylated 230 amino acid microglial membrane-spanning stimulatory and signaling sensor-receptor of the immune-globulin/lectin-like gene superfamily encoded in mice on chr17 and in humans on chr6p21.1. Along with the membrane-spanning linker protein TYROBP (DAP12), TREM2 directly participates in Aβ40 and Aβ42 peptide sensing, phagocytosis and removal, and microglial cytokine and reactive oxygen and nitrogen species (ROS, RNS) production [42–50] (Figure 1). TREM2’s critical importance in Aβ40 and Aβ42 peptide monomer clearance is underscored by eight recent observations: (i) that relatively rare mutations of TREM2 or of its coupling protein TYROBP (DAP12; see Figure 1) are currently associated with the progressive, presenile dementing diseases Nasu-Hakola syndrome, polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy (POSL), sporadic amyotrophic lateral sclerosis (ALS) and sporadic AD [51–54]; (ii) that the abundant environmental neurotoxin aluminum, via an NF-kB-mediated induction of microRNA-34a (miRNA-34a), can down-regulate TREM2 and stimulate amyloid accumulation and aggregation in cultured microglial cells [55]; (iii) that down-regulation in the ability of microglia to phagocytose and degrade Aβ42 peptides in AD, and down-regulation in TREM2 expression, is observed in sporadic AD brain tissues [55,56]; (iv) that TREM2 knock-down has been shown to exacerbate age-related neuroinflammatory signaling and induce cognitive deficits in senescence accelerated mouse prone-8 (SAMP8) mice [57]; (v) that microglial TREM2 gene expression in cell culture, both at the level of mRNA and protein, have been shown to be remarkably sensitive to external cytokine stressors such as tumor necrosis factor-alpha (TNFα), a pro-inflammatory adipokine known to be up-regulated in AD brain [50; unpublished observations]; (vi) that pro-inflammatory neurotoxins such as bacterial LPS strongly down-regulate TREM2 and the ability of microglial cells to phagocytose extracellular debris [40,58; unpublished observations]; (vii) that down-regulation in the expression of TREM2 appears to be regulated in part by the up-regulation of the microglial-enriched, NF-kB-sensitive miRNA-34a and perhaps other NF-kB-sensitive miRNAs may be involved [47–50]; and (viii) that both anti-NF-kB and anti-microRNA therapeutic strategies have been shown to be useful in the restoration of homeostatic TREM2 gene expression levels, and the neutralization of inflammatory signaling and amyloidogenesis, at least in vitro [48–50; unpublished observations].
It is clear that insufficient TREM2 would allow Aβ40 and Aβ42 peptide monomers to progressively accumulate and aggregate within the extracellular space, and this appears to be what occurs over time in the sporadic AD brain [1–11]. From what is currently known, and recently discovered, it is tempting to speculate: (i) that loss-of-function engendered by TREM2 mutations in familial forms of AD may have the same end effects on deficiencies in phagocytosis as a down-regulation of a fully functional TREM2 in sporadic AD; and (ii) that modest TREM2 over-expression might be useful in enhancing the sensing, scavenging, phagocytosis and removal of cellular debris in the aging CNS, including neurotoxic and self-aggregating Aβ42 monomeric peptides. However, once Aβ40 and Aβ42 monomeric peptides become organized into higher order structures such as oligomers and fibrils, TREM-2 mediated systems may have difficulty in the phagocytosis and removal of these larger, insoluble and pro-inflammatory amyloid aggregates. Importantly, TREM2 expression and signaling have been recently shown to be selectively inducible and manipulated from outside of the microglial cell, at least in vitro [47–49]. These findings suggest that the modulation of TREM2 expression may be effectively regulated using highly specific targeting via exogenously supplied drug-based pharmacological approaches including NF-kB inhibitors and/or stabilized anti-miRNA strategies [7,48–50].
Summary
The recognition of the potential contribution of microbiome-derived LPS and amyloid peptides to human neurodegenerative diseases with an amyloidogenic component, such as sporadic AD and prion disease, are relatively recent discoveries [24–28, 32–36]. Microbiome species and their secretory products are extremely powerful pro-inflammatory and innate-immune activators in the host. These, in turn, induce host complement proteins and inflammatory cytokines, which subsequently accelerate the generation of free radicals, up-regulate ROS and/or RNS, increase vascular permeability, immunogenicity and aberrant activation of the innate-immune system. These pathological actions have been shown to further intensify the aggregation of amyloids into SP lesions and thereby promote the inflammatory degeneration characteristic of AD neuropathology, thereby maintaining a progressively defective Aβ peptide clearance mechanism. Indeed, a more thorough understanding of the human ‘hologenome’ and the human microbial ecosystem and their secretory products should provide insight into their contribution to age-related neurological diseases associated with amyloidogenesis, CNS inflammation and progressive age-related neurodegeneration [24,49]. It would certainly be interesting to ascertain: (i) if microbiome-generated amyloids, LPS or other microbial-derived factors become more systemically available as humans age; (ii) if any microbiome-secreted amyloids or related signaling molecules co-localize with the amyloid-dense SP deposits or other insoluble lesions that characterize AD; (iii) if these microbial-sourced molecules can induce immunogenicity in the host, perhaps via molecular mimicry or related immunological mechanisms [29]; (iv) if these highly interactive factors impact the onset, development, propagation and/or course of age-related inflammatory neurodegenerative disorders such as AD; (v) what the nature and evolution of amyloid-related communication between the microbiome and the CNS has on the development or propagation of amyloidogenesis and inflammatory degeneration throughout the aging CNS; and (vi) how our increased knowledge of microbiome-mediated mechanisms of amyloidogenesis might lead to the advancement of more effective anti-amyloid therapeutic strategies.
While the transcriptional regulation of βAPP expression has been known for some time, the regulation of expression of this Aβ peptide-generating precursor, and Aβ peptide clearance by small non-coding RNAs and microRNAs including miRNA-34a is a relatively more recent discovery [47,49,50,59]. A highly schematicized depiction of the potential contribution of GI tract microbiome-derived amyloids and lipopolysaccharides (LPSs) to systemic and/or CNS amyloid burden is shown in Figure 1. Such pathways may become increasingly important during the course of aging when both the GI tract and blood-brain barriers become more ‘leaky’ to the passage of small signaling molecules [35–41]. It is not clear if a ‘homeostatic’ amount of TREM2 would be able to handle this presumptive extra amyloid peptide load progressively provided by the microbiome during the course of aging.
In conclusion, deficits in Aβ42 peptide phagocytosis, clearance and amyloidogenesis may be orchestrated: (i) through amyloid- or LPS-triggered cytokines or other small microbiome-sourced pro-inflammatory molecules which transit normally protective GI tract and blood-brain barriers; (ii) via direct amyloid, LPS or other microbiome-sourced biomolecular ‘leakage’ through age- compromised GI tract or blood-brain barriers; and/or (iii) via deficits in the abundance of the TREM2 sensor/receptor and/or the associated phagocytosis mechanism of the microglial cell. These actions might be expected to place a tremendous additional amyloid burden on homeostatic CNS structure and function. It is important to appreciate that collectively, microbiome-derived bacterial, fungal and other microbial-derived sectretory products constitute an extremely large class of very powerful pro-inflammatory, complement and innate-immune system activators that have enormous potential to further induce pro-inflammatory cytokines, complement proteins and altered immunogenicity in the host CNS. Such pathogenic actions might be expected to further trigger GI tract and blood-brain vascular permeability, up-regulate host innate-immunity, and induce amyloid aggregation and inflammation. These in turn would drive the generation of free radicals, including ROS, RNS, and NF-kB signaling in self-perpetuating neuropathogenic cycles that are characteristic of age-related CNS diseases such as AD, and other neurological disorders with an amyloidogenic component [10–12, 24–28, 56,60–63] (Figure 1).
Acknowledgments
This work was presented in part at the Autism-One Meeting 20–24 May 2015, Chicago IL, USA, the Society for Neuroscience (SFN) Annual Meeting 15–19 November 2014, Washington, USA and at the Association for Research in Vision and Ophthalmology (ARVO) Annual conference 3–7 May 2015 in Denver CO USA. Sincere thanks are extended to Drs. L. Carver, E. Head, W. Poon, H. LeBlanc, F. Culicchia, C. Eicken and C. Hebel for short post-mortem interval (PMI) human brain and/or retinal tissues or extracts, miRNA array work and initial data interpretation, and to D Guillot and AI Pogue for expert technical assistance. Thanks are also extended to the many neuropathologists, physicians and researchers of Canada and the US who have provided high quality, short post-mortem interval (PMI) human CNS and retinal tissues or extracted total brain and retinal RNA for scientific study. Research on miRNA in the Lukiw laboratory involving the innate-immune response in AD, AMD and in other forms of neurological or retinal disease, amyloidogenesis and neuro-inflammation was supported through an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN) and NIH grants NEI EY006311, NIA AG18031 and NIA AG038834.
Footnotes
Conflict of interest: No conflicts declared.
References
- 1.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Van Broeck B, Van Broeckhoven C, Kumar-Singh S. Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener Dis. 2007;4:349–365. doi: 10.1159/000105156. [DOI] [PubMed] [Google Scholar]
- 3.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J Neurochem. 2012;120:9–21. doi: 10.1111/j.1471-4159.2011.07519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukiw WJ. Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer’s disease (AD) Expert Opin Emerg Drugs. 2012 Mar 23; doi: 10.1517/14728214.2012.672559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merezhko M, Muggalla P, Nykänen NP, Yan X, Sakha P, Huttunen HJ. Multiplex assay for live-cell monitoring of cellular fates of Aβ precursor protein (APP) PLoS One. 2014;9:e98619. doi: 10.1371/journal.pone.0098619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. Neuroreport. 2011;22:623–7. doi: 10.1097/WNR.0b013e3283497334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jang H, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R. Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs. Chem Soc Rev. 2014;43(19):6750–64. doi: 10.1039/c3cs60459d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connelly L, Jang H, Arce FT, Capone R, Kotler SA, Ramachandran S, Kagan BL, Nussinov R, Lal R. Atomic force microscopy and MD simulations reveal pore-like structures of all-D-enantiomer of Alzheimer’s β-amyloid peptide: relevance to the ion channel mechanism of AD pathology. J Phys Chem B. 2012;116(5):1728–35. doi: 10.1021/jp2108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sindi IA, Dodd PR. New insights into Alzheimer’s disease pathogenesis: the involvement of neuroligins in synaptic malfunction. Neurodegener Dis Manag. 2015;5(2):137–45. doi: 10.2217/nmt.14.54. [DOI] [PubMed] [Google Scholar]
- 12.Tran L, Ha-Duong T. Exploring the Alzheimer Aβ peptide conformational ensemble: A review of molecular dynamics approaches. Peptides. 2015;69:86–91. doi: 10.1016/j.peptides.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 13.Ferrera D, Mazzaro N, Canale C, Gasparini L. Resting microglia react to Aβ42 fibrils but do not detect oligomers or oligomer-induced neuronal damage. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.05.023. S0197-4580(14)00386-8. [DOI] [PubMed] [Google Scholar]
- 14.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 15.Hammer ND, Wang X, McGuffie BA, Chapman MR. Amyloids: friend or foe? J Alzheimers Dis. 2008;13:407–419. doi: 10.3233/jad-2008-13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dasari M, Espargaro A, Sabate R, Lopez del Amo JM, Fink U, Grelle G. Bacterial inclusion bodies of Alzheimer’s disease β-amyloid peptides can be employed to study native-like aggregation intermediate states. Chembiochem. 2011;12:407–423. doi: 10.1002/cbic.201000602. [DOI] [PubMed] [Google Scholar]
- 17.Asti A, Gioglio L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J Alzheimers Dis. 2014;39:169–79. doi: 10.3233/JAD-131394. [DOI] [PubMed] [Google Scholar]
- 18.Le NT, Narkiewicz J, Aulić S, Salzano G, Tran HT, Scaini D, Moda F, Giachin G, Legname G. Synthetic prions and other human neurodegenerative proteinopathies. Virus Res. 2014 doi: 10.1016/j.virusres.2014.10.020. S0168-1702(14)00437-7. [DOI] [PubMed] [Google Scholar]
- 19.Watts JC, Condello C, Stöhr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc Natl Acad Sci USA. 2014;111:10323–8. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernández-Borges N, Eraña H, Venegas V, Elezgarai SR, Harrathi C, Castilla J. Animal models for prion-like diseases. Virus Res. 2015 doi: 10.1016/j.virusres.2015.04.014. pii:S0168-1702(15)00148-3. [DOI] [PubMed] [Google Scholar]
- 21.Hill JM, Lukiw WJ, Gebhardt BM, Higaki S, Loutsch JM, Myles ME, Thompson HW, Kwon BS, Bazan NG, Kaufman HE. Gene expression analyzed by microarrays in HSV-1 latent mouse trigeminal ganglion following heat stress. Virus Genes. 2001;23:273–280. doi: 10.1023/a:1012517221937. [DOI] [PubMed] [Google Scholar]
- 22.Higaki S, Gebhardt BM, Lukiw WJ, Thompson HW, Hill JM. Effect of immunosuppression on gene expression in the HSV-1 latently infected mouse trigeminal ganglion. Invest Ophthalmol Vis Sci. 2002;43:1862–1869. [PubMed] [Google Scholar]
- 23.Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour - epigenetic regulation of the gut-brain axis. Genes Brain Behav. 2014;13(1):69–86. doi: 10.1111/gbb.12109. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Dua P, Lukiw WJ. Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s disease (AD) J Alzheimers Dis Parkinsonism. 2015;5(1):177. doi: 10.4172/2161-0460.1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill JM, Lukiw WJ. Microbial-generated amyloids and Alzheimer’s disease (AD) Front Aging Neurosci. 2015;7:9. doi: 10.3389/fnagi.2015.000092015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill JM, Clement C, Pogue AI, Bhattacharjee S, Zhao Y, Lukiw WJ. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD) Front Aging Neurosci. 2014;6:127. doi: 10.3389/fnagi.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharjee S, Lukiw WJ. Alzheimer’s disease and the microbiome. Front Cell Neurosci. 2013;7:153. doi: 10.3389/fncel.2013.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill JM, Bhattacharjee S, Pogue AI, Lukiw WJ. The gastrointestinal tract microbiome and potential link to Alzheimer’s disease. Front Neurol. 2014;5:43. doi: 10.3389/fneur.2014.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedland RP. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J Alzheimers Dis. 2015;45(2):349–62. doi: 10.3233/JAD-142841. [DOI] [PubMed] [Google Scholar]
- 30.Catanzaro R, Anzalone M, Calabrese F, Milazzo M, Capuana M, Italia A, Occhipinti S, Marotta F. The gut microbiota and its correlations with the central nervous system disorders. Panminerva Med. 2015;57(3):127–43. [PubMed] [Google Scholar]
- 31.Burokas A, Moloney RD, Dinan TG, Cryan JF. Microbiota regulation of the Mammalian gut-brain axis. Adv Appl Microbiol. 2015;91:1–62. doi: 10.1016/bs.aambs.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Syed AK, Boles BR. Fold modulating function: bacterial toxins to functional amyloids. Front Microbiol. 2014;5:401. doi: 10.3389/fmicb.2014.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobley L, Harkins C, MacPhee CE, Stanley-Wall NR. Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol Rev. 2015 Apr 22; doi: 10.1093/femsre/fuv015. fuv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lei YM, Nair L, Alegre ML. The interplay between the intestinal microbiota and the immune system. Clin Res Hepatol Gastroenterol. 2014 doi: 10.1016/j.clinre.2014.10.008. S2210-7401(14)00277-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhee SH. Lipopolysaccharide: basic biochemistry, intracellular signaling, and physiological impacts in the gut. Intest Res. 2014;12:90–95. doi: 10.5217/ir.2014.12.2.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoemark DK, Allen SJ. The microbiome and disease: reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J Alzheimers Dis. 2015;43:725–38. doi: 10.3233/JAD-141170. [DOI] [PubMed] [Google Scholar]
- 37.Tran L, Greenwood-Van Meerveld B. Age-associated remodeling of the intestinal epithelial barrier. J Gerontol ABiol Sci Med Sci. 2013;68:1045–1056. doi: 10.1093/gerona/glt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques F, Sousa JC, Sousa N, Palha JA. Blood-brain-barriers in aging and in Alzheimer’s disease. Mol Neurodegener. 2013;8:38. doi: 10.1186/1750-1326-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oakley R, Tharakan B. Vascular hyperpermeability and aging. Aging Dis. 2014;5:114–25. doi: 10.14336/AD.2014.0500114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badtke MP, Hammer ND, Chapman MR. Functional amyloids signal their arrival. Sci Signal. 2009;2:pe43. doi: 10.1126/scisignal.280pe43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol. 2012;20:66–73. doi: 10.1016/j.tim.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malkki H. Alzheimer disease: The involvement of TREM2 R47H variant in Alzheimer disease confirmed, but mechanisms remain elusive. Nat Rev Neurol. 2015 doi: 10.1038/nrneurol.2015.84. [DOI] [PubMed] [Google Scholar]
- 43.Dempsey LA. TREM2 function in microglia. Nat Immunol. 2015 Apr 21;16(5):447. doi: 10.1038/ni.3166. [DOI] [Google Scholar]
- 44.Savage JC, Jay T, Goduni E, Quigley C, Mariani MM, Malm T, Ransohoff RM, Lamb BT, Landreth GE. Nuclear receptors license phagocytosis by TREM2+ myeloid cells in mouse models of AD. J Neurosci. 2015;35(16):6532–43. doi: 10.1523/JNEUROSCI.4586-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015;125:2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivest S. TREM2 enables amyloid β clearance by microglia. Cell Res. 2015;25(5):535–6. doi: 10.1038/cr.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharjee S, Zhao Y, Lukiw WJ. Deficits in the miRNA-34a-regulated endogenous TREM2 phagocytosis sensor-receptor in Alzheimer’s disease (AD); an update. Front Aging Neurosci. 2014;6:116. doi: 10.3389/fnagi.2014.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones BM, Bhattacharjee S, Dua P, Hill JM, Zhao Y, Lukiw WJ. Regulating amyloidogenesis through the natural triggering receptor expressed in myeloid/microglial cells 2 (TREM2) Front Cell Neurosci. 2014;8:94. doi: 10.3389/fncel.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y, Lukiw WJ. TREM2 signaling, miRNA-34a and the extinction of phagocytosis. Front Cell Neurosci. 2013;7:131. doi: 10.3389/fncel.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Bhattacharjee S, Jones BM, Dua P, Alexandrov PN, Hill JM, Lukiw WJ. Regulation of TREM2 expression by an NF-κB-sensitive miRNA-34a. Neuroreport. 2013;24(6):318–23. doi: 10.1097/WNR.0b013e32835fb6b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 52.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variantp.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2013.6237. Epubahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malkki H. Alzheimer disease: The involvement of TREM2 R47H variant in Alzheimer disease confirmed, but mechanisms remain elusive. Nat Rev Neurol. 2015 doi: 10.1038/nrneurol.2015.84. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki A, Kakita A, Yoshida K, Konno T, Ikeuchi T, Hayashi S, Matsuo H, Shioda K. Variable expression of microglial DAP12 and TREM2 genes in Nasu-Hakola disease. Neurogenetics. 2015 doi: 10.1007/s10048-015-0451-3. [DOI] [PubMed] [Google Scholar]
- 55.Zhao Y, Hill JM, Bhattacharjee S, Percy ME, Pogue AI, Lukiw WJ. Aluminum-induced amyloidogenesis and impairment in the clearance of amyloid peptides from the central nervous system in Alzheimer’s disease. Front Neurol. 2014;5:167. doi: 10.3389/fneur.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem Pharmacol. 2014;88:495–8. doi: 10.1016/j.bcp.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang T, Yu JT, Zhu XC, Tan MS, Gu LZ, Zhang YD, Tan L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014;35(6):1243–51. doi: 10.1016/j.neurobiolaging.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 58.Zhong L, Chen XF, Zhang ZL, Wang Z, Shi XZ, Xu K, Zhang YW, Xu H, Bu G. DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J Biol Chem. 2015 doi: 10.1074/jbc.M115.645986. jbc.M115.645986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lukiw WJ, Rogaev EI, Wong L, Vaula G, McLachlan DR, St George Hyslop P. Protein-DNA interactions in the promoter region of the amyloid precursor protein (APP) gene in human neocortex. Brain Res Mol Brain Res. 1994;22:121–131. doi: 10.1016/0169-328x(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 60.Lukiw WJ, Handley P, Wong L, Crapper McLachlan DR. BC200 RNA in normal human neocortex, non-Alzheimer dementia (NAD), and senile dementia of the Alzheimer type (AD) Neurochem Res. 1992;17:591–597. doi: 10.1007/BF00968788. [DOI] [PubMed] [Google Scholar]
- 61.Coleman BM, Hill AF. Extracellular vesicles - Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin Cell Dev Biol. 2015;40:89–96. doi: 10.1016/j.semcdb.2015.02.007. Epub 2015Feb 20. [DOI] [PubMed] [Google Scholar]
- 62.Morrone CD, Liu M, Black SE, McLaurin J. Interaction between therapeutic interventions for Alzheimer’s disease and physiological Aβ clearance mechanisms. Front Aging Neurosci. 2015;7:64. doi: 10.3389/fnagi.2015.00064.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Folch J, Ettcheto M, Petrov D, Abad S, Pedrós I, Marin M, Olloquequi J, Camins A. Review of the advances in treatment for Alzheimer disease: Strategies for combating β-amyloid protein. Neurologia. 2015 doi: 10.1016/j.nrl.2015.03.012. S0213-4853(15)00064-X. [DOI] [PubMed] [Google Scholar]