

Summary
Cancer pain is a debilitating disorder and a primary determinant of the poor quality of life. Here, we report a non-vascular role for ligands of the Vascular Endothelial Growth Factor (VEGF) family in cancer pain. Tumor-derived VEGF-A, PLGF-2, and VEGF-B augment pain sensitivity through selective activation of VEGF receptor 1 (VEGFR1) expressed in sensory neurons in human cancer and mouse models. Sensory-neuron-specific genetic deletion/silencing or local or systemic blockade of VEGFR1 prevented tumor-induced nerve remodeling and attenuated cancer pain in diverse mouse models in vivo. These findings identify a therapeutic potential for VEGFR1-modifying drugs in cancer pain and suggest a palliative effect for VEGF/VEGFR1-targeting anti-angiogenic tumor therapies.
Graphical Abstract
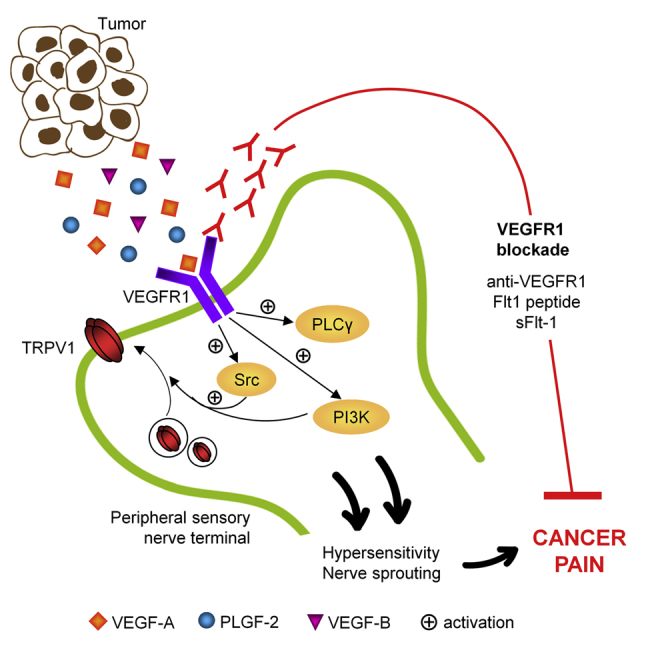
Highlights
-
•
VEGF induces nociceptive sensitization via VEGFR1 in peripheral sensory neurons
-
•
VEGFR1 is upregulated in sensory nerves in human cancer and mouse cancer models
-
•
Sensory-neuron-specific loss of VEGFR1 attenuates tumor-induced nerve remodeling
-
•
Local/systemic blockade of VEGFR1 or VEGF family ligands attenuates cancer pain
Selvaraj et al. show that ligands of the vascular endothelial growth factor (VEGF) family contribute to cancer-associated pain through selective activation of VEGF receptor 1 (VEGFR1) expressed in sensory neurons. Inhibition of VEGFR1 reduces cancer-associated pain.
Significance
Several types of cancer are associated with severe pain, which markedly reduces the quality of life in patients. Molecular mechanisms underlying cancer pain remain poorly understood. Despite close proximity between sensory nerves and blood vessels in tumor-affected tissues, neither mutual interactions nor angiogenic molecules have been studied in the context of cancer pain. Here, we report that VEGF signaling is operational in nerves as well as blood vessels at the tumor-nerve interface. Perturbing expression, activation, or signaling of VEGFR1, but not of VEGFR2, in peripheral sensory nerves disrupts attenuated cancer-induced pain and tumor-induced remodeling of nerves in mice in vivo. Thus, drugs targeting VEGFR1 as well as VEGF family ligands hold potential in treating cancer pain.
Introduction
Pain is one of the most severe and common symptoms of a variety of cancers and constitutes a primary determinant of the poor quality of life in cancer patients (Mantyh, 2013). Bone metastases and pancreatic ductal adeno carcinoma (PDAC) are among the most painful forms of cancer-associated pain. Apart from inflammatory and neuropathic components, bilateral interactions between tumor cells and nerve cells constitute the cornerstone of cancer (Cain et al., 2001; Mantyh, 2013; Schweizerhof et al., 2009). Therefore, molecular mechanisms underlying tumor-nerve interactions hold promise for understanding and treating cancer-associated pain.
Although nerves and blood vessels share physical proximity and wide set of signaling systems, regulators of vascular remodeling have not received attention in the context of tumor-associated pain so far. Vascular Endothelial Growth Factor (VEGF) family of factors-mediated signaling is not only crucial in vascular development during embryogenesis and angiogenesis (Olsson et al., 2006), but also operational in a variety of non-endothelial cells, including developing neurons.
VEGF family ligands can bind to and signal via two receptor tyrosine kinases, namely, VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2), which are highly homologous in overall structure, but have distinct biological functions in vivo (Ferrara et al., 2003). So far, VEGFR2 has been implicated in all aspects of physiological and pathological angiogenesis, e.g., in tumor angiogenesis, macular degeneration of the retina, whereas VEGFR1 has been mostly thought to act primarily as a decoy receptor that indirectly limits VEGFR2 activation (Ferrara et al., 2003; Hiratsuka et al., 1998). However, there is also evidence for VEGFR1 in transferring biological signals and enhancing the activity of VEGFR2 in vivo (Olsson et al., 2006), indicating that the relative contributions of VEGFR1 and VEGFR2 can vary in a context-dependent manner. Apart from VEGF-A, other VEGF family ligands, such as placental growth factor-2 (PLGF-2) and VEGF-B, are also released in the tumor microenvironment (Fischer et al., 2008).
This study investigates biological mechanisms and therapeutic potential of VEGF family of factors and their signaling mechanisms in cancer pain.
Results
VEGFRs Are Expressed in Peripheral Sensory Nerves and Their Cell Bodies in Dorsal Root Ganglia
As a first step toward characterizing a potential role for the VEGF signaling axis in the tumor-neuro-vascular link, we analyzed the expression of VEGFRs in mouse tissues using antibodies that have been tested for specificity in previous studies (Stefater et al., 2011) and in RNA interference experiments described below.
Confocal dual immunofluorescence analysis on sections of mouse dorsal root ganglia (DRG), where the somata of sensory nerves reside, broadly revealed immunoreactivity for anti-VEGFR1 distributed over the whole cell body of sensory neurons, whereas anti-VEGFR2 immunoreactivity was primarily localized to the nucleus (Figures 1A and S1A). All classes of sensory neurons, including peptidergic nociceptive (CGRP), non-peptidergic nociceptive (Isolectin-B4-binding), and tactile-sensitive neurons expressing Neurofilament 200 (NF200), broadly expressed VEGFR1 and VEGFR2 (Figures 1A, 1B, S1B, and S1C).
Figure 1.

Expression of VEGFR1 and VEGFR2 in Sensory Neurons and Nociceptive Sensitization by VEGF-A
(A and B) Typical examples (A) and quantitative representation (B) of large- and small-diameter sensory neurons (arrows and arrowheads, respectively) showing co-labeling of anti-VEGFR1 or anti-VEGFR2 with markers for subtypes of sensory neurons in sections of mouse DRG (n = 32 sections analyzed from five mice).
(C) Western blot analysis on mouse DRG or lysates of neuron-enriched DRG cultures.
(D) Co-immunostaining of anti-VEGFR1 or anti-VEGFR2 with a marker for peripheral nerves (PGP9.5) in mouse paw skin biopsies.
(E and F) Time course (E) and dose dependence (F) of modulation of mechanical sensitivity at the plantar surface of the hind paw following intraplantar application of VEGF-A. Shown in (F) is the integral of entire stimulus-response frequency curves (area under the curve [AUC]) to application of graded mechanical force (0.07–0.6 g) at different time points following administration of VEGF-A or saline in the hind paw (n = at least 4 mice per dose); an increase in the AUC is indicative of exaggerated pain sensitivity.
(G and H) Time course (G) and dose dependence (H) of effects of intraplantar injection of VEGF-A or vehicle on paw withdrawal latency to noxious heat (n = at least 4 mice per dose).
(I) Analysis of Evan’s blue extravasation in mouse skin explants after cutaneous injection of PBS (vehicle), histamine (1 μg), or various doses of VEGF-A (1–10 ng).
(J) Typical examples (upper traces) and summary (lower graph) of mean firing rates of mechanoreceptive nociceptors at 30 min after exposure to vehicle (PBS) or various doses of VEGF-A in the paw skin-saphenous nerve preparation (n = at least 10 fibers/dose). Negative control lacking primary antibodies and bright-field images of H&E staining showing tissue morphology in sections (not adjacent, but derived from the same animals as the immunostained sections) are shown in (A) and (D).
∗p < 0.05 as compared with basal, †p < 0.05 as compared with corresponding data point for the vehicle group, ANOVA followed by post hoc Fisher’s test. Data are presented as mean ± SEM. Scale bars represent 50 μm in (A) and (D). See also Figure S1.
Western blot analyses on lysates of mouse DRG revealed a band of the expected size for VEGFR1 at 180 kDa (Figures 1C and S1D), but not bands corresponding to soluble (short-form) VEGFR1. The unglycosylated form of VEGFR2 at 150 kDa (Huang et al., 2010) was detected in DRG lysates (Figures 1C and S1E). Expression of VEGFR1 and VEGFR2 in neuron-enriched cultured mouse DRG cultures was confirmed via western blotting as well as reverse transcriptase PCR analysis (Figures 1C and S1F). Anti-VEGFR1 immunoreactivity was also observed on sensory axons in DRG cultures (Figure S1G) as well as peripheral nerves in the mouse hind paw skin in vivo (Figure 1D).
In Vivo Modulation of Nociception by VEGF-A
To address potential function, we simulated VEGF receptor activation via unilateral intraplantar injections of VEGF-A in the hind paw of mice at doses comparable to concentrations of VEGF-A reported in the tumor microenvironment (Kut et al., 2007) and to doses of other nociceptive mediators studied previously (Schweizerhof et al., 2009). Although VEGF-A did not evoke spontaneous pain behavior, mice showed a significant hypersensitivity to graded von Frey mechanical stimuli and infrared noxious heat applied to the hind paw within 30 min of VEGF-A injection, as compared with saline-treated mice (Figures 1E–1H). VEGF-A-induced hypersensitivity was dose dependent, starting with doses as low as 1–10 pg and reaching maximal values at 100 pg, and lasted for up to 24 hr after a single injection (Figures 1F and 1H).
Direct Modulation of Sensory Nerve Function by VEGF-A
Doses of VEGF-A that elicited marked nociceptive hyperexcitability led to neither any obvious signs of vasodilation nor significant changes in neurogenic inflammation (Figure 1I), in contrast to the positive control histamine.
Furthermore, electrophysiological single-fiber recordings on an in vitro preparation of paw skin with attached saphenous nerve perfused in Ringer solution, single Aδ-nociceptive fibers exposed to VEGF-A (1 to 200 ng/ml for 30 min) demonstrated significantly higher discharge rates in response to graded mechanical stimuli than fibers exposed to the vehicle (Figure 1J), thereby demonstrating that VEGF-A directly sensitizes nociceptive axons.
VEGF-A Mediates Its Pronociceptive Effects via VEGFR1
Mechanical and thermal hyperalgesia evoked by intraplantar VEGF-A was blocked by intraplantar pretreatment with a neutralizing antibody against the extracellular domain of VEGFR1, but not by a VEGFR2-neutralizing antibody or control IgG (Figures 2A and 2B). Consistently, VEGF-A failed to induce mechanical sensitization of Aδ-nociceptors in electrophysiological recordings when pretreated with VEGFR1 antibody (Figure 2C). Furthermore, mice selectively lacking the tyrosine kinase signaling domain of VEGFR1 (VEGFR1-Tk−/−; Hiratsuka et al., 1998) did not develop mechanical and thermal sensitization upon intraplantar VEGF-A injection (Figures 2D and S2A). Thus, VEGFR1 acting via tyrosine kinase signaling, but not VEGFR2, is required for sensitization of nociceptors by VEGF-A.
Figure 2.

Delineating VEGF Family Ligands and Their Receptors in Nociceptive Sensitization
(A) Schematic representation of known ligand specificity and tools employed.
(B) Effects of intraplantar delivery of neutralizing antibodies against VEGFR1 or VEGFR2 on intraplantar VEGF-A-induced mechanical hypersensitivity (left) and thermal hyperalgesia (right) (n = 8 mice/group).
(C) Effects of a VEGFR1-neutralizing antibody on mechanoreceptive nociceptors in skin-nerve preparation (n = 8 fibers/group).
(D) Effects of intraplantar injection VEGF-A (100 pg) in VEGFR1-Tk−/− mice and WT controls (n = 8 mice/group).
(E) Effects of intraplantar neutralizing antibodies against NRP1 or NRP2 on VEGF-A-induced mechanical hypersensitivity (left) or thermal hyperalgesia (right) (n = 6 mice/group).
(F–H) Effects of intraplantar application of VEGF-E (F), VEGF-B (G), or PLGF-2 (H) on mechanical sensitivity in hind paw of mice (n = at least 4 mice per dose).
∗p < 0.05 as compared with vehicle in (D) and compared with basal in all other panels; †p < 0.05 as compared with corresponding control group; ANOVA followed by post hoc Fisher’s test. Data are presented as mean ± SEM. See also Figure S2.
Furthermore, intraplantar injection of neutralizing antibodies directed against NRP-1 or NRP-2, which function as VEGFR co-receptors in some systems (Olsson et al., 2006), did not inhibit VEGF-induced nociceptive sensitization (Figure 2E), indicating that these potential co-receptors do not contribute to VEGF-induced nociceptive sensitization.
Contributions of Other Ligands of the VEGF Family to Nociceptive Sensitization
Intraplantar application of VEGF-E, a specific agonist at VEGFR2 (Ferrara et al., 2003) (Figure 2A), at doses up to 100 ng, did not evoke any changes in mechanical or thermal sensitivity as compared with saline injection (Figures 2F and S2B). In contrast, intraplantar injection of VEGF-B or PLGF-2, which activate VEGFR1 but not VEGFR2 (Fischer et al., 2008), evoked dose-dependent mechanical and thermal hypersensitivity (Figures 2G, 2H, S2C, and S2D), with PLGF-2 being efficacious at doses as low as 1 pg. Thus, diverse peripheral VEGFR1-specific agonists can induce nociceptive hypersensitivity.
Signaling Mediators of VEGFR1 and Their Contribution to Nociceptive Hypersensitivity
VEGFRs activate diverse intracellular signaling targets (Figure 3A; Olsson et al., 2006). In neuron-enriched DRG cultures, VEGF-A induced phosphorylation of ERK1/2 (Figure 3B), indicating that VEGFRs expressed on sensory neurons are functionally active. To test mechanistic contributions, we administered diverse pharmacological inhibitors intraplantar, at doses reported to be specific in previous in vivo studies (Malik-Hall et al., 2005). Neither the vehicle (2% DMSO) nor any of the inhibitors affected nociceptive sensitivity when given in the absence of VEGF treatment (Figure S3A). Inhibition of phospholipase C gamma (PLCγ) with U71322, the nitric oxide synthase (NOS) with L-NAME, phosphotidyl inositol 3-kinase (PI3K) with LY294002, or the tyrosine kinase Src with PP2 either completely abolished or strongly attenuated mechanical and thermal hypersensitivity induced by VEGF-A given 30-min post-inhibitor treatment (Figure 3C). PP3, the inactive analog of PP2, did not affect VEGF-A-induced hypersensitivity (Figure S3B). Inhibition of MEK-dependent activation of ERK1/2 via the MEK inhibitor PD98059 significantly attenuated VEGF-A-induced mechanical hypersensitivity, but did not affect VEGF-A-induced thermal hyperalgesia (Figure 3C; p = 0.322). Finally, inhibition of the p38 MAPK pathway using SB203580 affected neither VEGF-A-induced mechanical hypersensitivity nor thermal hyperalgesia (Figure 3C; p = 0.75).
Figure 3.

Signaling Pathways Underlying Nociceptive Sensitization by VEGF-A
(A) A schematic overview of intracellular signaling mediators activated by VEGFRs (in oval symbols) and their respective pharmacological inhibitors in square boxes.
(B) Western blots showing VEGF-A-induced phosphorylation of ERK1/2 in neuron-enriched cultured DRG neurons (n = 3 independent experiments).
(C) Effects of hind-paw injection of pharmacological inhibitors on mechanical hypersensitivity (left) and thermal hypersensitivity (right) evoked by intraplantar injection of VEGF-A (1 ng). Shown are effects following single-dose intraplantar injection: L-NAME (18.5 nmoles, NOS inhibitor), U71322 (20 pmoles, PLCγ inhibitor), LY294002 (1 nmole, PI3K inhibitor), PP2 (200 pmoles, Src Kinase inhibitor), PD98059 (18.7 nmoles, MEK inhibitor), SB203580 (30 nmoles, p38 MAPK inhibitor), vehicle (1% DMSO).
(D) Experiments comparing the above data with effects of intraplantar combinations of half-maximal doses of PLCγ inhibitor and anti-VEGFR1 antibody on VEGF-A-induced mechanical hypersensitivity to 0.4 g von Frey force (upper) and thermal hyperalgesia (lower).
(E and F) Effects of VEGF-A (1 ng, intraplantar) in mice lacking Prkg1 selectively in nociceptive neurons of the DRG (SNS-Prkg1−/− mice; E) or in mice lacking Trpv1 (Trpv1−/− mice; F) (n = 6–8 mice/group).
(G) Western blots on Src phosphorylation in DRG cultures (three independent experiments).
(H) TRPV1 expression in membranes of distal branches of sciatic nerve in mice receiving intraplantar injections of VEGF-A or vehicle (n = 3 mice/group).
(I) Effects of VEGF-A (1 ng, intraplantar) in mice lacking Trpa1 (Trpa1−/− mice) (n = 8 mice/group).
∗p < 0.05 as compared with basal value, †p < 0.05 as compared with corresponding control, ANOVA followed by post hoc Fisher’s test. Data are presented as mean ± SEM. See also Figure S3.
Moreover, we observed identical modulation of PLGF-2-induced hyperalgesia with the aforementioned inhibitors as with VEGF-A-induced hyperalgesia (Figure S3C), indicating common VEGFR1-mediated mechanisms.
We then addressed whether the diverse pathways involved in VEGF-A/PLGF-2-dependent hypersensitivity all function downstream of VEGFR1 or act in parallel to VEGFR1. Doses of inhibitors that only partially inhibit VEGF-induced hypersensitivity were identified in experiments shown in Figures 3D, S3D, and S3E. We observed that half-maximal doses of the inhibitors of PLCγ, PI3K, or Src kinase, which represent the initiation points of three distinct VEGFR1-activated pathways, did not add to the pain-alleviating function of partial VEGFR1 blockade (Figures 3D, S3D, and S3E) suggesting serial, rather than parallel, relationship of these signaling cascades with VEGFR1.
Consistent with our observations with NO blockade (above), we observed that mice lacking Prkg1, a key target of the NO-cGMP pathway, specifically in peripheral nociceptors (Luo et al., 2012), did not develop mechanical and thermal hypersensitivity with VEGF-A (Figure 3E). We also found several lines of evidence linking VEGF-A signaling to modulation of TRPV1, the key final effector of thermal hyperalgesia (Basbaum et al., 2009). First, VEGF-A failed to evoke thermal hyperalgesia in mice genetically lacking Trpv1, although VEGF-A-induced mechanical hypersensitivity was fully preserved (Figure 3F). Second, Src, which is known to phosphorylate TRPV1 and increase its membrane targeting (Basbaum et al., 2009), was rapidly and significantly phosphorylated in cultured sensory neurons upon treatment with VEGF-A (Figure 3G). Third, in mice injected intraplantar with VEGF-A, the quantity of TRPV1, which could be pulled down from membranes of the distal sciatic nerve segments, was significantly increased as compared with vehicle-injected mice with a time course consistent with VEGF-A-induced thermal hyperalgesia (Figure 3H). These results indicate that VEGF promotes the trafficking and surface expression of TRPV1 in the sciatic nerve and thereby provides a mechanistic basis for thermal hyperalgesia. On the other hand, TRPA1 and P2X3 have been implicated in mechanical hypersensitivity (Brierley et al., 2009; Jarvis et al., 2002). VEGF-A-induced mechanical hypersensitivity was not affected by A317491 at the concentration reported to be efficacious in blocking P2X3 (Jarvis et al., 2002) (Figure S3F), but was abolished in mice lacking Trpa1 (Figure 3I). Thus, modulation of ion channels, such as TRPV1 and TRPA1, by VEGFR1-activated signaling pathways in peripheral neurons can account for nociceptive sensitization evoked by VEGFR1 agonists.
Altered Expression of VEGFR1 in Sensory Nerves of Mice Implanted with Osteolytic Sarcoma and Its Specific Contribution to Cancer Pain
To study tumor-associated pain, we first utilized a model based on injection of osteolytic sarcoma cells into the intermedullary space of the calcaneus bone of the mouse heel, which reflects pain caused by primary sensitization of nerves in the vicinity of the calcaneus bone and which has been reported to closely mimic tumor-induced sensory alterations in humans (Cain et al., 2001; Wacnik et al., 2001). Western blot analysis showed that VEGFR1, but not VEGFR2, was significantly upregulated in the lysates of ipsilateral L3–L4 lumbar DRGs isolated from tumor-bearing mice as compared with sham-treated animals (Figure 4A); no upregulation was observed in contralateral DRGs of tumor-bearing mice (Figure S4A). Furthermore, injection of non-tumorigenic cells, such as MEFs, in the calcaneus cavity neither led to an induction of VEGFR1 in the DRG (Figure S4B) nor elicited nociceptive hypersensitivity (Figure S4C), indicating specificity. Interestingly, TRPV1, which mediates VEGF-A-induced thermal hyperalgesia, (Figure 3F) was found to be also upregulated in DRGs of cancer-bearing mice (Figure S4D).
Figure 4.

Role of VEGFR1 in Cancer Pain following Osteolytic Sarcoma Cell Implantation in the Calcaneus Bone
(A) Western blot analyses for VEGFR1 or VEGFR2 expression in ipsilateral L3–L4 DRG of tumor-bearing mice or sham-treated mice and their quantitative densitometric analysis (n = 3 independent experiments; ∗p = 0.02; Student’s t test).
(B) Typical examples and quantitative summary of VEGFR1 expression in PGP9.5-positive peripheral nerves overlying bone metastases in tumor-affected or sham hind paw (n = 6 mice/group). Negative controls for immunostaining and H&E-stained sections from same animals (not adjacent sections) are included to judge morphology. epi, epidermis; der, dermis.
(C and D) Whole-mount images (C) or cryosection (D) of a DRG 3 weeks after injection of lentivirions expressing EGFP and shRNA.
(E) Western blot analysis of VEGFR1 expression in L3–L4 DRGs injected ipsilaterally (ipsi) with lentivirions, using contralateral DRGs as control.
(F–H) Tumor-induced mechanical hypersensitivity (F) and tumor-induced hypertrophy and sprouting of epidermal sensory nerves expressing the marker protein PGP9.5 (G and H) in mice injected with lenti-VEGFR1-shRNA as compared with lenti-non targeting shRNA in the DRG (n = 5 mice/group).
∗p < 0.05 as compared with sham in (B), (H) and compared with basal in (F); †p < 0.05 as compared with lenti-non-targeting control; ANOVA followed by post hoc Fisher’s test. Scale bars represent 50 μm in (B), (D), and (G) and 250 μm in (C). Data are presented as mean ± SEM. See also Figure S4.
Furthermore, we observed that immunoreactivity for anti-VEGFR1 was significantly increased in paw sections derived from tumor-bearing mice as compared with sham-treated mice, including PGP9.5-positive peripheral nerves (Figure 4B).
A key functional contribution of VEGFR1 expressed in DRG neurons to pain was observed in experiments involving specific lentiviral knockdown of VEGFR1 expression in L3–L4 DRGs (Figure 4C; infection rate, 76% ± 5.3% of all DRG neurons shown in Figure 4D; lack of infection of blood vessels in the DRG confirmed via co-staining with anti-CD31 in Figure S4E). The efficacy of VEGFR1 downregulation in ipsilateral L3–L4 DRGs was ascertained by western blotting (Figure 4E) and behavioral experiments showing a marked reduction in VEGF-A-induced mechanical and thermal hypersensitivity (Figure S4F). Importantly, as compared with mice expressing non-targeting shRNA, development of tumor-induced mechanical hypersensitivity was significantly and markedly attenuated in mice expressing VEGFR1-shRNA (Figure 4F). Furthermore, tumor-induced structural changes of nerves in the paw skin overlaying the growing tumor, such as hypertrophy and epidermal sprouting (Cain et al., 2001; Schweizerhof et al., 2009), were evident in control shRNA-expressing mice, but not in VEGFR1-shRNA-expressing mice (Figures 4G and 4H). These observed differences between tumor pain and nerve remodeling did not result from any differences in tumor growth (Figure S4G).
Evidence from Genetic Models and an Independent Cancer Pain Model
To further obtain evidence from genetic models and to specifically delineate the contribution of VEGFR1 expressed in nociceptive neurons of the DRG, we generated a line of transgenic mice conditionally lacking VEGFR1 in nociceptive neurons by mating Vegfr1fl/fl mice (Ambati et al., 2006) with SNS-Cre mice (Agarwal et al., 2004) (SNS-Vegfr1−/− mice). Western blot analysis (Figure 5A) confirmed a decrease in DRG expression of VEGFR1 (55% ± 2.8% of the value in Vegfr1fl/fl mice). Intraplantar VEGF-A-induced mechanical and thermal hypersensitivity was nearly entirely lost in SNS-Vegfr1−/− mice as compared with their WT littermates, indicating a requirement for VEGFR1 expressed in peripheral nociceptors (Figures 5B and 5C).
Figure 5.

Role of VEGFR1 in Cancer Pain following Hind Paw Implantation of C57BL6-Isogenic Lewis Lung Carcinoma Cells
(A) Western blot analysis on DRGs from SNS-Vegfr1−/− and Vegfr1fl/fl (control) mice.
(B–G) Analysis of SNS-Vegfr1−/− and Vegfr1fl/fl mice (n = 6–8 mice/group) with respect to intraplantar VEGF-A-induced mechanical (B) and thermal hypersensitivity (C), tumor-associated hypersensitivity (D), tumor-induced hypertrophy and sprouting of PGP-9.5-immunoreactive sensory nerves (E and F), and tumor growth (G), including negative control for immunostaining and an H&E-stained section.
(H and I) Tumor-induced mechanical hypersensitivity (H) and tumor growth (I) in VEGFR1-Tk−/− and WT mice.
∗p < 0.05 as compared with sham in (F) and compared with basal in other panels, ANOVA followed post hoc Fisher’s test; in (D) and (H), ANOVA for repeated-measures was performed; †p < 0.05 as compared with corresponding control group, ANOVA for random measures followed by post hoc Fisher’s test. Scale bar represents 50 μm in (E). Data are presented as mean ± SEM.
The C57Bl6 genetic background of these genetically modified mice necessitated employing a model of cancer pain involving isogenic tumor cells. We therefore employed a previously described model involving subcutaneous injection of isogenic LL2 lung carcinoma cells, which results in behavioral, structural, and functional changes similar to the above-described calcaneus model (Constantin et al., 2008). LL2 carcinoma-induced mechanical hypersensitivity (Figure 5D) and tumor-induced remodeling of neighboring PGP9.5-positive nerves (Figures 5E and 5F) were significantly decreased in SNS-Vegfr1−/− mice as compared with Vegfr1fl/fl mice, without any observable difference in tumor growth across genotypes (Figure 5G). Moreover, tumor-bearing VEGFR1-Tk−/− mice showed a significantly attenuated hypersensitivity as compared with tumor-bearing WT littermates without showing changes in tumor growth (Figures 5H and 5I).
VEGFR1 Expression in Sensory Nerves in Human Cancer with Pain and Its Contribution to Pain in a Mouse Model of Pancreatic Ductal Adenocarcinoma
Given the abundance of VEGF-A and PLGF-2 in the tumor milieu of human pancreatic carcinoma (Chang et al., 2008), we analyzed pancreatic biopsies derived from human pancreatic ductal adenocarcinoma (PDAC) patients and observed anti-VEGFR1 immunoreactivity not only in blood vessels, but also in 66.3% ± 6.5% of PGP9.5-positive peripheral nerves (Figures 6A and S5A). Furthermore, anti-VEGFR1 immunoreactivity was observed in more than 70% of each of the following fiber types analyzed from 21 sections derived from 13 PDAC biopsies: CGRP-positive nociceptive nerves, NF200-positive large-diameter (tactile) sensory nerves, TH-expressing sympathetic nerves, and a subpopulation of tactile sensory fibers and ChAT-expressing parasympathetic nerves (Figures 6B and S5B).
Figure 6.

Role of VEGFR1 in Pain Associated with PDAC
(A and B) Examples (A) and quantitative analyses (B) of nerves (arrows) showing anti-VEGFR1 immunoreactivity in PDAC biopsies from patients. Arrowheads indicate blood vessels. n = 21 sections analyzed from 13 biopsies.
(C and D) Typical examples (C) and quantitative summary (D) of VEGFR1 expression in PGP9.5-positive nerves in human PDAC and pancreas of healthy donors (n = 95 sections from 30 patients and 7 donors).
(E and F) Typical examples (E) and quantitative summary (F) of the relation between VEGFR1 immunoreactivity in human PDAC biopsies and subjective pain rating reported by the patients (n = 79 sections from 30 patients).
(G) Western blot analysis of VEGFR1 expression in DRGs of mice with Advillin-Cre-mediated pan-DRG VEGFR1 deletion (Adv-Vegfr1−/−) and controls (Vegfr1fl/fl).
(H) VEGF-A-induced mechanical hypersensitivity (left) and thermal hyperalgesia (right) in Adv-Vegfr1−/− and Vegfr1fl/fl mice (n = at least 4 mice/group).
(I and J) Comparison of early post-operative pain (red arrow), tumor-associated hypersensitivity (blue arrow) to 0.008 g of abdominal von Frey application (I) and integral of responses to all von Frey forces tested (right) and tumor mass (J) between Adv-Vegfr1−/− mice, SNS-Vegfr1−/− mice, and Vegfr1fl/fl mice (n = at least 8 mice/group).
∗p < 0.05, t test in (D) and (F). ∗p < 0.05 as compared with basal in (H) and (I); ANOVA for repeated-measures followed post hoc Fisher’s test; †p < 0.05 as compared with corresponding control group, ANOVA for random measures followed by post hoc Fisher’s test. Scale bar represents 100 μm in (A) and (C) and 50 μm in (E). Data are presented as mean ± SEM. See also Figure S5.
Importantly, nerve fibers showed a significant increase in the intensity as well as the area of anti-VEGFR1 immunoreactivity in biopsies of PDAC patients as compared with healthy donors (Figures 6C and 6D), which was significantly proportional to perceived pain intensity in PDAC patients (severe, moderate, or mild) based upon subjective pain ratings (Figures 6E and 6F).
We then undertook functional experiments to address the role of VEGFR1 in PDAC-related pain. First, because VEGFR1 was observed to be broadly expressed in the DRG, including non-nociceptive and nociceptive neurons, we generated transgenic mice conditionally lacking VEGFR1 specifically in all neurons of the DRG (Adv-Vegfr1−/− mice) by using Advillin-Cre mice (Zurborg et al., 2011). Adv-Vegfr1−/− mice were studied in western blot analysis (Figure 6G; 69% ± 3.5% loss over control mice) and showed a complete loss of mechanical and thermal sensitization with intraplantar VEGF-A (Figure 6H). Second, we established behavioral analysis of pain associated with PDAC by employing a model based on orthotopic implantation of mouse PanCO2 PDAC tumor cells or vehicle (sham) in the pancreas of C57Bl6 mice (Zhu et al., 2008). Upon testing sensitivity to abdominal application of low force-von Frey hairs, we observed that post-operative pain was comparable across PanCO2-injected Adv-Vegfr1−/− mice and control littermates (also evident in sham-injected mice; data not shown), but tumor-induced mechanical hypersensitivity starting from day 13 (specific to PanCO2-injected mice) was significantly reduced in Adv-Vegfr1−/− mice as compared with control littermates (Figure 6I). Importantly, SNS-Vegfr1−/− mice demonstrated a similar degree of attenuation of tumor-induced nociceptive hypersensitivity as Adv-Vegfr1−/− mice (Figure 6I), indicating that VEGFR1 expressed on nociceptive neurons can largely account for the key role observed for VEGFR1 to tumor pain. Pancreatic tumor size was not significantly different across genotypes (Figure 6J).
Availability of Diverse VEGFR1 Ligands in the Context of Cancer Pain and Their Functional Contributions
ELISA-based analysis demonstrated a high level of availability of VEGF-A as well as PLGF-2 in paw tissue surrounding the tumor, including sensitized skin, in the calcaneus osteolytic sarcoma implantation model (Figure 7A). Injection of specific function-blocking antibodies directed against VEGF-A or PLGF-2 in the vicinity of the tumor led to a strong reduction in tumor-associated mechanical hypersensitivity (Figure 7B). Anti-VEGF-A, but not anti-PLGF-2, significantly reduced tumor growth at the doses tested (Figure 7B). Moreover, an antibody neutralizing VEGF-B significantly reduced tumor-induced hypersensitivity, but to a lesser degree than anti-VEGF-A or anti-PLGF-2 (Figure S6A), without affecting tumor growth (Figure S6B). Thus, diverse VEGFR1 ligands are locally available in the tumor milieu and contribute collectively to tumor-induced hypersensitivity.
Figure 7.

Role of Diverse VEGFR1 Ligands and Sequestering Agents in Cancer Pain in the Calcaneus Osteolytic Sarcoma Implantation Model
(A) ELISA-based analysis of VEGF-A and PLGF-2 in hind paw (n = 4 mice each).
(B) Effects of intraplantar application of VEGF-A-sequestering antibody (upper), PLGF-2-sequestering antibody (lower), or control IgGs on tumor-induced mechanical hypersensitivity and tumor growth (n = at least 6 mice/group).
(C) Effects of intraplantar application of soluble-VEGFR1 (sFlt1) or control protein on tumor-induced mechanical hypersensitivity and tumor size (n = 5–6 mice/group).
(D) Typical example (left) and densitometric quantification (right) of tyrosine phosphorylation of VEGFR1 in mouse hind paw in sham or tumor-bearing mice and its modulation by local application of anti-Flt1 or reverse peptide 8 days post-tumor implantation (n = 3 mice/group).
(E) Effects of intraplantar application of anti-Flt1 or reverse peptide on tumor-induced mechanical hypersensitivity and tumor growth (n = 6–8 mice).
∗p < 0.05, ANOVA followed post hoc Fisher’s test as compared with basal, ANOVA of repeated-measures performed in (B), (C), and (E); †p < 0.05 as compared with corresponding control group and #p < 0.01 as compared with reverse peptide in (D), ANOVA for random measures followed by post hoc Fisher’s test. Data are presented as mean ± SEM. See also Figure S6.
We further employed two biological tools to bind and sequester VEGFR1 ligands and/or prevent them from binding to VEGFR1. First, in comparison to injection of a control Fc protein, local injection of soluble form of VEGFR1 lacking the C terminus and the kinase domain (sFlt1) (Ferrara et al., 2003) blocked VEGF-A-induced hyperalgesia (Figures S6C and S6D), significantly attenuated tumor-associated hypersensitivity (Figure 7C), and reduced tumor growth to a small but significant extent (Figure 7C). Second, we employed a small anti-VEGFR1 hexapeptide (GNQWFI, anti-Flt1 peptide), which prevents binding of all ligands to the extracellular domain of VEGFR1 (Bae et al., 2005). However, because this peptide has only been used in a few in vivo studies so far (Bae et al., 2005), we validated in vivo efficacy by testing impact on VEGFR1 activation judged via tyrosine phosphorylation on VEGFR1 on position 1213, which was significantly enhanced in tumor-bearing paw compared with sham-treated paw (Figure 7D). Upon administration of a single intraplantar injection of the anti-Flt1 peptide at a dose of 25 μg, tumor-associated tyrosine phosphorylation of VEGFR1 was significantly attenuated for 24 hr (Figure 7D). This dose of the anti-Flt1 peptide blocked intraplantar VEGF-A-induced nociceptive hypersensitivity (Figures S6E and S6F) and also significantly attenuated tumor-associated mechanical hypersensitivity when given over multiple time points (Figure 7E). These differences did not arise from differences in tumor growth between groups (Figure 7E). Furthermore, infiltration of GR-1-positive neutrophils in the tumor vicinity was not significantly different across the above groups (Figure S6G).
Effects of Local Immunological Blockade of VEGFR1 or VEGFR2 Signaling on Cancer Pain and Nerve Remodeling
Given their growing therapeutic potential in cancer treatment, we tested immunologicals for efficacy in repressing VEGFR1-mediated nociceptive hypersensitivity and tumor-associated pain as well as the accompanying nerve remodeling.
First, in the calcaneus osteolytic sarcoma implantation model, local administration of anti-VEGFR1, but not anti-VEGFR2 or control IgG, in the vicinity of the tumor on days 1, 3, 5, 7, 9, 11, and 13 post-tumor cell implantation significantly blocked tumor-induced mechanical hypersensitivity (Figure 8A) and significantly attenuated tumor-associated sprouting and hypertrophy of PGP9.5-positive nerve fibers in the epidermis (Figure 8B). No differences were observed with respect to local tumor growth or local inflammation with anti-VEGFR1 at the dose applied (Figures S7A–S7C). Interestingly, however, in tumor-bearing mice, locally administered anti-VEGFR2 suppressed tumor angiogenesis to a much stronger extent than anti-VEGFR1, as compared with mice that received control IgG (Figure S7D), indicating that VEGFR1 and VEGFR2 mediate differential aspects of remodeling in the tumor-nerve-blood vessel milieu.
Figure 8.

Effects of Local or Systemic Immunological Blockade of VEGFR1 or VEGFR2 on Cancer Pain
(A and B) Effects of a regime of local, intraplantar injections of either IgG or anti-VEGFR1 or anti-VEGFR2 antibodies (5 μg/dose) on tumor-induced mechanical hypersensitivity (A) and tumor-induced hypertrophy and sprouting of PGP9.5-immunoreactive epidermal sensory nerves (B) in the calcaneus osteolytic sarcoma implantation model (n = at least 6 mice/group).
(C–E) Analysis of tumor-induced mechanical hypersensitivity in the ipsilateral mouse paw (C), spontaneous pain (D), and sprouting of the femur periosteal peptidergic nociceptive fibers (E) induced by implantation of mammary cancer cells in the mouse femur bone in mice receiving a regime of systemic injections of either control IgG or the VEGFR1-neutralizing MF-1 clone or the VEGFR2-neutralizing DC101 clone (40 mg/kg body weight per injection, n = 8 mice/group).
In all panels, ∗p < 0.05 as compared with basal; ANOVA of repeated-measures, †p < 0.05 as compared with control IgG, #p < 0.01 as compared with sham; only in (D), p < 0.05 between MF-1 and DC101 groups is indicated by $ sign; ANOVA followed by post hoc Fisher’s test. Scale bars represent 50 μm and 25 μm in (B) and (E), respectively. Data are presented as mean ± SEM. See also Figure S7 and Movies S1, S2, S3, and S4.
Efficacy of Systemically Administered Anti-VEGFR Immunologicals in a Model of Cancer-Induced Bone Pain as a Potential Therapeutic Strategy
Finally, we endeavored to test the efficacy of systemically administered immunologicals using a clinically relevant model that simulates pain induced by metastases in large skeletal bones in cancer patients (Bloom et al., 2011; Mantyh, 2013). Upon unilateral implantation of mammary carcinoma cells in the intermedullary cavity of mouse femur bone, mice not only show nociceptive hypersensitivity in the ipsilateral hind paw (secondary hyperalgesia), but also signs of spontaneous ongoing pain, such as lifting or flicking of the affected limb and altered gait (Bloom et al., 2011; Mantyh, 2013; Movies S1, S2, S3, and S4). High-quality function-blocking antibodies against VEGFR1 (MF-1) or VEGFR2 (DC101), which have been validated in vivo previously upon intraperitoneal delivery in mice (Carmeliet et al., 2001; Fischer et al., 2007), were administered at every 3 days until day 29 post-tumor cell implantation.
Systemically delivered MF-1 strongly attenuated the development of nociceptive hypersensitivity to von Frey force applied to the ipsilateral hind paw (Figure 8C) as compared with control IgG or DC101 application. Similar results were observed with respect to tumor-induced spontaneous nocifensive pain behavior (Figure 8D; Movies S1, S2, S3, and S4). At late stages only (30-days post-tumor implantation), a partial suppression of nociceptive hypersensitivity or spontaneous pain behavior was observed with the systemically delivered DC101.
Furthermore, mice receiving systemic treatment with the MF1, but not DC101 or control IgG, showed a marked reduction in bone metastases-induced sprouting of periosteal nerves (Figure 8E). Both clones significantly reduced the growth rate of mammary carcinoma cells in mice upon systemic delivery (Figure S7E); however, DC101 suppressed tumor growth to a higher extent than MF1, indicating again a mechanistic dichotomy of VEGFR2 and VEGFR1 in the modulation of tumor growth and nociceptive sensitization in cancer pain.
Discussion
VEGF signaling has already gained major significance in the pathophysiology and therapy of cancer; this study extends its functional and therapeutic repertoire beyond tumor angiogenesis and metastases to the avenue of cancer pain by acting at the interface between tumor cells, blood vessels, and nerves to regulate pain sensitivity via functional and structural modulation. We present several converging lines of evidence showing that peripherally acting VEGF ligands induce nociceptive sensitization via direct effects on peripheral nerves, rather than non-neuronal modulation, and that the contribution of nociceptors is paramount.
Our analyses indicate that VEGFR1 activation in sensory neurons triggers both functional and structural remodeling. VEGFR1 activated diverse kinases, such as PLCγ, PI3K, and Src kinase, that sensitize transducers of heat, pressure, and chemical stimuli in nociceptive terminals, such as TRPV1 and TRPA1, as well as amplifiers of afferent excitability, such as Nav1.8 (Basbaum et al., 2009; Hucho and Levine, 2007). Our observation that VEGFR1 modulates TRPV1 function via Src and promotes its trafficking in peripheral nerves is interesting because TRPV1 senses local acidosis and algogens, such as eicosanoids and lipids, which are abundant in the tumor environment (Mantyh, 2013).
Our findings on the role of VEGFR1 in cancer-related structural remodeling of nerves and its upregulation in sensory nerves within the tumor milieu in mouse models and PDAC patients indicates a strong link between VEGF signaling in sensory nerves, structural remodeling, and pain; however, a causal relationship between nerve sprouting and pain in cancer cannot yet be derived. Of note, VEGF-mediated signaling converges with that of NGF, another major player in nerve remodeling (Mantyh et al., 2011), on common intracellular effectors, e.g., the PI3K-Akt pathway.
Remarkably, our results suggest a functional dichotomy of the two main lines of VEGF signaling in sensory nerves and blood vessels. Although VEGFR2 regulates tumor angiogenesis (Ferrara et al., 2003), it does not contribute to sensitization of nerves: this may be accounted for by the observed nuclear localization for VEGFR2 in sensory neurons, the higher level of distribution of VEGFR1 at the cell surface of sensory neurons and the 10-fold higher affinity of VEGF-A for VEGFR1 over VEGFR2.
The efficacy of targeting PLGF-2 in cancer therapy is a much debated topic of acute interest and high relevance in the cancer field (e.g., Bais et al., 2010; Van de Veire et al., 2010). Our observation that PLGF-2 is even more potent and efficacious than VEGF-A in inducing nociceptive hypersensitivity may stem from proposed differences in the VEGFR1 binding/activation mode between these agonists (Carmeliet et al., 2001). Furthermore, on the background of reports on synergistic activation of VEGFR1 by PLGF-2 and VEGF-A in angiogenesis and plasma extravasation (Carmeliet et al., 2001), our results suggest that a co-release of these factors in the tumor microenvironment may cooperate in evoking exaggerated pain sensitivity.
Administration of Bevacizumab, a humanized monoclonal immunological sequestering VEGF-A, enhances the quality of life in cancer patients (Garassino et al., 2007; Johnson et al., 2004), which would be generally consistent with this study since pain is an important parameter for quality of life in cancer patients. Our results therefore suggest that pain should be directly and objectively tested as a clinical end point in clinical studies involving VEGF-targeted therapies in cancer patients. Furthermore, given our results showing roles for VEGF-A, PLGF-2, and VEGF-B in bone metastatic pain, we suggest that targeting individual ligands may be less efficacious than targeting the receptor (VEGFR1) they converge upon. Moreover, our results on the efficacy of locally applied small anti-Flt1 peptide in tumor pain models provide scope for limiting side effects attributed to immunologicals and global intervention in VEGF signaling.
Taken together, our findings from several animal models of cancer pain, human cancer material, and mechanistic analyses indicate therapeutic potential for VEGFR1-modifying drugs in cancer pain and suggest that combined targeting of VEGFR1 and VEGFR2 may enable modulation of distinct aspects of cancer at the tumor-neuro-vascular interface.
Experimental Procedures
Sensory-Neuron-Specific Knockout Mice
Mice lacking VEGFR1 in a sensory neuron-specific manner were generated by mating mice carrying floxed alleles of the Flt1 (vegfr1) gene (Ambati et al., 2006) with mice expressing the Cre recombinase selectively in nociceptors (SNS-Cre; Agarwal et al., 2004) or with mice expressing the Cre recombinase selectively in all neurons of the DRG (Advillin-Cre; Zurborg et al., 2011).
Antibodies Used for Immunohistochemistry and Western Blotting
The following are used for immunohistochemistry on mouse paw skin: PGP9.5 (1:1000, Ultraclone), and on human pancreatic biopsies: (1) anti-PGP9.5 (1:1000; Ultraclone), (2) anti-VEGFR1 (1:200, #AF321, R&D Systems), (3) anti-VEGFR2 (1:200; MAB3571; R&D Systems), (4) anti-choline acetyl transferase (1:400; Neuromics), (5) anti-tyrosine hydroxylase (#22941; Immunostar), (6) anti-CGRP (1:300; 24112; Immunostar), (7) anti-neurofilament-200 antibody (1:500, #N4142; Sigma Aldrich), and (8) anti-CD31 (clone JC70A, #M0823; DAKO Deutschland GmBH).
The following are used for immunofluorescence analysis on mouse DRGs: anti-VEGFR1 (1:100; MAB471; R&D Systems); anti-VEGFR2 (1:100; sc-505; Santa Cruz Biotechnology SCBT), anti-CGRP (1:300; 24112; Immunostar) and biotinylated IB4 (1:200; B-1205; Vector), anti-neurofilament-200 antibody (1:500, #N2912, #N4142; Sigma Aldrich), anti-CD31 (1:500, #550274; BD PharMingen) (see Supplemental Experimental Procedures); western blotting: anti-VEGFR1 (1:1000; #1301-1; Epitomics), anti-VEGFR2 (1:1000; sc-505; SCBT), a phospho-VEGFR1 (Tyr1213) antibody (07-758; Merck Millipore), anti-Src and anti-phospho-Src antibodies (1:1000; #2208; #2101; Cell Signaling Technologies), anti-TRPV1 (1:2500; sc-12498; SCBT), and anti-alpha-tubulin (1:2500, T9026; Sigma Aldrich).
Four Independent Mouse Models of Cancer-Associated Pain
All animal experiments were approved by the local governing board on animal protection laws (Regierungspräsidium) and were performed in accordance with their regulatory standards. All behavioral measurements were done in awake, unrestrained, age-matched adult (3 months old) male mice.
-
(1)
Osteolytic sarcoma model of cancer pain in the calcaneus bone: osteolytic sarcoma cells NCTC clone 2472 (National Collection of Type Cultures clone 2472; ATCC) (2 × 106 per injection) were unilaterally injected into and around the calcaneus bone of C3H/HeNCrl mice (Wacnik et al., 2001). Blocking antibodies, soluble VEGFR1 (sFlt1), anti-Flt1 peptide, or their respective controls were injected ipsilaterally intraplantar in the vicinity of the calcaneus bone. Antibodies against VEGFR1 (AF471; R&D Systems), VEGFR2 (AF644; R&D Systems), PLGF-2 (MAB465; R&D Systems), VEGF-B (AF590; R&D Systems), VEGF-A (AF-493-NA; R&D Systems) or normal goat IgG or rat IgG (AB-108-C, MAB006; R&D Systems) (each 5 μg diluted in 25 μl) were applied on days 1, 3, 5, 7, 9, 11, and 13 post-tumor implantation. An anti-Flt1 peptide, GNQWFI (25 μg diluted in 25 μl, RB-PP-0245; Ray Biotech), reverse peptide sequence, IFWQNG (25 μg diluted in 25 μl; Ray Biotech), soluble VEGFR1 (sFlt-1, SFC-M06; 10 μg diluted in 25 μl; Reliatech), or mouse IgG-Fc fragment (10 μg diluted in 25 μl, 4460-MG; R&D Systems) were administered every other day from days 1 to 13 post-tumor implantation.
-
(2)
Femur model of cancer-induced bone pain: Cells of the murine osteolytic breast carcinoma cell line, 4T1-Luc, which express the luciferase reporter gene (Srivastava et al., 2014) or PBS (sham) were in mice of the Balb/c strain directly into the intermedullary space of the mouse femur following an arthrotomy (1.5 × 105 cells/injection), as described previously (Bloom et al., 2011). Different cohorts of mice treated with MF-1 (ImClone Systems), DC101 (ImClone Systems), or control IgG (DivBioScience) were injected systemically (each 40 mg/kg body weight) every third day over 4 weeks post-tumor implantation (Carmeliet et al., 2001; Fischer et al., 2007).
-
(3)
In gene-targeted mice, we injected lung carcinoma cells of the ETCC clone 1642 (European Collection of Cell Cultures), isogenic with the C57Bl6 mouse strain, subcutaneously in the plantar and dorsal side of the mouse hind paw (7 × 105 cells/injection) (Constantin et al., 2008).
-
(4)
Orthotopic injection model of PDAC: Mouse pancreatic carcinoma cells (PanCO2, 5000 cells resuspended in 5 μl) or PBS (sham) were injected into the pancreas of deeply anesthetized C57Bl6 mice (Zhu et al., 2008), keeping the abdominal surgical cut to a minimum. Abdominal sensitivity to graded von Frey stimuli was assessed in awake, freely moving mice, and withdrawal behavior was analyzed.
In all models, all behavioral analyses were performed in a blinded manner; i.e., the investigator was unaware of the experimental condition tested.
Ethics Statement for Human Tissues
The use of archived pancreatic tissues was approved by the institutional review board, and written informed consent was obtained from the patients prior to the surgical procedure (ethics committee, University of Heidelberg; #301/2001).
Additional details on experimental procedures are given in Supplemental Information.
Author Contributions
D.S. designed and performed the majority of experiments and analyzed data. V.G. performed several experiments and analyzed data. C.W.M, M.K., S.S., K.S., and M.S. performed and helped with some experiments. J.W., N.F., P.H., and M.S. provided the various mouse lines. H.G.A. provided important tools and conceptual input on the manuscript. R.K. conceived, designed, and supervised the study. The paper was written by D.S. and R.K.
Acknowledgments
The authors thank Rose LeFaucheur for secretarial help and Hans-Joseph Wrede, Karin Meyer, and Dunja Baumgartner-Ahlert for technical assistance. We acknowledge support from the Interdisciplinary Neurobehavioral Core for the behavioral experiments performed here. This work was supported by grants from the Sander Stiftung to R.K., the Association of International Cancer Research to R.K., an ERC Advanced Investigator Grant (294293) from the European research Council to R. K., and the Deutsche Forschungsgemeinschaft SFB-TR23, A3, to H.G.A. R.K. is principal investigator in the Excellence Cluster “CellNetworks” of Heidelberg University and the Molecular Medicine Partnership Unit, Heidelberg. D.S. was supported by a Fellowship from the Hartmut Hoffmann-Berling International Graduate School for Molecular and Cellular Biology.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
This depicts the nature and extent of nocifensive behavior and weight bearing in mice 30 days after sham surgery.
{kind=link}
This depicts the nature and extent of nocifensive behavior, such as guarding of the tumor-bearing limb and lifting of the affected paw above the wire-mesh bottom, reduction in weight-bearing of the affected paw, sporadic hopping behavior during ambulation in tumor-bearing mice 30 days after implantation of breast cancer cells unilaterally in the femur bone cavity; in addition, the mouse shown in the video received intraperitoneal injections of control IgG.
{kind=link}
This depicts the nature and extent of nocifensive behavior in tumor-bearing mice 30 days after implantation of breast cancer cells unilaterally in the femur bone cavity; in addition, the mouse received intraperitoneal injections of an anti-VEGFR1 antibody (MF1 clone).
{kind=link}
This depicts the nature and extent of nocifensive behavior in tumor-bearing mice 30 days after implantation of breast cancer cells unilaterally in the femur bone cavity; in addition, the mouse received intraperitoneal injections of an anti-VEGFR2 antibody (DC101 clone).
{kind=link}
References
- Agarwal N., Offermanns S., Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis. 2004;38:122–129. doi: 10.1002/gene.20010. [DOI] [PubMed] [Google Scholar]
- Ambati B.K., Nozaki M., Singh N., Takeda A., Jani P.D., Suthar T., Albuquerque R.J., Richter E., Sakurai E., Newcomb M.T. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae D.G., Kim T.D., Li G., Yoon W.H., Chae C.B. Anti-flt1 peptide, a vascular endothelial growth factor receptor 1-specific hexapeptide, inhibits tumor growth and metastasis. Clin. Cancer Res. 2005;11:2651–2661. doi: 10.1158/1078-0432.CCR-04-1564. [DOI] [PubMed] [Google Scholar]
- Bais C., Wu X., Yao J., Yang S., Crawford Y., McCutcheon K., Tan C., Kolumam G., Vernes J.M., Eastham-Anderson J. PlGF blockade does not inhibit angiogenesis during primary tumor growth. Cell. 2010;141:166–177. doi: 10.1016/j.cell.2010.01.033. [DOI] [PubMed] [Google Scholar]
- Basbaum A.I., Bautista D.M., Scherrer G., Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom A.P., Jimenez-Andrade J.M., Taylor R.N., Castañeda-Corral G., Kaczmarska M.J., Freeman K.T., Coughlin K.A., Ghilardi J.R., Kuskowski M.A., Mantyh P.W. Breast cancer-induced bone remodeling, skeletal pain, and sprouting of sensory nerve fibers. J. Pain. 2011;12:698–711. doi: 10.1016/j.jpain.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley S.M., Hughes P.A., Page A.J., Kwan K.Y., Martin C.M., O’Donnell T.A., Cooper N.J., Harrington A.M., Adam B., Liebregts T. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology. 2009;137:2084–2095.e3. doi: 10.1053/j.gastro.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain D.M., Wacnik P.W., Turner M., Wendelschafer-Crabb G., Kennedy W.R., Wilcox G.L., Simone D.A. Functional interactions between tumor and peripheral nerve: changes in excitability and morphology of primary afferent fibers in a murine model of cancer pain. J. Neurosci. 2001;21:9367–9376. doi: 10.1523/JNEUROSCI.21-23-09367.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Moons L., Luttun A., Vincenti V., Compernolle V., De Mol M., Wu Y., Bono F., Devy L., Beck H. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001;7:575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- Chang Y.T., Chang M.C., Wei S.C., Tien Y.W., Hsu C., Liang P.C., Tsao P.N., Jan I.S., Wong J.M. Serum vascular endothelial growth factor/soluble vascular endothelial growth factor receptor 1 ratio is an independent prognostic marker in pancreatic cancer. Pancreas. 2008;37:145–150. doi: 10.1097/MPA.0b013e318164548a. [DOI] [PubMed] [Google Scholar]
- Constantin C.E., Mair N., Sailer C.A., Andratsch M., Xu Z.Z., Blumer M.J., Scherbakov N., Davis J.B., Bluethmann H., Ji R.R., Kress M. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J. Neurosci. 2008;28:5072–5081. doi: 10.1523/JNEUROSCI.4476-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N., Gerber H.P., LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Fischer C., Jonckx B., Mazzone M., Zacchigna S., Loges S., Pattarini L., Chorianopoulos E., Liesenborghs L., Koch M., De Mol M. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Fischer C., Mazzone M., Jonckx B., Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat. Rev. Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- Garassino M.C., Hollander L., Torri V. Bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2007;356:1373–1375. doi: 10.1056/NEJMc070042. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S., Minowa O., Kuno J., Noda T., Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA. 1998;95:9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Tan Y., Tang Q., Liu X., Guan X., Feng Z., Zhu J. A high-affinity human/mouse cross-reactive monoclonal antibody, specific for VEGFR-2 linear and conformational epitopes. Cytotechnology. 2010;62:61–71. doi: 10.1007/s10616-010-9262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T., Levine J.D. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Jarvis M.F., Burgard E.C., McGaraughty S., Honore P., Lynch K., Brennan T.J., Subieta A., Van Biesen T., Cartmell J., Bianchi B. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc. Natl. Acad. Sci. USA. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.H., Fehrenbacher L., Novotny W.F., Herbst R.S., Nemunaitis J.J., Jablons D.M., Langer C.J., DeVore R.F., 3rd, Gaudreault J., Damico L.A. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2004;22:2184–2191. doi: 10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Kut C., Mac Gabhann F., Popel A.S. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br. J. Cancer. 2007;97:978–985. doi: 10.1038/sj.bjc.6603923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C., Gangadharan V., Bali K.K., Xie R.G., Agarwal N., Kurejova M., Tappe-Theodor A., Tegeder I., Feil S., Lewin G. Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol. 2012;10:e1001283. doi: 10.1371/journal.pbio.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik-Hall M., Dina O.A., Levine J.D. Primary afferent nociceptor mechanisms mediating NGF-induced mechanical hyperalgesia. Eur. J. Neurosci. 2005;21:3387–3394. doi: 10.1111/j.1460-9568.2005.04173.x. [DOI] [PubMed] [Google Scholar]
- Mantyh P. Bone cancer pain: causes, consequences, and therapeutic opportunities. Pain. 2013;154(Suppl 1):S54–S62. doi: 10.1016/j.pain.2013.07.044. [DOI] [PubMed] [Google Scholar]
- Mantyh P.W., Koltzenburg M., Mendell L.M., Tive L., Shelton D.L. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology. 2011;115:189–204. doi: 10.1097/ALN.0b013e31821b1ac5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A.K., Dimberg A., Kreuger J., Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Schweizerhof M., Stösser S., Kurejova M., Njoo C., Gangadharan V., Agarwal N., Schmelz M., Bali K.K., Michalski C.W., Brugger S. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat. Med. 2009;15:802–807. doi: 10.1038/nm.1976. [DOI] [PubMed] [Google Scholar]
- Srivastava K., Hu J., Korn C., Savant S., Teichert M., Kapel S.S., Jugold M., Besemfelder E., Thomas M., Pasparakis M., Augustin H.G. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell. 2014;26:880–895. doi: 10.1016/j.ccell.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Stefater J.A., 3rd, Lewkowich I., Rao S., Mariggi G., Carpenter A.C., Burr A.R., Fan J., Ajima R., Molkentin J.D., Williams B.O. Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature. 2011;474:511–515. doi: 10.1038/nature10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Veire S., Stalmans I., Heindryckx F., Oura H., Tijeras-Raballand A., Schmidt T., Loges S., Albrecht I., Jonckx B., Vinckier S. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell. 2010;141:178–190. doi: 10.1016/j.cell.2010.02.039. [DOI] [PubMed] [Google Scholar]
- Wacnik P.W., Eikmeier L.J., Ruggles T.R., Ramnaraine M.L., Walcheck B.K., Beitz A.J., Wilcox G.L. Functional interactions between tumor and peripheral nerve: morphology, algogen identification, and behavioral characterization of a new murine model of cancer pain. J. Neurosci. 2001;21:9355–9366. doi: 10.1523/JNEUROSCI.21-23-09355.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Tibensky I., Schmidt J., Ryschich E., Märten A. Interferon-alpha enhances antitumor effect of chemotherapy in an orthotopic mouse model for pancreatic adenocarcinoma. J. Immunother. 2008;31:599–606. doi: 10.1097/CJI.0b013e3181818769. [DOI] [PubMed] [Google Scholar]
- Zurborg S., Piszczek A., Martínez C., Hublitz P., Al Banchaabouchi M., Moreira P., Perlas E., Heppenstall P.A. Generation and characterization of an Advillin-Cre driver mouse line. Mol. Pain. 2011;7:66. doi: 10.1186/1744-8069-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This depicts the nature and extent of nocifensive behavior and weight bearing in mice 30 days after sham surgery.
This depicts the nature and extent of nocifensive behavior, such as guarding of the tumor-bearing limb and lifting of the affected paw above the wire-mesh bottom, reduction in weight-bearing of the affected paw, sporadic hopping behavior during ambulation in tumor-bearing mice 30 days after implantation of breast cancer cells unilaterally in the femur bone cavity; in addition, the mouse shown in the video received intraperitoneal injections of control IgG.
This depicts the nature and extent of nocifensive behavior in tumor-bearing mice 30 days after implantation of breast cancer cells unilaterally in the femur bone cavity; in addition, the mouse received intraperitoneal injections of an anti-VEGFR1 antibody (MF1 clone).
This depicts the nature and extent of nocifensive behavior in tumor-bearing mice 30 days after implantation of breast cancer cells unilaterally in the femur bone cavity; in addition, the mouse received intraperitoneal injections of an anti-VEGFR2 antibody (DC101 clone).