

Abstract
Abnormal neuronal signaling caused by metabolic changes characterizes several neurological disorders, and in some instances metabolic interventions provide therapeutic benefits. Indeed, altering metabolism either by fasting or by maintaining a low-carbohydrate (ketogenic) diet might reduce epileptic seizures and offer neuroprotection in part because the diet increases mitochondrial biogenesis and brain energy levels. Here we focus on a novel hypothesis that a ketogenic diet-induced change in energy metabolism increases levels of ATP and adenosine, purines that are critically involved in neuron–glia interactions, neuromodulation and synaptic plasticity. Enhancing brain bioenergetics (ATP) and increasing levels of adenosine, an endogenous anticonvulsant and neuroprotective molecule, might help with understanding and treating a variety of neurological disorders.
Introduction
Key factors influencing normal and abnormal signaling in the brain include neuron–glia interactions as well as cellular and mitochondrial bioenergetics. Clinically, the relationship between metabolism and altered brain activity is well documented and, although this relationship has been exploited therapeutically, the mechanisms underlying these effects are not well understood. One example of this relationship is the change in brain activity precipitated by a ketogenic diet, a low-carbohydrate high-fat protocol designed to mimic fasting that was introduced in the 1920s as a strategy to manage and reduce epileptic seizures [1]. Historical observations noted that fasting decreased the incidence of seizures in persons with epilepsy, but because fasting cannot be maintained, seizures inevitably return. Like fasting, the ketogenic diet results in limited glucose availability due to the very low carbohydrate intake and forces the body to use ketones as an alternate source of acetyl-CoA to generate ATP. Also similar to fasting, a ketogenic diet does reduce the incidence of seizures significantly and it is particularly useful in pediatric cases of medically intractable epilepsy [2,3]. Conservatively, adherence to a ketogenic diet results in more than a 50% reduction in seizures in more than 50% of cases, and the neuroprotective benefits of this diet extend to a variety of acute and chronic neurological disorders [4].
Although therapeutic benefits and metabolic changes associated with a ketogenic diet have been described, to date the specific mechanism(s) by which a ketogenic diet alters neural activity is not well understood [5]. This gap between clinical efficacy and mechanistic understanding deters widespread use of the diet and hampers development of alternate approaches to treating seizure disorders. Currently, antiepileptic drugs are associated with side effects of headaches, weight gain, drowsiness and dizziness, as well as hormonal and fertility disruptions [6], and up to 35% of persons with epilepsy are not treated successfully with available antiepileptic drugs (AEDs). By contrast, ketogenic diets are largely devoid of these major side effects [7]. Overall, the efficacy of ketogenic diet therapy is at least comparable to AEDs [3], and the success of the ketogenic diet in cases of pharmacoresistant epilepsy suggests that altered metabolism is not targeting solely the same neural mechanisms as the AEDs [7]. New therapeutic strategies are needed that lack significant side effects and avoid compliance issues that restrict the use of this strict and unpalatable dietary regimen [3,7,8].
In general, AEDs act to either decrease neuronal excitability and/or increase neuronal inhibition, and some of the major mechanisms targeted by AEDs include voltage-gated ion channels and GABA-mediated inhibition [9]. In a clear distinction from AEDs, the anticonvulsant properties of the ketogenic diet have been attributed most often to mechanisms such as direct cellular effects of ketone bodies, alterations in neurotransmitter levels or circulating factors that act as neuromodulators, and transformations of cerebral energy metabolism [5,10]. However, these underlying mechanisms for ketogenic diets do not explain fully the changes in forebrain neuronal activity manifest clinically by anticonvulsant and neuroprotective outcomes [4]. Based on previous [11,12] and recent [13,14] research, it appears likely that altered brain bioenergetics and purinergic signaling explain most of the changes in neuronal activity observed with a ketogenic diet. Specifically, an increase in the purines ATP and adenosine integrates many observed experimental results and provides a direct neuronal mechanism underlying the anticonvulsant and neuroprotective properties of ketogenic diets. In this article, we present and discuss evidence that favors the hypothesis (see Figure 1) that purinergic mechanisms help explain the anticonvulsant and neuroprotective effects of ketogenic diets.
Figure 1.
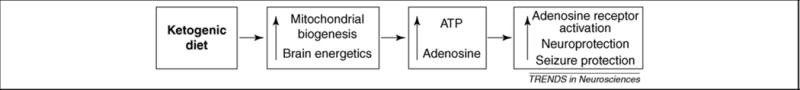
A purinergic signaling hypothesis for the neuroprotective and antiseizure actions of the ketogenic diet. During maintenance on a ketogenic diet, increased mitochondrial biogenesis, brain energetics and brain ATP levels are observed. Increased intracellular ATP enhances brain energy stores and homeostasis, and increased extracellular ATP is rapidly dephosphorylated into extracellular adenosine with subsequent signaling through adenosine receptors. Adenosine, acting at adenosine A1 receptors, exerts an inhibitory and anticonvulsant influence thereby providing neuroprotection and antiseizure effects. Enhanced signaling although A1 receptors provides a mechanism directly linking cellular energy molecules (ATP and adenosine) with decreased neuronal activity (A1 receptor activation).
Bioenergetics, purines and neuronal signaling
Bioenergetic status in the brain is a carefully orchestrated balance between energy needs—demanded mainly by Na+/K+ ATPase enzymes to maintain ionic homeostasis—and energy supplies such as glucose, ATP, creatine phosphate, glycogen, ketone bodies and various amino acids. Seizure disorders and insults related to acute and chronic neurodegenerative disorders greatly increase cellular energy demand. Without adequate supplies of primary and alternate sources of energy coupled with strong feedback and constraints on energy expenditure—particularly neuronal excitation—neuron dysfunction and death are likely. Therefore, increasing brain energy supplies and/or reducing energy demand could help prevent a bioenergetic crisis.
In brain, adenosine and ATP participate interdependently to balance energy supplies against demands and both purines regulate neuronal signaling directly via cell-surface receptors [15]. Adenosine activates four subtypes of G-protein-coupled receptors (A1, A2A, A2B and A3), and tonic levels of adenosine present extracellularly are sufficient to activate A1 and A2A receptors that have high affinity (~100 nM) for adenosine [16]. In hippocampus and cortex, adenosine A1 receptors are strongly inhibitory and their activation confers neuroprotection and elevates seizure thresholds [17]. Presynaptic adenosine A1 receptors reduce glutamate release, and postsynaptic adenosine A1 receptors hyperpolarize neurons through G-protein-coupled potassium channels.
Accordingly, augmenting adenosine A1 receptor activity can stop [18] or limit the spread of seizures [19] and enhance neuronal survival [17]. Conversely, reducing the influence of adenosine A1 receptor activation increases neuronal damage after an insult such as stroke or hypoxia and increases and strengthens persistent neuronal bursting activity patterns in both in vivo [20] and in vitro models of epilepsy [21]. Akin to a ketogenic diet’s success in refractory epilepsy, adenosine is an effective anticonvulsant even in animal models of drug-resistant epilepsy [22]. Thus, adenosine can reduce seizures by mechanisms independent of those implicated in the actions of AEDs.
ATP acts at its own purine receptors—a large family of both ionotropic (P2X) and metabotropic (P2Y) receptor subtypes with diverse pharmacological profiles and cellular effects [23]. Cell-surface ATP receptors are located presynaptically, where they can act to increase neuronal excitation [24] and postsynaptically, where they help control a variety of neuronal functions [25]. Intracellular ATP can also link directly to neuronal excitability via ATP-sensitive K+ channels, and reports have shown neuroprotective effects after augmenting [26] or blocking [27] their activity. Importantly, in addition to ATP providing cellular energy, signaling intracellularly, regulating ion channels and activating ATP receptors, extracellular ATP is dephosphorylated rapidly to adenosine and thereby contributes significantly to the ongoing influence of adenosine on synaptic transmission [28].
Basal endogenous extracellular adenosine is derived mainly from ATP released from neurons and glia [29], and adenosine levels are regulated by various enzymes including ecto- and endo-nucleotidases, adenosine kinase, adenosine deaminase, purine nucleoside phosphorylase and S-adenosylhomocysteine hydrolase. Whether formed intra- or extracellularly, adenosine can be transported across the plasma membrane by a family of nucleoside transporters and, under normal conditions, extracellular adenosine is removed constantly from the extracellular space via equilibrative transporters [30]. During energetically stressful conditions such as hypoxia, hypoglycemia or seizures, cellular energy demands outstrip ATP salvage, intracellular adenosine levels rise and adenosine levels rise rapidly in the extracellular space [31,32]. A sufficient increase in extracellular adenosine in many brain regions, including hippocampus and cerebral cortex, halts synaptic transmission entirely. In addition to pathological conditions such as stroke or seizure, diverse nonpathological stimuli such as NMDA receptor activation and small changes in temperature, pH and free radicals can regulate adenosine and/or ATP [33], and physiologically relevant changes in these variables alter seizure propensity and synaptic transmission significantly [34,35]. Likewise, we propose that a shift in metabolism, such as that precipitated by a ketogenic diet or fasting, might be a primary mechanism where altered bioenergetics influences neural activity and signaling significantly via changes in ATP and adenosine.
Glial cells, specifically astrocytes, regulate purine signaling and metabolism and participate strongly in controlling physiological and pathophysiological conditions [17,36,37]. Astrocytes modulate local neuronal activity and ongoing synaptic plasticity by releasing a variety of gliotransmitters [38] including ATP [39]. ATP in the extracellular space is dephosphorylated rapidly and the resulting adenosine contributes significantly to ongoing inhibitory influences exerted by activated A1 receptors [39]. This relationship is illustrated in Figure 2. Of special interest is the significantly increased size, differentiation and diverse localization of astrocytes in the human brain as compared to brains of other species [40]. The complexity of the human brain astrocytic network might be critical for ongoing neuromodulation and synaptic plasticity, but also greatly increases the propensity for pathological changes in astrocytes and results in abnormal purine signaling after a seizure or injury [41]. Thus, current research reveals the parallel ongoing and dynamic links between metabolism and neuronal activity through the purines ATP and adenosine, with critical regulation of extracellular purine signaling by astrocytes, purine receptors, adenosine transporters and ecto-enzymes.
Figure 2.
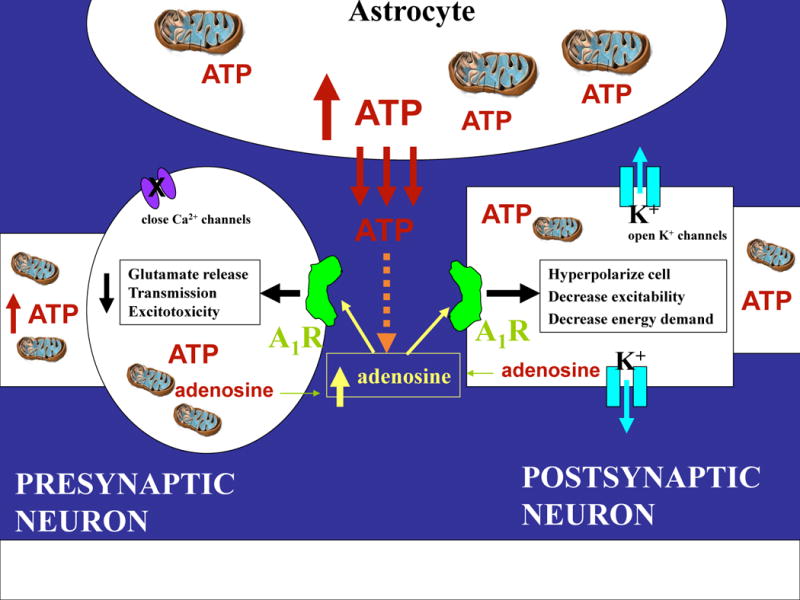
Proposed mechanisms underlying the therapeutic effects of a ketogenic diet. Metabolic alterations by ketogenic diets can lead to increased levels and actions of ATP and adenosine. Under normal conditions, ATP is released from astrocytes, and ATP and adenosine can also be ‘released’ from neurons. ATP released from astrocytes contributes to an ongoing influence on neural excitability and synaptic plasticity via adenosine, and any change in intracellular ATP significantly influences adenosine owing to the large gradient between ATP and adenosine inside the cell. Extracellular adenosine, whether it is released or originates from ATP, can bind to and activate inhibitory adenosine A1 receptors both pre- and postsynaptically. Together, ATP and adenosine can mimic many of the actions of ketogenic diets including hyperpolarizing neurons, closing calcium channels and opening potassium channels, decreasing glutamate release, decreasing neurotransmission, preventing excitotoxicity and averting cellular energy crisis.
The ketogenic diet, ATP and adenosine
Metabolic equilibrium and energy demands during compromised conditions are spearheaded by the relationship between glia and neurons, and the brain prioritizes its energy demands and ATP supply under conditions of stress [42]. The normal human brain increases its levels of high-energy molecules in response to an acute episode of either hyper- or hypoglycemia; this response in the brain to changes in glucose contrasts sharply with that in skeletal muscle [43]. Similarly, a more prolonged change in metabolism, either with fasting or with a ketogenic diet, is capable of changing the levels of purines and other energy molecules. Previous reports have suggested that a ketogenic diet increases ATP [11,12], and phosphocreatine to ATP ratio [44], and although it did not reach significance, doubles ADP levels [13].
Several physiological consequences of a ketogenic diet are similar to fasting and include hypoglycemia and increased levels of ketone bodies and free fatty acids [5]. There is little evidence that the ketone bodies themselves—β-hydroxybutyrate, acetoacetate and acetone—are responsible for the anticonvulsant influence. Direct application of β-hydroxybutyrate or acetoacetate did not affect synaptic responses [45], and levels of either these ketones [46], acetone [47] or its metabolites [4,48] do not correlate with the level of seizure control. Overall, acetone shows the best anticonvulsant effects in either in vivo or in vitro models, but a direct neuronal mechanism underlying the effects of acetone is unclear. In terms of excitatory neurotransmitters, an increase in glutamate uptake and transport has been discounted recently as a mechanism underlying the ketogenic diet’s success [49].
Both hypoglycemia and decreased pH are physiological changes that have been shown to increase adenosine and/or ATP levels. A decrease in glucose and changes in acid–base balance are observed with either a severe ketogenic diet or fasting [50], and although there is little evidence for a global change in brain pH with a ketogenic diet [51], there could be smaller changes in pH in restricted microdomains [34] or an increased magnitude of activity-dependent pH changes. Furthermore, astrocyte metabolism increases with a ketogenic diet and astrocytes are a major contributor to extracellular purines; astrocytes likely contribute to the metabolic changes by providing ketones to neurons when glucose is scarce [52].
Recently, there have been several new mechanisms proposed to underlie the anticonvulsant success of the ketogenic diet. One intriguing synaptic mechanism is increased activity at ATP-sensitive K+ channels, a mechanism which directly hyperpolarizes neuronal membranes [53]. Notably, adenosine A1 receptors regulate the activity of ATP-sensitive K+ channels [54], and a combination of increased activity at A1 receptors as well as increased ATP-sensitive K+ channels offers a more comprehensive anticonvulsant mechanism involving both purines. Inhibiting glycolytic enzymes with 2-deoxy-D-glucose (2-DG) is a metabolic manipulation other than a ketogenic diet or fasting which offers significant anticonvulsant activity. In addition, 2-DG might exert its anticonvulsant effects through changes in growth and transcription factors [14]. Although such mechanisms might play a crucial role, it should be noted that 2-DG and a host of metabolic inhibitors also serve to release adenosine, both at the synaptic [55] and the behavioral [56] level, and increased adenosine could be a complementary mechanism for both acute and chronic seizure protection and neuroprotection.
Enhanced mitochondrial biogenesis and a concerted upregulation of mitochondrial genes occur with a ketogenic diet [13]. These changes might be the substrate for increases in high-energy molecules such as ATP and ADP; accordingly, an increase in adenine nucleotides offers the potential for increased adenosine, particularly because the intracellular concentrations of ATP are ~10,000-fold higher than adenosine. Although no single mechanism can explain the anticonvulsant effects of a ketogenic diet [5], we propose that changes in ATP and adenosine are critical players in developing metabolic strategies to reduce seizures and, as outlined above, note overlap in mechanisms known to regulate purines and the mechanisms proposed to underlie the success of a ketogenic diet. As schematized in Figure 1 and illustrated in Figure 2, the actions of a ketogenic diet might be twofold: (1) enhanced energy status (mitochondrial biogenesis, high-energy molecules and ATP levels) helps maintain cellular homeostasis, and (2) enhanced adenosine levels acting at A1 receptors reduce neuronal activity and metabolic demand—directly increasing seizure threshold and reducing neuronal excitability.
Adenosine is best known as a sleep-promoting [57] and neuroprotective [58] molecule, released consequent to any type of mechanical brain injury, hypoxia, seizures or hypoglycemia. In addition to its success with epilepsy, the ketogenic diet shares clinical parallels with the physiological actions of adenosine in that the diet has been shown to improve sleep quality in children with epilepsy [59] and offer neuroprotection [4] in models of ischemia, traumatic brain injury [60] and insulin-induced hypoglycemia [61]. Although we currently propose that both changes occur—increased bioenergetics and increased adenosine—the endpoint of increased adenosine via another intermediary mechanism of regulation (decreased pH in a critical intra- or extracellular compartment, hypoglycemia, net intracellular ATP dephosphorylation and/or changes in enzymes that metabolize adenosine) remains an alternate explanation. Increased adenosine in forebrain regions critical for seizure initiation and propagation would still provide an anticonvulsant (and neuroprotective) influence, regardless of whether it is caused by or causal to the enhanced bioenergetics.
Food for thought: therapeutic implications
The recent popularity of and expanded food choices for low-carbohydrate diets, the ongoing challenges of pediatric and medically refractory epilepsy sustain ketogenic diets as a topic of intense research interest. Despite clinical success for more than 80 years, a poor understanding of the mechanisms underlying the therapeutic efficacy of a ketogenic diet limits more widespread acceptance of the diet. In addition, an incomplete understanding of mechanisms makes it impossible thus far to develop analogous and specific therapies which could eliminate the need for such strict compliance to a restrictive, controversial and unpalatable diet [62].
In parallel, more than 20 years ago, adenosine was noted as the brain’s natural anticonvulsant [63], and adenosine is a long-coveted therapeutic target for conditions ranging from epilepsy to stroke to chronic pain [17]. To date, however, efforts to capture its therapeutic and neuroprotective potential have been thwarted by unacceptable side effects of otherwise very specific and potent adenosine receptor agonists. The short half-life of adenosine in the blood, and the widespread distribution of adenosine receptors, including identical receptors in both brain and heart, makes it difficult to administer adenosine directly and avoid these complications. As a result, recent adenosine-based therapies have moved toward placing local adenosine-releasing grafts as polymers, encapsulated cells or engineered adenosine-releasing stem cells directly into the brain [22]. Knockout strategies to manipulate enzymes involved in adenosine regulation, such as adenosine kinase, are also being explored [20,41]. Other stimuli which regulate adenosine, such as decreased pH [34,64,65], also have clinical implications and might lead to novel therapies. Underscoring adenosine’s strong anticonvulsant potential are studies that have shown good seizure control under a variety of in vivo and in vitro conditions, including in animal models of pharmacoresistant epilepsy [22].
Interactions among bioenergetics, glial function and neuronal activity are gaining recognition for their paramount importance in influencing the ongoing physiology and pathophysiology of the brain. It is well known that ATP and adenosine play multiple roles in energy cycles and in glial and neuronal signaling [15]. Because of its direct connection between energy metabolism and neuronal signaling, adenosine has been described as a ‘retaliatory metabolite’ [66]. Alternate therapeutic strategies could exploit this unique and direct link between metabolism and neuronal activity for significant clinical potential. Recent evidence of the release of ATP from glia, its degradation to adenosine, and the specific metabolic and neuromodulatory neighborhood created by this relationship offers the perspective of a local anticonvulsant and neuroprotective zone created by the relationship between neurons, glia and purines (Figure 2). Progress in understanding the regulation of purines and the importance of the ‘tripartite synapse’ highlights the interrelationship between metabolism and synaptic transmission via purine molecules.
Here we present ATP and adenosine as key molecules underlying the success of a ketogenic diet (Figure 1). As illustrated in Figure 2 and demonstrated experimentally [28,67], either ATP or adenosine in the extracellular space will increase activation of A1 receptors and decrease neuronal excitability. To date, we and others have noted increases in purine levels with a ketogenic diet, but their direct role in signaling and the anticonvulsant and neuroprotective properties of the diet have not been elucidated. Still to be evaluated are possible changes in receptor levels and the relative role of glia versus neurons. Understanding how to alter metabolism to modulate ATP and adenosine reliably, and potentially in a brain region-specific manner, could yield major new therapeutic opportunities. Furthermore, the significance of understanding the critical mechanisms underlying the anticonvulsant and neuroprotective benefits of the diet extends far beyond epilepsy to other conditions, particularly chronic or neurodegenerative conditions as well as acute conditions with a prolonged recovery time. As examples, head injury and stroke share a sudden onset but offer an extended window during which enhanced survival of neurons could be pivotal in increasing long-term quality of life and recovery of function.
Notably, conditions that are influenced by a ketogenic diet are also those where adenosine is known to have therapeutic potential [4,68,69]. At this time, until a better pharmacological approach is developed, a testable, inexpensive and nontoxic modification to current treatment protocols postinjury or postseizure could be to avoid glucose and administer an alternate energy source such as lactate [70], ketones [60] or related compounds as an adjuvant neuroprotective strategy which upregulates ATP and adenosine. This approach would capitalize on the relationship between metabolism and neuronal signaling, augment endogenous levels of purines and thus enhance their endogenous anticonvulsant and neuroprotective potential. These types of readily testable and implementable therapeutic options are needed more urgently than ever, and understanding the key anticonvulsant and neuroprotective mechanisms underlying the ketogenic diet is viewed as a research imperative.
Acknowledgments
The work in the Masino laboratory was supported by National Institutes of Health grants NS29173 and NS61290, a NASA EPSCoR grant to the Connecticut Space Grant College Consortium and a Howard Hughes Medical Institute institutional grant to Trinity College. The work in the Geiger laboratory was supported by grant RR017699 from the National Center for Research Resources, a component of the National Institutes of Health.
References
- 1.Wilder RM. The effects of ketonemia on the course of epilepsy. Mayo Clin Proc. 1921;2:307–308. [Google Scholar]
- 2.Hallbook T, et al. Effects of ketogenic diet on epileptiform activity in children with therapy resistant epilepsy. Epilepsy Res. 2007;77:134–140. doi: 10.1016/j.eplepsyres.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Freeman JM, et al. The ketogenic diet: one decade later. Pediatrics. 2007;119:535–543. doi: 10.1542/peds.2006-2447. [DOI] [PubMed] [Google Scholar]
- 4.Gasior M, et al. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. 2006;17:431–439. doi: 10.1097/00008877-200609000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48:43–58. doi: 10.1111/j.1528-1167.2007.00915.x. [DOI] [PubMed] [Google Scholar]
- 6.Kim JY, Lee HW. Metabolic and hormonal disturbances in women with epilepsy on antiepileptic drug monotherapy. Epilepsia. 2007;48:1366–1370. doi: 10.1111/j.1528-1167.2007.01052.x. [DOI] [PubMed] [Google Scholar]
- 7.Greene AE, et al. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J Neurochem. 2003;86:529–537. doi: 10.1046/j.1471-4159.2003.01862.x. [DOI] [PubMed] [Google Scholar]
- 8.Hartman AL, Vining EP. Clinical aspects of the ketogenic diet. Epilepsia. 2007;48:31–42. doi: 10.1111/j.1528-1167.2007.00914.x. [DOI] [PubMed] [Google Scholar]
- 9.Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartzkroin PA. Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res. 1999;37:171–180. doi: 10.1016/s0920-1211(99)00069-8. [DOI] [PubMed] [Google Scholar]
- 11.Nakazawa M, et al. Effects of ketogenic diet on electroconvulsive threshold and brain contents of adenosine nucleotides. Brain Dev. 1983;5:375–380. doi: 10.1016/s0387-7604(83)80042-4. [DOI] [PubMed] [Google Scholar]
- 12.DeVivo DC, et al. Chronic ketosis and cerebral metabolism. Ann Neurol. 1978;3:331–337. doi: 10.1002/ana.410030410. [DOI] [PubMed] [Google Scholar]
- 13.Bough KJ, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- 14.Garriga-Canut M, et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9:1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- 15.Kato F, et al. ATP- and adenosine-mediated signaling in the central nervous system: synaptic purinoceptors: the stage for ATP to play its “dual-role. J Pharmacol Sci. 2004;94:107–111. doi: 10.1254/jphs.94.107. [DOI] [PubMed] [Google Scholar]
- 16.Klotz KN. Adenosine receptors and their ligands. N–S Arch Pharmacol. 2000;362:382–391. doi: 10.1007/s002100000315. [DOI] [PubMed] [Google Scholar]
- 17.Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Gouder N, et al. Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia. 2003;44:877–885. doi: 10.1046/j.1528-1157.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- 19.Fedele DE, et al. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- 20.Gouder N, et al. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thummler S, Dunwiddie TV. Adenosine receptor antagonists induce persistent bursting in the rat hippocampal CA3 region via an NMDA receptor-dependent mechanism. J Neurophysiol. 2000;83:1787–1795. doi: 10.1152/jn.2000.83.4.1787. [DOI] [PubMed] [Google Scholar]
- 22.Boison D. Adenosine-based cell therapy approaches for pharmacoresistant epilepsies. Neurodegener Dis. 2007;4:28–33. doi: 10.1159/000100356. [DOI] [PubMed] [Google Scholar]
- 23.Volonte C, et al. P2 receptor web: complexity and fine-tuning. Pharmacol Ther. 2006;112:264–280. doi: 10.1016/j.pharmthera.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Khakh BS, et al. ATP modulation of excitatory synapses onto interneurons. J Neurosci. 2003;23:7426–7437. doi: 10.1523/JNEUROSCI.23-19-07426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franke H, Illes P. Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol Ther. 2006;109:297–324. doi: 10.1016/j.pharmthera.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Sun XL, et al. K(ATP) channel openers facilitate glutamate uptake by GluTs in rat primary cultured astrocytes. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301501. [DOI] [PubMed] [Google Scholar]
- 27.Nistico R, et al. The blockade of K(+)-ATP channels has neuroprotective effects in an in vitro model of brain ischemia. Int Rev Neurobiol. 2007;82:383–395. doi: 10.1016/S0074-7742(07)82021-6. [DOI] [PubMed] [Google Scholar]
- 28.Dunwiddie TV, et al. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fellin T, et al. Astrocytes coordinate synaptic networks: balanced excitation and inhibition. Physiology (Bethesda) 2006;21:208–215. doi: 10.1152/physiol.00161.2005. [DOI] [PubMed] [Google Scholar]
- 30.Dunwiddie TV, Diao LH. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J Pharmacol Exp Ther. 1994;268:537–545. [PubMed] [Google Scholar]
- 31.Lloyd HGE, et al. Intracellular formation and release of adenosine from rat hippocampal slices evoked by electrical stimulation or energy depletion. Neurochem Int. 1993;23:173–185. doi: 10.1016/0197-0186(93)90095-m. [DOI] [PubMed] [Google Scholar]
- 32.Frenguelli BG, et al. Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J Neurochem. 2007;101:1400–1413. doi: 10.1111/j.1471-4159.2006.04425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- 34.Dulla CG, et al. Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron. 2005;48:1011–1023. doi: 10.1016/j.neuron.2005.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong ZQ, et al. Activity-dependent intracellular acidification correlates with the duration of seizure activity. J Neurosci. 2000;20:1290–1296. doi: 10.1523/JNEUROSCI.20-04-01290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halassa MM, et al. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Tian GF, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bains JS, Oliet SH. Glia: they make your memories stick! Trends Neurosci. 2007;30:417–424. doi: 10.1016/j.tins.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Pascual O, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 40.Oberheim NA, et al. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006;29:547–553. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Li T, et al. Adenosine kinase is a target for the prediction and prevention of epileptogenesis. J Clin Invest. 2008;118:571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters A, et al. The selfish brain: competition for energy resources. Neurosci Biobehav Rev. 2004;28:143–180. doi: 10.1016/j.neubiorev.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Oltmanns KM, et al. Differential energetic response of brain vs. skeletal muscle upon glycemic variations in healthy humans. Am J Physiol Regul Integr Comp Physiol. 2008;294:R12–R16. doi: 10.1152/ajpregu.00093.2007. [DOI] [PubMed] [Google Scholar]
- 44.Pan JW, et al. Ketosis and epilepsy: 31P spectroscopic imaging at 4.1 T. Epilepsia. 1999;40:703–707. doi: 10.1111/j.1528-1157.1999.tb00766.x. [DOI] [PubMed] [Google Scholar]
- 45.Thio LL, et al. Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology. 2000;54:325–331. doi: 10.1212/wnl.54.2.325. [DOI] [PubMed] [Google Scholar]
- 46.Musa-Veloso K, et al. Breath acetone predicts plasma ketone bodies in children with epilepsy on a ketogenic diet. Nutrition. 2006;22:1–8. doi: 10.1016/j.nut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Seymour KJ, et al. Identification of cerebral acetone by 1H-MRS in patients with epilepsy controlled by ketogenic diet. MAGMA. 1999;8:33–42. doi: 10.1007/BF02590633. [DOI] [PubMed] [Google Scholar]
- 48.Gasior M, et al. The anticonvulsant activity of acetone, the major ketone body in the ketogenic diet, is not dependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvic acid. Epilepsia. 2007;48:793–800. doi: 10.1111/j.1528-1167.2007.01026.x. [DOI] [PubMed] [Google Scholar]
- 49.Bough KJ, et al. Evidence against enhanced glutamate transport in the anticonvulsant mechanism of the ketogenic diet. Epilepsy Res. 2007;74:232–236. doi: 10.1016/j.eplepsyres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Hood VL. pH regulation of endogenous acid production in subjects with chronic ketoacidosis. Am J Physiol. 1985;249:F220–F226. doi: 10.1152/ajprenal.1985.249.2.F220. [DOI] [PubMed] [Google Scholar]
- 51.al-Mudallal AS, et al. Diet-induced ketosis does not cause cerebral acidosis. Epilepsia. 1996;37:258–261. doi: 10.1111/j.1528-1157.1996.tb00022.x. [DOI] [PubMed] [Google Scholar]
- 52.Guzman M, Blazquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12:169–173. doi: 10.1016/s1043-2760(00)00370-2. [DOI] [PubMed] [Google Scholar]
- 53.Ma W, et al. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andoh T, et al. A1 adenosine receptor-mediated modulation of neuronal ATP-sensitive K channels in rat substantia nigra. Brain Res. 2006;1124:55–61. doi: 10.1016/j.brainres.2006.09.085. [DOI] [PubMed] [Google Scholar]
- 55.Zhao YT, et al. 2-Deoxy-D-glucose-induced changes in membrane potential, input resistance, and excitatory postsynaptic potentials of CA1 hippocampal neurons. Can J Physiol Pharmacol. 1997;75:368–374. [PubMed] [Google Scholar]
- 56.Minor TR, et al. Escape deficits induced by inescapable shock and metabolic stress are reversed by adenosine receptor antagonists. Behav Brain Res. 2001;120:203–212. doi: 10.1016/s0166-4328(00)00376-4. [DOI] [PubMed] [Google Scholar]
- 57.Basheer R, et al. Adenosine and sleep-wake regulation. Prog Neurobiol. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 58.Fredholm BB. Adenosine and neuroprotection. Int Rev Neurobiol. 1997;40:259–280. [PubMed] [Google Scholar]
- 59.Hallbook T, et al. Ketogenic diet improves sleep quality in children with therapy-resistant epilepsy. Epilepsia. 2007;48:59–65. doi: 10.1111/j.1528-1167.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- 60.Prins ML, et al. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res. 2005;82:413–420. doi: 10.1002/jnr.20633. [DOI] [PubMed] [Google Scholar]
- 61.Yamada KA, et al. Ketogenic diet reduces hypoglycemia-induced neuronal death in young rats. Neurosci Lett. 2005;385:210–214. doi: 10.1016/j.neulet.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 62.Stafstrom CE. Dietary approaches to epilepsy treatment: old and new options on the menu. Epilepsy Curr. 2004;4:215–222. doi: 10.1111/j.1535-7597.2004.46001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dragunow M. Adenosine: the brain’s natural anticonvulsant? Trends Pharmacol Sci. 1986;7:128–130. [Google Scholar]
- 64.Otsuguro K, et al. Involvement of adenosine in depression of synaptic transmission during hypercapnia in isolated spinal cord of neonatal rats. J Physiol. 2006;574:835–847. doi: 10.1113/jphysiol.2006.109660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gourine AV, et al. ATP is a mediator of chemosensory transduction in the central nervous system. Nature. 2005;436:108–111. doi: 10.1038/nature03690. [DOI] [PubMed] [Google Scholar]
- 66.Newby AC. Adenosine and the concept of “retaliatory metabolites. Trends Biochem Sci. 1984;9:42–44. [Google Scholar]
- 67.Masino SA, et al. Modulation of hippocampal glutamatergic transmission by ATP is dependent on adenosine a(1) receptors. J Pharmacol Exp Ther. 2002;303:356–363. doi: 10.1124/jpet.102.036731. [DOI] [PubMed] [Google Scholar]
- 68.Hartman AL, et al. The neuropharmacology of the ketogenic diet. Pediatr Neurol. 2007;36:281–292. doi: 10.1016/j.pediatrneurol.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holloway R, et al. Effect of lactate therapy upon cognitive deficits after traumatic brain injury in the rat. Acta Neurochir (Wien) 2007;149:919–927. doi: 10.1007/s00701-007-1241-y. [DOI] [PubMed] [Google Scholar]