

Abstract
The nematode Caenorhabditis elegans secretes ascarosides, structurally diverse derivatives of the 3,6-dideoxysugar ascarylose, and uses them in chemical communication. At high population densities, specific ascarosides, which are together known as the dauer pheromone, trigger entry into the stress-resistant dauer larval stage. In order to study the structure-activity relationships for the ascarosides, we synthesized a panel of ascarosides and tested them for dauer-inducing activity. This panel includes a number of natural ascarosides that were detected in crude pheromone extract, but as yet have no assigned function, as well as many unnatural ascaroside derivatives. Most of these ascarosides, some of which have significant structural similarity to the natural dauer pheromone components, have very little dauer-inducing activity. Our results provide a primer to ascaroside structure-activity relationships and suggest that slight modifications to ascaroside structure dramatically influence binding to the relevant G protein-coupled receptors that control dauer formation.
Keywords: ascarosides, Caenorhabditis elegans, nematodes, dauer formation
1. Introduction
Nematode (roundworm) species are extremely diverse and inhabit virtually every type of environment on the planet,1 but only recently has progress been made in identifying the chemical signals that these species use in communication.2-4 Although many nematodes are free-living (eating bacteria and/or fungi), approximately a third are parasites of plants, insects, animals, or humans.5 Both free-living and parasitic nematode species have been shown to use ascarosides, derivatives of the unusual 3,6-dideoxy-L-sugar ascarylose, in chemical signaling.2-4 However, the structures of ascarosides produced vary from species to species. The free-living model organism, Caenorhabditis elegans, secretes ascarosides to control its development and behavior.2,3 In order to monitor its population density, C. elegans secretes the “dauer pheromone ascarosides” into its environment and senses their concentration using chemosensory neurons in its head (Figure 1A, B).3a,b,d At high population densities, these dauer pheromone ascarosides induce C. elegans to enter a stress-resistant, non-feeding dispersal larval stage called the dauer.6 Many parasitic nematode species also have a dauer-like larval stage, called the infective L3 stage, which is stress-resistant and allows the nematode to survive in the environment as it seeks a new host.4b,7 An understanding of the ascaroside chemical language used by C. elegans and other nematode species to control developmental decisions and behavior may lead to new chemical methods to control parasitic species. An important step in developing an understanding of this chemical language is to determine the structure-activity relationships for the ascarosides.
Figure 1.
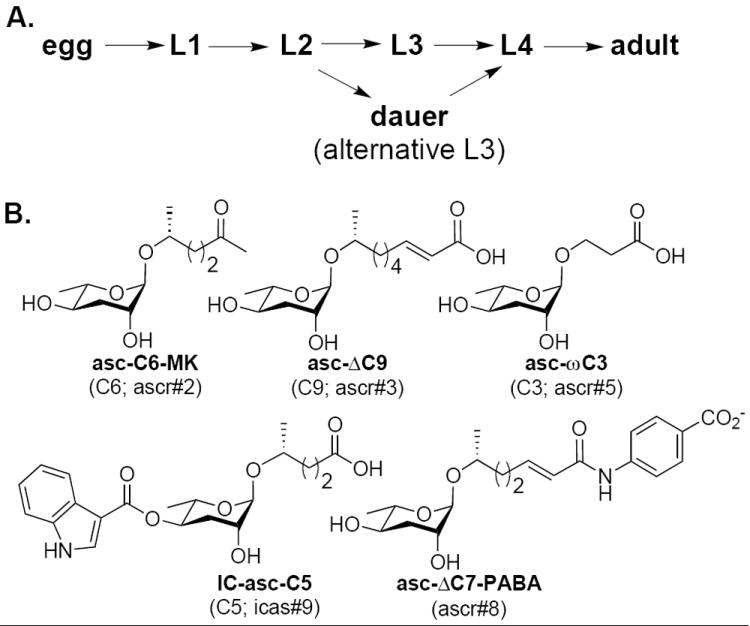
The dauer stage and the dauer pheromone ascarosides. A. Under favorable conditions (low population density, adequate food), C. elegans hatches from an egg, and progresses through four larval stages (L1-L4) before becoming a reproductive adult. If, on the other hand, L1 or L2 larvae are met with unfavorable conditions (e.g. high population density, inadequate food supply), they will instead enter the long-lived dauer larval stage. B. In order to sense its population density, C. elegans secretes and monitors the concentration of the dauer pheromone ascarosides. See the legend of Figure 2 for a description of the rubric used to name the ascarosides.
The ascarosides in C. elegans are connected via an α-linkage to fatty acid-derived side chains of varying lengths (see Fig. 2 for a general structure of the ascarosides). The fatty acid side chain is attached to the sugar at either the (ω-1)- or ω-position and sometimes has unsaturation adjacent to the chain terminal carbonyl. Additional modifications (head groups) can occur on the sugar, such as indole-3-carbonyl (IC),2a,b,e 4-hydroxybenzoyl (HB),2a (E)-2-methyl-2-butenoyl (MB)2a and β-glucosyl (glu).2f Additional modifications (terminal groups) can also occur at the end of the side chain, such as methyl ketone (MK)2h or para-aminobenzoic acid (PABA).2c Although the nomenclature for the ascarosides varies in the literature, we name the ascarosides according to the following rubric: (head group)-asc-(ω)(Δ)C#(terminal group). This rubric considers an ascaroside with a saturated fatty acid side chain oxygenated at the (ω-1)-position as the canonical structure and notes deviations from that structure.
Figure 2.
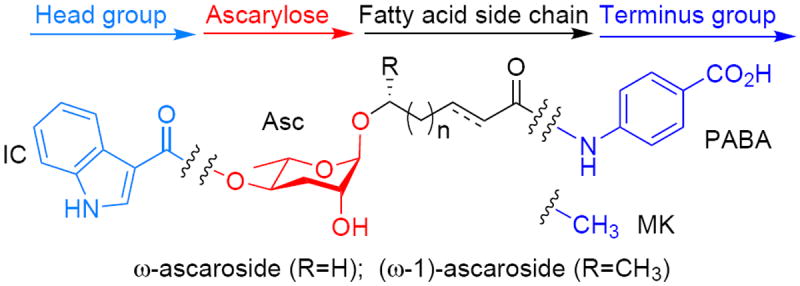
We describe the ascarosides here using the following structure-based nomenclature: (head group-)asc-(ω)(Δ)C#(terminal group). The canonical structure is an ascaroside with a saturated fatty acid side chain oxygenated at the (ω-1)-position. Deviations from this structure, including α-β unsaturation (Δ) or oxygenation at the ω-position (ω), are indicated in the name. Head group abbreviations: indole-3-carbonyl (IC). Terminus group abbreviations: para-aminobenzoic acid (PABA) and methyl ketone (MK).
The components of the dauer pheromone work in the nM-μM range and include asc-C6-MK (alternative names: C6; ascr#2),2h asc-ΔC9 (C9; ascr#3),2h asc-ωC3 (C3; ascr#5),2g IC-asc-C5 (C5, icas#9),2e and asc-ΔC7-PABA (ascr#8)2c (Figure 1B). The first four of these components were identified using activity-guided fractionation of C. elegans-conditioned culture medium and structure elucidation by NMR spectroscopy and mass spectrometry. The last of these components, asc-ΔC7-PABA (ascr#8), was identified through comparative metabolomics using NMR spectroscopy. IC-asc-C5 is perhaps the most potent of the dauer pheromone ascarosides in terms of its ability to induce dauer formation, but displays a bell-shaped activity curve where at high concentrations it inhibits its own activity. Ascaroside asc-ωC3 is the only dauer pheromone component with the fatty acid side chain that is attached at the ω-position, and interestingly, this ascaroside works synergistically with other components, including asc-C6-MK, asc-ΔC9, and IC-asc-C5, to induce dauer formation.2g At much lower concentrations (fM-pM), some of the dauer pheromone ascarosides work in conjunction with additional ascarosides to influence certain behaviors, including mating attraction and aggregation.2f,3e
The dauer pheromone ascarosides have been shown to target members of several families of G protein-coupled receptors (GPCRs) expressed in chemosensory neurons that are directly exposed to the environment through pores in the head of the worm.3a,b,d For example, asc-C6-MK and asc-ΔC9 promote dauer formation by inhibiting two GPCRs belonging to the srbc family of GPCRs, SRBC-64 and -66.3d Ascaroside asc-ωC3, on the other hand, promotes dauer formation by inhibiting members of the srg family of GPCRs (SRG-36 and -37).3b The synergism seen between asc-ωC3 and the other dauer pheromone ascarosides reflect the fact that they target different families of GPCRs.3b The fact that various ascarosides target completely different families of GPCRs suggests that the diversity in ascarosides produced by C. elegans is matched by a diversity of GPCRs that respond to them. Indeed, GPCRs have undergone recent expansion in C. elegans and related nematodes,8 perhaps reflecting the importance of chemical signaling in C. elegans survival and reproduction.
C. elegans produces a variety of ascarosides with different side-chain lengths, with either saturation or α-β unsaturation, and with oxygenation at either the ω- or (ω-1)-position.2 The biological role of many of these naturally produced ascarosides has not been established. It is possible that some of these ascarosides are simply intermediates in the biosynthetic pathway to the relevant ascaroside pheromones. In addition, the relationship between various fatty acid side chain structural attributes, including length, unsaturation, oxygenation at the ω- or (ω-1)-position, head group functionalities, and terminus group functionalities and the corresponding potency in triggering dauer formation and modulating other physiological processes in C. elegans is poorly understood. To address these issues, we have initiated a program aimed at the synthesis and evaluation in dauer formation and other assays of a series of ascarosides with varying side chain lengths, unsaturation adjacent to carbonyl, oxygenation patterns, and placement of the indole-3-carbonyl moiety (Figure 3). In this paper, we will discuss the chemical synthesis and evaluation of a panel of ascarosides for their dauer-inducing ability.
Figure 3.
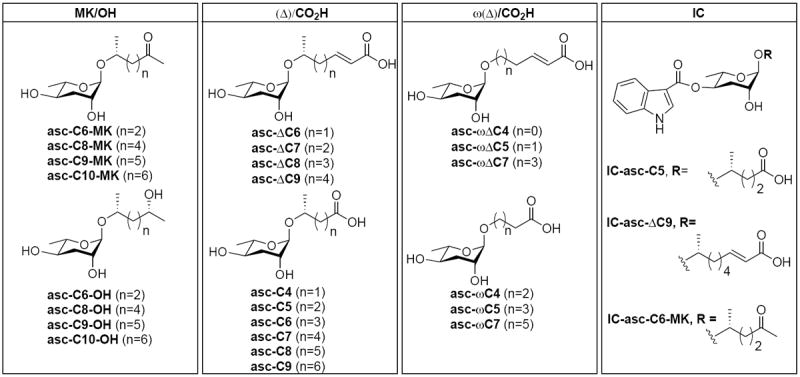
Ascarosides targeted for synthesis and biological evaluation in the dauer formation assay.
2. Results and Discussion
We envisioned the synthesis and biological evaluation of four differing sets of compounds: MK/OH, (Δ)/CO2H, ω(Δ)/CO2H and IC (Figure 3). Importantly, the compounds asc-ΔC7, asc-C5, asc-C9, asc-ωΔC5, asc-ωΔC7, asc-ωC5, asc-ωC7, and IC-asc-ΔC9 have been identified through metabolomics or bioassay-guided fractionation and NMR2a-c but have not been tested in dauer formation.
Our general approach (Scheme 1) began with the glycosylation of a series of 1° and 2° alcohols with the donor dibenzoyl ascarylose (1, Scheme 1).2i As we had successfully used cross metathesis in a previous synthesis,2d we employed this same strategy for the elaboration of intermediates 2 to 3. Subsequent hydrogenation (for the synthesis of 4) and saponification resulted in the formation of all ascarosides in the (Δ)/CO2H and ω(Δ)/CO2H series. An optional oxidation of 2° alcohol-containing intermediates 4 (in the synthesis of asc-C6-10-MK) after glycosylation pre-empted saponification for the synthesis of these compounds. The general synthetic scheme for the IC analogs involved partial saponification of generic intermediates 10 accessed via the common intermediates 8 that are acylated with an excess of acyl chloride 99 (acylation with 1 equiv. results in predominant esterification at the axial C2 oxygen2e) followed by an additional saponification that results in faster hydrolysis of the axial C2 ester.2e
Scheme 1.
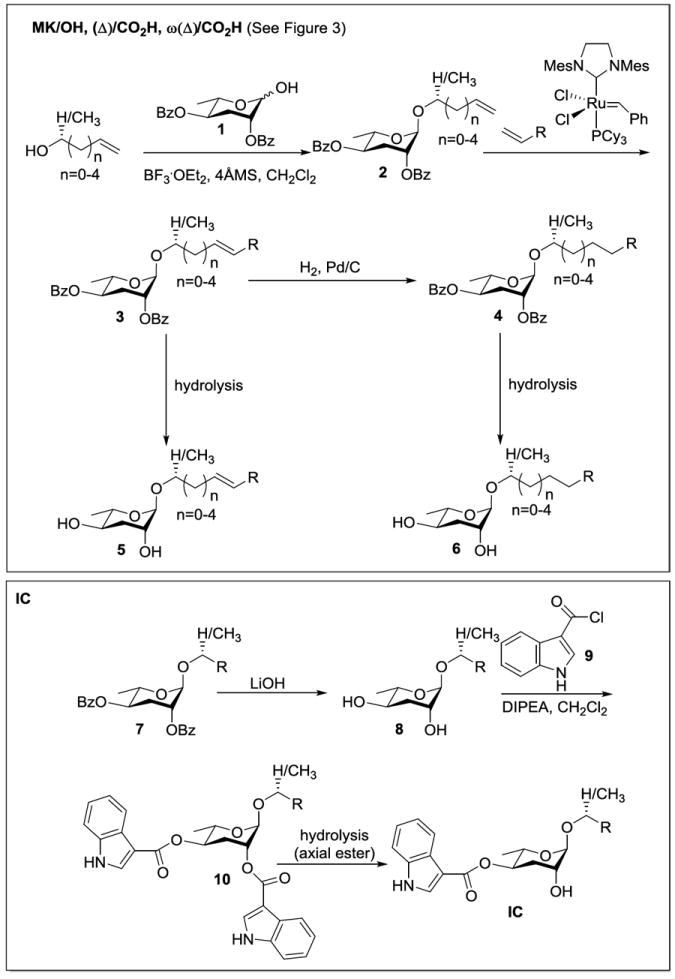
Synthetic Strategy
Synthesis of the (Δ)/CO2H analogs was carried out as detailed in Scheme 2. Glycosylation of alcohols 11-14 resulted in moderate to high yields (52-90%) of glycosylated products 15-18. Elaboration of 15-18 to the (Δ)/CO2H series involved cross metathesis with 5 equiv. of methyl acrylate in the presence of the Grubbs second generation ruthenium catalyst to stereoselectively generate 19-22 (75-93%). Hydrogenation (73-98%) to 23-26 then preceded saponification (1M LiOH, tBuOH) of the intermediates 19-26 to generate the (Δ)/CO2H series.
Scheme 2.
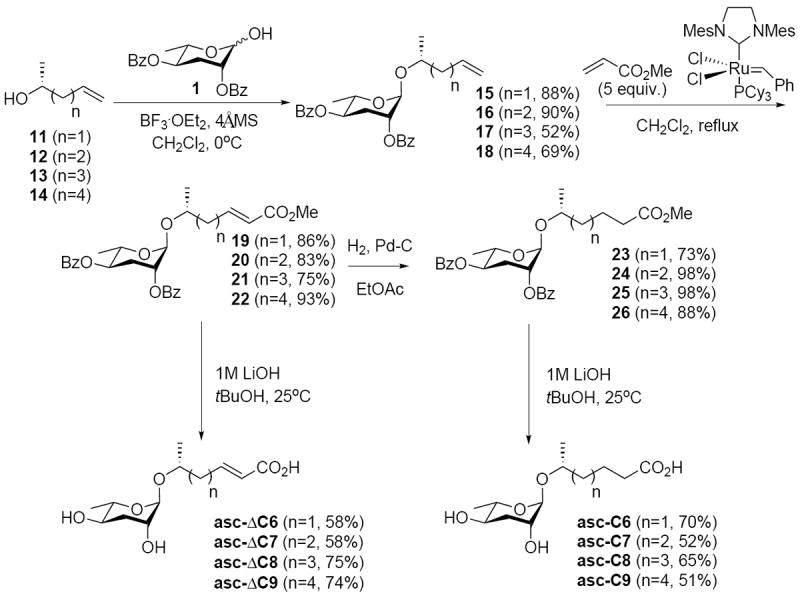
Synthesis of (Δ)/CO2H Ascarosides
Synthesis of the final two analogs in the (Δ)/CO2H series, asc-C4 and asc-C5, did not necessitate the use of cross metathesis (Scheme 3). In this instance, alkenes 15 and 16 were ozonolyzed to generate intermediate aldehydes that were subjected to Pinnick oxidation and then hydrolysis to afford target structures asc-C4 and asc-C5.
Scheme 3.
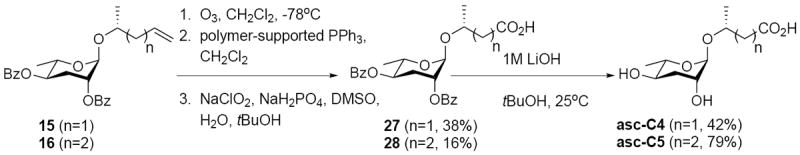
Synthesis of (Δ)/CO2H Ascarosides
Synthesis of the ω(Δ)/CO2H series proceeded in a manner analogous to that of the (Δ)/CO2H series (Scheme 4) with the cross metathesis of 32-34 in low to moderate yields (37-54%) followed by hydrogenation (81-93%) and saponification (23-43%).
Scheme 4.
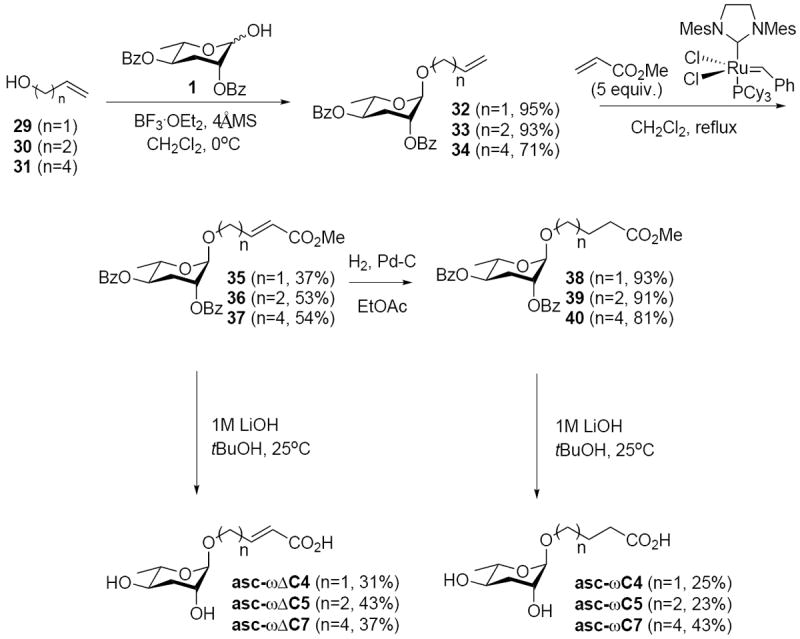
Synthesis of ω(Δ)/CO2H Ascarosides
The synthesis of the MK/OH series (Scheme 5) employed 10-fold excesses of cross-metathesis coupling partners 11 and 12 with the glycosylation products 15 and 16 to obtain high yields (75-93%) of products 41-43. Cross-metathesis, in this case, afforded inseparable mixtures of E and Z products, though this was not detrimental as the next step involved hydrogenation of alkene to afford products 44-46 (94-99%). Oxidation of 44-46 with PCC afforded ketone intermediates 47-49 (97-99%). Finally, saponification afforded the desired MK/OH series (44-94%).
Scheme 5.
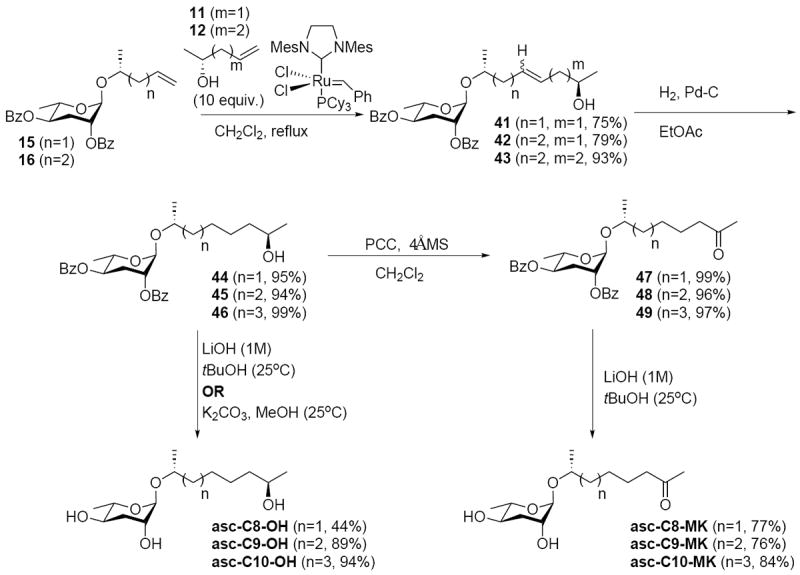
Synthesis of MK/OH Ascarosides
Synthesis of the IC analogs proved, as expected, to be a more difficult task. Synthesis of IC-asc-C6-MK (Scheme 6) proceeded via the intermediacy of naturally-occurring asc-C6-MK which was synthesized according to a shorter route relative to the previously reported synthesis.2h Monoglycosylation of (2R,5R)hexane-2,5-diol (50) with 1 preceded PCC oxidation of intermediate secondary alcohol 51 (that was itself converted to naturally-occurring asc-C6-OH via saponification for evaluation in the dauer formation assay) and saponification to afford asc-C6-MK. Diacylation with an excess of indole-3-carbonyl chloride (9) afforded the diester 53. Hydrolysis of the axial indole-3-carboxylate ester provided mixtures that were most easily purified when hydrolysis was run in iPrOH as cosolvent. While the target product IC-asc-C6-MK was produced in only 35% yield after multiple purifications, none of the product of monohydrolysis of the equatorial indole-3-carboxylate ester was ever isolated.
Scheme 6.
Synthesis of IC-asc-C6-MK
Synthesis of IC analog IC-asc-ΔC9 (Scheme 7) proceeded from intermediate 18. Saponification preceded double esterification with 9 and then cross metathesis with methyl acrylate. Hydrolysis of methyl ester and axial indole-3-carboxylate ester proceeded with tBuOH as cosolvent in a satisfactory yield of 65% after careful purification to afford IC-asc-ΔC9.
Scheme 7.
Synthesis of Ascaroside IC-asc-ΔC9
29 synthetic ascarosides, including the five known components of the dauer pheromone, were tested in parallel in the dauer formation assay at two concentrations, 220 nM and 6000 nM (Fig. 4). Of these ascarosides, 13 (indicated by an asterisk in Fig. 4) had previously been shown to be produced by C. elegans and had been chemically synthesized to verify their structures (see supplemental information for details).2a-c,2e,2g-i However, to our knowledge, the dauer formation activity of asc-C4, asc-C5, asc-ωC5, asc-C9, and IC-asc-ΔC9 have not been previously published. Five of the 29 ascarosides (indicated by a double asterisk in Fig. 4) had been detected in C. elegans culture medium extracts by LC-MS/MS, but had not yet been chemically synthesized to verify their structures. By analyzing the ascarosides secreted by C. elegans by LC-MS/MS, as well as the synthetic standards for asc-C6, asc-ΔC8, and asc-C8 that we synthesized, we were able to confirm that these three ascarosides are secreted by C. elegans (Figure S1).
Figure 4.
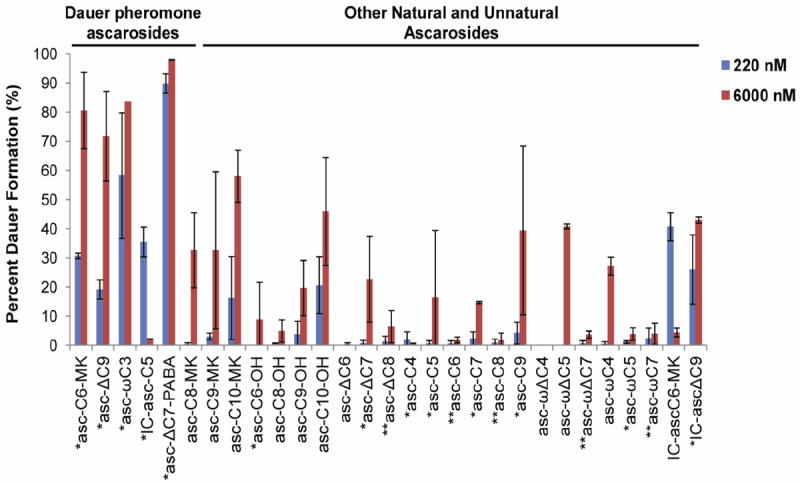
Percent dauer formation in the presence of ascarosides. Compounds were tested at two concentrations (220 nM and 6000 nM). Data represent the average of two independent experiments (± standard deviation). A single asterisk (*) indicates an ascaroside that is produced naturally by C. elegans and that has previously been synthesized chemically. A double asterisk (**) indicates an ascaroside that has been detected by LC-MS/MS and was synthesized chemically herein.
Other than the confirmed dauer pheromone ascarosides (i.e., asc-C6-MK, asc-ΔC9, asc-ωC3, IC-asc-C5, and asc-ΔC7-PABA), very few of the ascarosides demonstrated potent activity in the dauer formation assay despite their structural similarities to the dauer pheromone ascarosides. For example, asc-C6-MK and asc-C5 are very similar, differing only by the presence of a methyl ketone versus carboxylic acid at the side chain terminus. Unlike asc-C6-MK, asc-C5 has virtually no activity. In another example, asc-ωC3 and asc-C4 differ only by the presence of a methyl group at the ascarylose-proximal terminus of the side chain. While asc-ωC3 is a potent activator of dauer formation, asc-C4 has virtually no activity. In yet another example, asc-C9 and asc-ΔC9 differ only by the presence of α,β-unsaturation in the latter, but this unsaturation appears to be relatively important for dauer formation activity. Previous work has shown that IC-asc-C5 has a bell-shaped activity curve in which it inhibits its own activity at high concentrations. Interestingly, the IC-asc-C6-MK derivative, which differs from IC-asc-C5 in having a methyl ketone instead of a carboxylic acid, mimics this activity. This strongly suggests that the higher concentration inhibition of dauer formation requires indole-3-carbonyl moiety (asc-C5 does not exhibit self-inhibition at higher concentrations). In contrast, IC-ascΔC9, which was previously shown to induce aggregation, has a significant amount of dauer formation activity, but does not appear to display a bell-shaped activity curve based on the two concentrations tested. Thus, there is also a side chain component to the self-inhibition exhibited by IC-asc-C6-MK and IC-asc-C5. Since the dauer-like infective L3 stage is an essential stage in the life cycle of parasitic nematodes, compounds that inhibit the formation of this stage could potentially be developed as novel anthelmintics. Our data also confirm previous work which has shown that asc-C6-OH, unlike asc-C6-MK, has very little activity and that asc-ΔC7, unlike asc-ΔC7-PABA, also has very little activity.2c
3. Conclusion
By profiling the dauer-inducing activity of ascarosides and ascaroside analogs, we have determined that the activity is highly dependent on structure. Although C. elegans produces a number of ascarosides of varying length, unsaturation and side chain functionality, only specific ones have dauer-inducing activity. Our results suggest that the GPCRs that bind to the ascarosides to induce dauer formation are highly specific to their cognate ascaroside ligands. This high degree of specificity could be due to the fact that many nematode species using ascarosides for signaling occupy the same environments,10 and it could therefore be in the interest of the different species to use non-overlapping sets of ascarosides in signaling to avoid interspecies ‘cross-talk.’ Structural elucidation of the ascaroside-binding GPCRs should yield insight into the observed high degree of specificity and explain the differences in activity seen with asc-C6-MK and asc-C5, asc-ωC3 and asc-C4, and IC-asc-C5 and IC-asc-ΔC9. This knowledge will in turn provide a framework for the understanding of ascaroside-GPCR interactions in other nematode species that use ascarosides in chemical signaling, including parasitic ones.
4. Experimental
4.1. Chemistry
4.1.1. General
Flash column chromatography was performed using 60Å silica gel. 1H NMR and 13C NMR spectroscopy were performed on a Bruker AV-400, DPX 400, DPX 250 or Varian 500 spectrometer. HPLC purifications were conducted with a Waters Breeze 2 system equipped with an XBridge C18 semi-preparative column (5 μm, 10×100 mm) with gradient runs of H2O in CH3CN. Mass spectra were obtained using an Agilent 6210 electrospray time-of-flight mass spectrometer. Optical rotation measurements were obtained using a JASCO P-2000 polarimeter. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Analytical TLC was conducted on aluminum sheets (Merck, silica gel 60, F254). Compounds were visualized by UV absorption (254 nm) and staining with anisaldehyde or KMnO4. All glassware (save for hydrolysis reactions) was flame-dried under vacuum and backfilled with dry nitrogen prior to use. Deuterated solvents were obtained from Cambridge Isotope Labs. All solvents were purified according to the method of Grubbs.11
4.1.2. Synthesis of dibenzoyl ascarylose (1)
See ref. 2i
4.1.3. General procedure for synthesis of alcohols 13, 14
See ref. 2i
4.1.4. Synthesis of (R)-hept-6-en-2-ol (13)
Began with 156.8 mg (R)-(+)-propylene oxide, (no purification necessary) yielded 175.4 mg (57%) of a light yellow liquid. [α]D25 = 38.3, c 0.73 (CH2Cl2); 1H NMR (250 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.09 – 4.88 (m, 2H), 3.79 (h, J = 6.0 Hz, 1H), 2.16 – 1.95 (m, 2H), 1.95 (s, 1H), 1.60 – 1.31 (m, 4H), 1.18 (d, J = 6.2 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ 138.63, 114.50, 67.87, 38.65, 33.62, 24.97, 23.42.
4.1.5. Synthesis of (R)-oct-7-en-2-ol (14)
Began with 596.1 mg (R)-(+)-propylene oxide, (no purification necessary) yielded 322.5 mg (88%) of a light yellow liquid. [α]D25 = 7.1, c 0.59 (CH2Cl2); 1H NMR (250 MHz, CDCl3) δ 5.77 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.04 – 4.84 (m, 2H), 3.75 (q, J = 5.9 Hz, 1H), 2.01 (h, J = 5.7 Hz, 3H), 1.47 – 1.22 (m, 8H), 1.14 (d, J = 6.2 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ 138.70, 114.18, 67.79, 38.98, 33.57, 28.74, 25.09, 23.26.
4.1.6. Representative glycosylation procedure: synthesis of (2S,3R,5R,6R)-2-methyl-6-((R)-pent-4-en-2-yloxy)tetrahydro-2H-pyran-3,5-diyl dibenzoate (15)
A suspension of 1.027 g (2.884 mmol) dibenzoyl ascarylose (1), 0.46 mL (4.5 mmol) alcohol 11 and 302.9 mg 4Å molecular sieves in 30 mL CH2Cl2 was cooled to 0°C. To this suspension was added 1.54 mL (12.2 mmol) BF3·OEt2 at once. The resulting suspension was stirred at 0°C for 2 h. 26 mL saturated NaHCO3 solution was then added. Upon cessation of effervescence, the aqueous layer was separated from the organic layer and then extracted with 2×50mL CH2Cl2. The resulting organic extracts were dried over MgSO4 and filtered to obtain 1.3094 g of oil after evaporation of solvent. Silica gel column chromatography (70 g silica gel, eluted with 30% hexanes in CH2Cl2) afforded 0.8915 g (73%) of a colorless syrup. [α]D25 = -10.7, c 1.015 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C28H34O7Na 447.1778, found 447.1790; 1H NMR (250 MHz, CDCl3): δ 8.12 (d, J = 7.6 Hz, 2H), 8.05 (d, J = 7.9 Hz, 2H), 7.57 (m, 2H), 7.47 (m, 4H), 5.89 (ddt, J = 17.3, 10.3, 7.1 Hz, 1H), 5.16 (m, 4H), 4.96 (s, 1H), 4.15 (dq, J = 12.7, 6.2 Hz, 1H), 3.92 (m, 1H), 2.32 (m, 4H), 1.28 (d, J = 6.2 Hz, 3H), 1.21 (d, J = 6.0 Hz, 3H); 13C NMR (62.5 MHz, CDCl3): δ 165.8; 165.7; 135.0; 133.31; 133.25; 130.1; 129.9; 129.7; 128.5; 117.3; 94.0; 72.5; 71.3; 70.7; 70.0; 41.7; 29.8; 19.0; 17.9.
4.1.7. Synthesis of (2R,3R,5R,6S)-2-((R)-hex-5-en-2-yloxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (16)
The title compound was prepared in a manner similar to that of 15. Started with 1.0228 g (2.8701 mmol) dibenzoyl ascarylose (1), 0.54 mL (4.5 mmol) alcohol 12, 325.8 mg 4Å molecular sieves, and 1.54 mL (12.2 mmol) BF3·OEt2 in 32 mL CH2Cl2. Silica gel chromatography afforded 1.1364 g (90%) of a colorless oil. [α]D25 = -15.0, c 1.74 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C26H30O6Na 461.1935, found 461.1946; 1H NMR (250 MHz, CDCl3): δ 8.13 (d, J = 7.0 Hz, 2H), 8.06 (d, J = 7.0 Hz, 2H), 7.56 (m, 2H), 7.45 (m, 4H), 5.89 (ddt, J = 16.8, 10.2, 6.5 Hz, 1H), 5.09 (m, 5H), 4.16 (dq, J = 9.8, 6.2 Hz, 1H), 3.89 (m, 1H), 2.45 (dt, J = 13.5, 4.0 Hz, 1H), 2.24 (m, 3H), 1.70 (m, 2H), 1.30 (d, J = 6.2 Hz, 3H), 1.22 (d, J = 6.1 Hz, 3H); 13C NMR (62.5 MHz, CDCl3): δ 166.0; 165.9; 138.6; 133.43; 133.37; 130.2; 130.1; 129.8; 128.6; 115.0; 93.9; 72.2; 71.4; 70.8; 67.2; 36.5; 30.2; 29.9; 19.3; 18.1.
4.1.8. Synthesis of (2R,3R,5R,6S)-2-((R)-hept-6-en-2-yloxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (17)
The title compound was prepared in a manner similar to that of 16. Began with 320.5 mg dibenzoyl ascarylose, column chromatography (30.5 g silica gel, gradient run from 5% ethyl acetate in hexanes to 10% ethyl acetate in hexanes) yielded 213.2 mg (52%) of a colorless oil. [α]D25 = -55.7, c 0.60 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C27H32O6Na 475.2091, found 475.2083; 1H NMR (250 MHz, CDCl3) δ 8.18 – 8.08 (m, 2H), 8.08 – 7.98 (m, 2H), 7.66 – 7.53 (m, 2H), 7.53 – 7.39 (m, 4H), 5.86 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H), 5.26 – 5.11 (m, 2H), 5.11 – 4.91 (m, 3H), 4.19 – 4.04 (m, 1H), 3.87 (m, 1H), 2.42 (dt, J = 13.0, 4.0 Hz, 1H), 2.30 – 2.00 (m, 3H), 1.74 – 1.39 (m, 4H), 1.28 (d, J = 6.1 Hz, 3H), 1.20 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 165.78, 165.65, 138.68, 133.21, 133.15, 130.01, 129.86, 129.84, 129.60, 128.42, 114.58, 93.76, 72.43, 71.24, 70.65, 66.97, 36.54, 33.60, 29.72, 24.95, 19.14, 17.86.
4.1.9. Synthesis of (2S,3R,5R,6R)-2-methyl-6-((R)-oct-7-en-2-yloxy)tetrahydro-2H-pyran-3,5-diyl dibenzoate (18)
The title compound was prepared in a manner similar to that of 16. Began with 486.1 mg dibenzoyl ascarylose, column chromatography (20 g silica gel, 30% hexanes in CH2Cl2) yielded 440.0 mg (69%) of a colorless oil. [α]D25 = 89.4, c 0.085 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C28H34O6Na 489.2248, found 489.2250; 1H NMR (250 MHz, CDCl3) δ 8.19 – 8.09 (m, 2H), 8.09 – 7.99 (m, 2H), 7.61 – 7.49 (m, 2H), 7.51 – 7.37 (m, 4H), 5.85 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.29 – 5.12 (m, 2H), 5.10 – 4.93 (m, 3H), 4.15 (dq, J = 12.3, 6.2 Hz, 1H), 3.87 (h, J = 5.9 Hz, 1H), 2.44 (dt, J = 13.4, 4.0 Hz, 1H), 2.23 (ddd, J = 13.8, 11.3, 3.1 Hz, 1H), 2.11 (d, J = 6.5 Hz, 2H), 1.70 – 1.41 (m, 6H), 1.31 (d, J = 6.3 Hz, 3H), 1.20 (d, J = 6.0 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ 165.57, 165.45, 138.64, 133.06, 133.01, 129.89, 129.77, 129.71, 129.50, 129.45, 128.28, 114.34, 93.62, 72.35, 71.13, 70.53, 66.83, 36.85, 33.65, 29.60, 28.65, 25.10, 19.06, 17.76.
4.1.10. Representative cross metathesis procedure
see ref. 2d (the cross metathesis procedure reported therein is representative of all cross metathesis procedures involving methyl acrylate)
4.1.11. Synthesis of (2R,3R,5R,6S)-2-(((R,E)-6-methoxy-6-oxohex-4-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (19)
See ref. 2d for representative procedure. Began with 177.2 mg 15, column chromatography (20.5 g silica gel, gradient run from 2% Et2O in hexanes to 10% Et2O in hexanes) yielded 173.6 mg (86%) of a colorless oil. [α]D25 = 75.9, c 1.27 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C27H30O8Na 505.1833, found 505.1838; 1H NMR (250 MHz, CDCl3) δ 8.11 (d, J = 7.5 Hz, 2H), 8.04 (d, J = 7.8 Hz, 2H), 7.66 – 7.52 (m, 2H), 7.50 – 7.39 (m, 4H), 7.14 – 6.94 (m, 1H), 5.96 (d, J = 15.8 Hz, 1H), 5.23 – 5.10 (m, 2H), 4.95 (s, 1H), 4.13 – 3.94 (m, 2H), 3.74 (s, 3H), 2.57 – 2.34 (m, 3H), 2.17 (t, J = 12.1 Hz, 1H), 1.32 – 1.19 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 166.64, 165.73, 165.59, 145.42, 133.25, 133.23, 133.15, 129.90, 129.81, 129.72, 129.61, 129.57, 128.41, 128.39, 123.29, 93.89, 71.47, 70.95, 70.37, 67.06, 51.43, 39.74, 29.58, 19.19, 17.70.
4.1.12. Synthesis of (2R,3R,5R,6S)-2-(((R,E)-7-methoxy-7-oxohept-5-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (20)
See ref. 2d for representative procedure. Began with 670.0 mg 16, column chromatography (107 g silica gel, gradient run from 2% Et2O in hexanes to 20% Et2O in hexanes) yielded 630.5 mg (83%) of a colorless oil. [α]D25 = 86.4, c 1.03 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C28H32O8Na 519.1989, found 519.1990; 1H NMR (250 MHz, CDCl3) δ 8.11 (d, J = 7.3 Hz, 2H), 8.05 (d, J = 7.4 Hz, 2H), 7.59 (t, J = 7.0 Hz, 2H), 7.53 – 7.40 (m, 4H), 7.15 – 6.95 (m, 1H), 5.91 (d, J = 15.7 Hz, 1H), 5.27 – 5.09 (m, 2H), 4.96 (s, 1H), 4.06 (dq, J = 12.8, 6.3 Hz, 1H), 3.88 (h, J = 11.8, 6.3 Hz, 1H), 3.73 (s, 3H), 2.51 – 2.29 (m, 3H), 2.19 (ddd, J = 13.9, 10.9, 3.1 Hz, 1H), 1.90 – 1.61 (m, 2H), 1.28 (d, J = 6.2 Hz, 3H), 1.22 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 167.01, 165.80, 165.68, 148.95, 133.29, 133.22, 129.95, 129.87, 129.81, 129.65, 128.47, 121.28, 93.73, 71.73, 71.14, 70.56, 67.20, 51.47, 35.36, 29.71, 28.40, 19.07, 17.87.
4.1.13. Synthesis of (2R,3R,5R,6S)-2-(((R,E)-8-methoxy-8-oxooct-6-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (21)
See ref. 2d for representative procedure. Began with 202.0 mg 17, column chromatography (32 g silica gel, gradient run from 5% ethyl acetate in hexanes to 15% ethyl acetate in hexanes) yielded 170.8 mg (75%) of a colorless oil. [α]D25 = -1.3, c 2.22 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C29H34O8Na 533.2146, found 533.2143; 1H NMR (250 MHz, CDCl3) δ 8.15 – 8.08 (m, 2H), 8.08 – 8.00 (m, 2H), 7.66 – 7.53 (m, 2H), 7.46 (m, 4H), 7.02 (dt, J = 15.7, 6.9 Hz, 1H), 5.88 (d, J = 15.7 Hz, 1H), 5.25 – 5.10 (m, 2H), 4.95 (s, 1H), 4.16 – 4.01 (m, 1H), 3.93 – 3.79 (m, 1H), 3.72 (s, 3H), 2.42 (dt, J = 13.5, 4.0 Hz, 1H), 2.35 – 2.10 (m, 3H), 1.77 – 1.49 (m, 3H), 1.28 (d, J = 6.2 Hz, 3H), 1.20 (d, J = 6.0 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ 167.04, 165.74, 165.63, 149.08, 133.21, 133.14, 129.92, 129.81, 129.59, 128.41, 121.16, 93.69, 72.20, 71.15, 70.56, 67.05, 51.39, 36.49, 32.05, 29.69, 24.12, 19.08, 17.86.
4.1.14. Synthesis of (2R,3R,5R,6S)-2-(((R)-9-methoxy-9-oxonon-7-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (22)
See ref. 2d for representative procedure. Began with 513.6 mg 18, column chromatography (50g silica gel, isocratic run of 100% CH2Cl2) yielded 534.4 mg (93%) of a colorless oil. [α]D25 = -7.0, c 7.58 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C30H36O8Na 547.2302, found 547.2317; 1H NMR (250 MHz, CDCl3) δ 8.16 – 8.09 (m, 2H), 8.09 – 8.02 (m, 2H), 7.64 – 7.56 (m, 2H), 7.48 (m, 4H), 7.01 (dt, J = 15.7, 7.0 Hz, 1H), 5.91 – 5.81 (m, 1H), 5.25 – 5.14 (m, 2H), 4.96 (s, 1H), 4.17 – 4.05 (m, 1H), 3.85 (t, J = 8.4 Hz, 1H), 3.72 (s, 3H), 2.44 (dt, J = 13.4, 4.0 Hz, 1H), 2.32 – 2.15 (m, 3H), 1.60 – 1.47 (m, 6H), 1.29 (d, J = 6.2 Hz, 3H), 1.21 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 167.17, 165.87, 165.77, 149.43, 133.36, 133.31, 130.10, 129.97, 129.73, 128.57, 121.18, 93.91, 72.63, 71.35, 70.76, 67.15, 51.49, 36.97, 32.28, 29.84, 28.09, 25.38, 19.28, 18.01.
4.1.15. Representative hydrogenation procedure: synthesis of (2R,3R,5R,6S)-2-(((R)-6-methoxy-6-oxohexan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (23)
A suspension of 90.4 mg (0.187 mmol) unsaturated ester 19 and 6.5 mg 10% palladium on activated carbon in 5.2 mL EtOAc was vacuum purged and back filled with H2 from a balloon. The resulting suspension was allowed to stir at 23°C for 19 h. The reaction mixture was filtered through Celite® and concentrated to obtain 85.5 mg of colorless oil. Column chromatography (2.5 g silica gel, CH2Cl2) yielded 66.1 mg (73%) of a colorless oil. HRMS (m/z): [M+Na]+ calcd. for C27H32O8Na 507.1989, found 507.1990; 1H NMR (250 MHz, CDCl3) δ 8.12 (d, J = 7.2 Hz, 2H), 8.06 (d, J = 7.1 Hz, 2H), 7.66 – 7.53 (m, 2H), 7.53 – 7.39 (m, 4H), 5.29 – 5.10 (m, 2H), 4.96 (s, 1H), 4.21 – 4.02 (m, 1H), 3.88 (h, J = 6.0 Hz, 1H), 3.70 (s, 3H), 2.51 – 2.30 (m, 3H), 2.21 (ddd, J = 13.9, 11.3, 3.1 Hz, 1H), 1.95 – 1.47 (m, 4H), 1.29 (d, J = 6.2 Hz, 3H), 1.22 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.89, 165.73, 165.62, 133.20, 133.13, 129.94, 129.81, 129.59, 128.39, 93.72, 72.12, 71.17, 70.56, 67.05, 51.48, 36.41, 33.90, 29.68, 21.14, 19.01, 17.82.
4.1.16. Synthesis of (2R,3R,5R,6S)-2-(((R)-7-methoxy-7-oxoheptan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (24)
The title compound was prepared in a manner similar to that of 46. Began with 324.0 mg 20, (no purification necessary) yielded 318.4 mg (98%) of a colorless oil. [α]D25 = -36.8, c 1.01 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C28H34O8Na 521.2146, found 521.2148 1H NMR (250 MHz, CDCl3) δ 8.11 (d, J = 7.2 Hz, 2H), 8.05 (d, J = 7.3 Hz, 2H), 7.59 (t, J = 7.3 Hz, 2H), 7.53 – 7.39 (m, 4H), 5.27 – 5.09 (m, 2H), 4.95 (s, 1H), 4.10 (dq, J = 12.1, 6.4 Hz, 1H), 3.90 – 3.77 (m, 1H), 3.68 (s, 3H), 2.48 – 2.30 (m, 3H), 2.19 (ddd, J = 13.7, 11.3, 3.1 Hz, 1H), 1.69 (m, 2H), 1.61 – 1.37 (m, 4H), 1.28 (d, J = 6.2 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.82, 165.58, 165.49, 133.08, 133.02, 129.87, 129.75, 129.69, 129.47, 128.29, 93.65, 72.26, 71.10, 70.51, 66.89, 51.30, 36.61, 33.87, 29.58, 25.11, 24.78, 18.98, 17.73.
4.1.17. Synthesis of (2R,3R,5R,6S)-2-(((R)-8-methoxy-8-oxooctan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (25)
The title compound was prepared in a manner similar to that of 46. Began with 83.1 mg 21, (no purification necessary) yielded 81.3 mg (98%) of a colorless oil. [α]D25 = -1.1, c 0.355 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C29H36O8Na 535.2302, found 535.2305; 1H NMR (250 MHz, CDCl3) δ 8.14 – 8.07 (m, 2H), 8.07 – 7.99 (m, 2H), 7.60 – 7.50 (m, 2H), 7.50 – 7.39 (m, 4H), 5.25 – 5.10 (m, 2H), 4.95 (s, 1H), 4.18 – 4.03 (m, 1H), 3.91 – 3.76 (m, 1H), 3.64 (s, 3H), 2.42 (dt, J = 13.6, 4.0 Hz, 1H), 2.33 (t, J = 7.4 Hz, 2H), 2.20 (ddd, J = 13.7, 11.3, 3.1 Hz, 1H), 1.77 – 1.57 (m, 3H), 1.57 – 1.32 (m, 5H), 1.28 (d, J = 6.2 Hz, 3H), 1.18 (d, J = 6.0 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ 173.97, 165.61, 165.51, 133.09, 133.03, 129.87, 129.75, 129.71, 129.48, 128.30, 93.64, 72.39, 71.13, 70.53, 66.87, 51.31, 36.80, 33.87, 29.60, 28.98, 25.28, 24.78, 19.02, 17.76.
4.1.18. Synthesis of (2R,3R,5R,6S)-2-(((R)-9-methoxy-9-oxononan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (26)
The title compound was prepared in a manner similar to that of 46. Began with 100.0 mg methyl ester 22, column chromatography (2.5 g silica gel, CH2Cl2) yielded 88.5 mg (88%) of a colorless oil. [α]D25 = 35.5, c 0.80 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C30H38O8Na 549.2459, found 549.2475; 1H NMR (250 MHz, CDCl3) δ 8.11 (d, J = 7.1 Hz, 2H), 8.04 (d, J = 7.1 Hz, 2H), 7.65 – 7.51 (m, 2H), 7.51 – 7.38 (m, 4H), 5.24 – 5.06 (m, 2H), 4.95 (s, 1H), 4.11 (dq, J = 12.5, 6.2 Hz, 1H), 3.84 (q, J = 5.9 Hz, 1H), 3.65 (s, 3H), 2.42 (dt, J = 13.4, 4.0 Hz, 1H), 2.32 (t, J = 7.4 Hz, 2H), 2.27 – 2.11 (m, 1H), 1.74 – 1.32 (m, 10H), 1.28 (d, J = 6.2 Hz, 3H), 1.18 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 174.17, 165.75, 165.63, 133.19, 133.13, 129.99, 129.84, 129.82, 129.57, 128.40, 93.76, 72.58, 71.23, 70.65, 66.93, 51.42, 37.04, 34.04, 29.70, 29.22, 29.10, 25.53, 24.89, 19.12, 17.84.
4.1.19. Representative procedure for hydrolysis with 1M LiOH in tBuOH
see ref. 4b.
4.1.20. Synthesis of (R,E)-5-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)hex-2-enoic acid (asc-ΔC6)
See ref. 4b for procedure. Began with 17.6 mg 19, HPLC purification yielded 5.5 mg (58%) of a colorless oil. [α]D25 = -59.5, c 0.82 (MeOH); HRMS (m/z): [M-H]- calcd. for C12H19O6 259.1187, found 259.1181 1H NMR (400 MHz, MeOD) δ 6.82 (dt, J = 15.0, 7.3 Hz, 1H), 5.87 (d, J = 15.6 Hz, 1H), 4.64 (s, 1H), 3.90 (h, J = 6.1 Hz, 1H), 3.72 (s, 1H), 3.68 – 3.56 (m, 1H), 3.47 (td, J = 10.7, 9.8, 4.6 Hz, 1H), 2.48 – 2.31 (m, 2H), 1.93 (dt, J = 13.1, 4.0 Hz, 1H), 1.77 (ddd, J = 13.3, 11.4, 3.1 Hz, 1H), 1.20 (d, J = 6.2 Hz, 3H), 1.15 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 170.79, 146.60, 125.71, 98.08, 72.42, 71.44, 69.91, 68.39, 40.81, 35.92, 19.51, 18.15.
4.1.21. Synthesis of (R,E)-6-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)hept-2-enoic acid (asc-ΔC7)
See ref. 4b for procedure. Began with 258.1 mg 20, column chromatography (32 g silica gel, gradient run from 5% iPrOH in CH2Cl2 to 50% iPrOH in CH2Cl2, then 10% 1:1:1 H2O:CH3CN:MeOH in ethyl acetate) yielded 83.3 mg (58%) of a colorless oil. [α]D25 = -77.0, c 2.39 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C13H22O6Na 297.1309, found 297.1302; 1H NMR (400 MHz, MeOD) δ 6.97 (dt, J = 15.5, 6.8 Hz, 1H), 5.83 (d, J = 15.6 Hz, 1H), 4.65 (s, 1H), 3.86 – 3.77 (m, 1H), 3.72 (d, J = 3.7 Hz, 1H), 3.66 – 3.54 (m, 1H), 3.51 (td, J = 10.1, 9.3, 4.6 Hz, 1H), 2.44 – 2.25 (m, 2H), 1.95 (dt, J = 12.8, 4.0 Hz, 1H), 1.76 (ddd, J = 17.2, 8.7, 3.4 Hz, 1H), 1.73 – 1.59 (m, 2H), 1.21 (d, J = 6.0 Hz, 3H), 1.15 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 170.42, 150.66, 123.06, 97.49, 71.72, 71.41, 70.02, 68.44, 36.86, 36.08, 29.51, 19.29, 18.24.
4.1.22. Synthesis of (R,E)-7-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)oct-2-enoic acid (asc-ΔC8)
See ref. 4b for procedure. Began with 63.5 mg 21, HPLC purification yielded 26.7 mg (75%) of a colorless oil. [α]D25 = -60.2, c 0.165 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C14H24O6Na 311.1465, found 311.1469; 1H NMR (400 MHz, MeOD) δ 6.94 (dt, J = 14.7, 6.9 Hz, 1H), 5.81 (d, J = 15.6 Hz, 1H), 4.64 (s, 1H), 3.79 (q, J = 5.8 Hz, 1H), 3.71 (s, 1H), 3.66 – 3.56 (m, 1H), 3.51 (td, J = 10.2, 4.2 Hz, 1H), 2.30 – 2.18 (m, 2H), 1.94 (dt, J = 13.2, 3.8 Hz, 1H), 1.81 – 1.69 (m, 1H), 1.68 – 1.43 (m, 4H), 1.21 (d, J = 6.1 Hz, 3H), 1.12 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 170.27, 150.91, 122.94, 97.65, 72.39, 71.36, 70.05, 68.45, 37.94, 36.07, 36.04, 33.15, 25.57, 19.46, 18.26.
4.1.23. Synthesis of (R,E)-8-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)non-2-enoic acid (asc-ΔC9)
See ref. 4b for procedure. Began with 98.5 mg methyl ester 22, HPLC purification yielded 40.9 mg (72%) of a colorless oil. [α]D25 = -61.0, c 0.53 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C15H26O6Na 325.1622, found 325.1633; 1H NMR (400 MHz, MeOD) δ 6.93 (dt, J = 14.2, 6.8 Hz, 1H), 5.80 (d, J = 15.8 Hz, 1H), 4.63 (s, 1H), 3.84 – 3.73 (m, 1H), 3.71 (s, 1H), 3.61 (dq, J = 11.3, 5.5 Hz, 1H), 3.51 (td, J = 10.2, 9.5, 4.5 Hz, 1H), 2.23 (q, J = 6.5 Hz, 2H), 1.94 (dt, J = 12.9, 4.0 Hz, 1H), 1.81 – 1.69 (m, 1H), 1.62 – 1.37 (m, 6H), 1.20 (d, J = 6.1 Hz, 3H), 1.11 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, MeOD) δ 170.55, 150.86, 123.04, 97.69, 72.54, 71.35, 70.10, 68.56, 68.47, 38.21, 36.09, 33.21, 29.31, 26.57, 19.49, 18.26.
4.1.24. Synthesis of (R)-5-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)hexanoic acid (asc-C6)
See ref. 4b for procedure. Began with 35.0 mg 23, HPLC purification yielded 13.3 mg (70%) of a colorless oil. [α]D25 = -74.9, c 0.81 (MeOH); HRMS (m/z): [M-H]- calcd. for C12H21O6 261.1344, found 261.1342; 1H NMR (400 MHz, MeOH) δ 4.64 (s, 1H), 3.79 (d, J = 6.1 Hz, 1H), 3.71 (td, J = 3.1, 1.4 Hz, 1H), 3.62 (dq, J = 9.4, 6.2 Hz, 1H), 3.50 (ddd, J = 11.1, 9.3, 4.5 Hz, 1H), 2.34 – 2.25 (m, 2H), 1.94 (dt, J = 13.1, 3.7 Hz, 2H), 1.82 – 1.70 (m, 2H), 1.70 – 1.45 (m, 2H), 1.21 (d, J = 6.2 Hz, 3H), 1.12 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 174.92, 94.54, 69.23, 68.29, 67.00, 65.42, 34.81, 33.00, 32.23, 19.58, 16.29, 15.14.
4.1.25. Synthesis of (R)-6-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)heptanoic acid (asc-C7)
See ref. 4b for procedure. Began with 279.5 mg 24, column chromatography (32 g silica gel, gradient run from 2% iPrOH in CH2Cl2 to 50% iPrOH in CH2Cl2) yielded 80.0 mg (52%) of a colorless oil. [α]D25 = -90.9, c 1.06 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C13H24O6Na 299.1465, found 299.1472; 1H NMR (400 MHz, MeOD) δ 4.63 (s, 1H), 3.82 – 3.73 (m, 1H), 3.71 (s, 1H), 3.67 – 3.56 (m, 1H), 3.56 – 3.45 (m, 1H), 2.29 (t, J = 7.4 Hz, 2H), 1.94 (dt, J = 13.0, 3.8 Hz, 1H), 1.75 (ddd, J = 13.4, 11.3, 3.1 Hz, 1H), 1.68 – 1.35 (m, 6H), 1.21 (d, J = 6.1 Hz, 3H), 1.11 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 177.88, 97.63, 72.44, 71.30, 70.08, 68.48, 38.17, 36.08, 35.15, 26.52, 26.21, 19.45, 18.22.
4.1.26. Synthesis of (R)-7-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)octanoic acid (asc-C8)
See ref. 4b for procedure. Began with 68.0 mg 25, HPLC purification yielded 25.2 mg (65%) of a colorless oil. [α]D25 = -81.5, c 0.955 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C14H26O6Na 313.1622, found 313.1627; 1H NMR (250 MHz, MeOD) δ 4.63 (s, 1H), 3.81 – 3.66 (m, 2H), 3.66 – 3.44 (m, 2H), 2.28 (t, J = 7.4 Hz, 2H), 1.94 (dt, J = 13.0, 3.9 Hz, 1H), 1.75 (ddd, J = 13.3, 11.0, 3.0 Hz, 1H), 1.68 – 1.31 (m, 8H), 1.21 (d, J = 6.0 Hz, 3H), 1.11 (d, J = 6.0 Hz, 3H); 13C NMR (63 MHz, MeOD) δ 177.83, 97.61, 72.53, 71.27, 70.07, 68.47, 38.35, 36.07, 35.13, 30.33, 26.70, 26.18, 19.47, 18.23.
4.1.27. Synthesis of (R)-8-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)nonanoic acid (asc-C9)
See ref. 4b for procedure. Began with 70.0 mg 26, column chromatography (30 g silica gel, gradient run from 50% ethyl acetate in CH2Cl2 to 100% ethyl acetate, then from 2% 1:1:1 H2O:CH3CN:MeOH in ethyl acetate to 10% 1:1:1 H2O:CH3CN:MeOH in ethyl acetate) yielded 20.5 mg (51%) of a colorless oil. [α]D25 = -58.4, c 0.23 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C15H28O6Na 327.1778, found 327.1778; 1H NMR (400 MHz, MeOD) δ 4.63 (s, 1H), 3.81 – 3.73 (m, 1H), 3.71 (s, 1H), 3.62 (dq, J = 11.7, 6.0 Hz, 1H), 3.50 (td, J = 10.4, 9.6, 4.6 Hz, 1H), 2.27 (t, J = 7.2 Hz, 2H), 1.94 (dt, J = 12.8, 4.0 Hz, 1H), 1.81 – 1.70 (m, 1H), 1.65 – 1.29 (m, 10H), 1.20 (d, J = 6.2 Hz, 3H), 1.11 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 178.19, 97.70, 72.67, 71.32, 70.11, 68.49, 38.47, 36.09, 35.38, 30.53, 30.36, 26.87, 26.34, 19.50, 18.24.
4.1.28. Representative procedure for ozonolysis/Pinnick oxidation: synthesis of (R)-3-(((2R,3R,5R,6S)-3,5-bis(benzoyloxy)-6-methyltetrahydro-2H-pyran-2-yl)oxy)butanoic acid (27)
To a solution of 220.0 mg (0.512 mmol) 15 in 80 mL dichloromethane was added a stream of O3 at -78°C. After 4 min the solution turned blue, at which time the flow of O3 was ceased. O2 was then allowed to bubble through the mixture for an additional 4 minutes until the blue color had completely disappeared, and 518.7 mg Janda Jel™ polymer-supported PPh3 (~1.555 mmol phosphine) was then added. The reaction mixture was then allowed to warm to 23°C and stir for 16h under an atmosphere of nitrogen. 50 mL H2O was then added and the layers were separated. The aqueous layer was then extracted with 2 × 50 mL dichloromethane, and the organic layers were combined, dried over anhydrous Na2SO4 and filtered. Evaporation of volatiles afforded 240.0 mg of an oil that was redissolved in 10 mL tBuOH and 2.5 mL dimethyl sulfoxide. To this mixture was added dropwise a solution of 440.2 mg (4.867 mmol) 80% NaClO2 and 599.4 mg (3.842 mmol) NaH2PO4·H2O in 4.5 mL H2O over the course of 2 min. The resulting solution was stirred at 23°C for 90 min after which the volatiles were evaporated in vacuo. The crude mixture was then suspended in 20 mL H2O, and the pH was adjusted to 3 via the addition of 1N HCl. 25 mL EtOAc was then added and the layers were separated. The aqueous layer was then extracted with 3 × 25 mL EtOAc and the organic layers were combined, dried over anhydrous Na2SO4 and filtered. Evaporation of volatiles afforded 390 mg of an oil. Column chromatography (35 g silica gel, gradient run from 100% CH2Cl2 to 25% EtOAc in CH2Cl2) afforded 190 mg of a residue that was suspended in 30 mL hexanes and filtered in order to remove contaminant dimethyl sulfone. Evaporation afforded 85.8 mg (38 %, 2 steps) of a colorless oil. [α]D25 = 12.5, c 0.815 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C24H26O8Na 465.1520, found 465.1535; 1H NMR (500 MHz, CDCl3) δ 9.61 (s, br, 1H), 8.11 (d, J = 7.8 Hz, 2H), 8.04 (d, J = 7.7 Hz, 2H), 7.63 – 7.53 (m, 2H), 7.49 – 7.41 (m, 4H), 5.17 (td, J = 10.6, 4.6 Hz, 1H), 5.11 (s, 1H), 4.98 (s, 1H), 4.35 (h, J = 5.3 Hz, 1H), 4.15 (dq, J = 12.0, 6.2 Hz, 1H), 2.70 (dd, J = 15.5, 8.1 Hz, 1H), 2.52 (dd, J = 15.5, 5.0 Hz, 1H), 2.39 (dt, J = 13.4, 3.9 Hz, 1H), 2.16 (ddd, J = 13.9, 11.3, 3.1 Hz, 1H), 1.28 (d, J = 2.5 Hz, 3H), 1.26 (d, J = 2.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 177.25, 165.70, 165.60, 133.25, 133.15, 129.88, 129.80, 129.67, 129.57, 128.40, 128.38, 93.48, 70.90, 70.44, 68.88, 67.09, 42.10, 29.52, 18.96, 17.63.
4.1.29. Synthesis of (R)-4-(((2R,3R,5R,6S)-3,5-bis(benzoyloxy)-6-methyltetrahydro-2H-pyran-2-yl)oxy)pentanoic acid (28)
The title compound was prepared in a manner similar to that of 27. Began with 300.0 mg 16. Column chromatography (10 g silica gel, gradient run from 100% CH2Cl2 to 10% EtOAc in CH2Cl2) yielded 51.2 mg (16%, 2 steps) of a colorless oil. [α]D25 = -8.6, c 1.105 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C25H28O8Na 479.1676, found 479.1670; 1H NMR (250 MHz, CDCl3) δ 8.17 – 7.99 (m, 4H), 7.66 – 7.52 (m, 2H), 7.52 – 7.38 (m, 4H), 5.24 – 5.09 (m, 2H), 4.96 (s, 1H), 4.19 – 4.01 (m, 1H), 3.93 (h, J = 5.9 Hz, 1H), 2.55 (t, J = 7.4 Hz, 2H), 2.42 (dt, J = 13.6, 3.9 Hz, 1H), 2.18 (ddd, J = 13.9, 11.3, 3.2 Hz, 1H), 1.92 (q, J = 7.1 Hz, 2H), 1.28 (d, J = 6.1 Hz, 3H), 1.22 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 179.10, 165.90, 165.80, 133.40, 133.31, 130.01, 129.97, 129.87, 129.77, 128.57, 128.55, 93.60, 71.25, 71.20, 70.59, 67.32, 31.87, 30.30, 29.79, 18.94, 17.92.
4.1.30. Synthesis of (R)-3-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)butanoic acid (asc-C4)
See ref. 4b for procedure. Began with 35.7 mg 27, HPLC purification yielded 8.0 mg (42%) of a colorless oil. [α]D25 = -98.8, c 0.25 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C10H18O6Na 257.0996, found 257.1006; 1H NMR (250 MHz, MeOD) δ 4.65 (s, 1H), 4.24 (h, J = 6.0 Hz, 1H), 3.76 – 3.55 (m, 2H), 3.48 (td, J = 10.0, 4.6 Hz, 1H), 2.60 – 2.33 (m, 2H), 1.92 (dt, J = 13.1, 3.9 Hz, 1H), 1.74 (ddd, J = 13.2, 11.2, 3.0 Hz, 1H), 1.21 (d, J = 2.5 Hz, 3H), 1.18 (d, J = 2.6 Hz, 3H); 13C NMR (126 MHz, cd3od) δ 175.88, 97.54, 71.28, 70.02, 70.00, 68.47, 43.97, 35.99, 19.35, 18.16.
4.1.31. Synthesis of (R)-4-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)pentanoic acid (asc-C5)
See ref. 4b for procedure. Began with 20.6 mg 28, HPLC purification yielded 8.8 mg (79%) of a colorless oil. [α]D25 = -62.2, c 0.51 (MeOH) HRMS (m/z): [M+Na]+ calcd. for C11H19O6Na 271.1152, found 271.1150; 1H NMR (250 MHz, MeOD) δ 4.64 (s, 1H), 3.93 – 3.75 (m, 1H), 3.71 (q, J = 2.7 Hz, 1H), 3.68 – 3.41 (m, 2H), 2.41 (t, J = 7.4 Hz, 2H), 1.95 (dt, J = 13.1, 3.9 Hz, 1H), 1.87 – 1.67 (m, 3H), 1.21 (d, J = 5.9 Hz, 3H), 1.14 (d, J = 6.0 Hz, 3H).
4.1.32. Synthesis of (2R,3R,5R,6S)-2-(allyloxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (32)
The title compound was prepared in a manner similar to that of 16. Started with 400 mg (1.122 mmol) dibenzoyl ascarylose (1). Silica gel column chromatography (39 g silica gel, gradient run from 5% EtOAc in hexanes to 15% EtOAc) afforded 420.8 mg (95 %) of colorless oil. [α]D25= 2.6, c 1.33; HRMS (m/z): [M+Na] calcd. For C23H24O6Na 419.1465, found 419.1473; 1H NMR (250 MHz, Chloroform-d) δ 8.13 (d, J = 7.0 Hz, 2H), 8.05 (d, J = 7.0 Hz, 2H), 7.65 – 7.52 (m, 2H), 7.52 – 7.37 (m, 4H), 6.10 – 5.87 (m, 1H), 5.38 (dq, J = 17.2, 1.6 Hz, 1H), 5.31 – 5.13 (m, 3H), 4.91 (s, 1H), 4.29 (ddt, J = 12.9, 5.2, 1.5 Hz, 1H), 4.21 – 4.02 (m, 2H), 2.46 (dt, J = 14.3, 4.0 Hz, 1H), 2.26 (ddd, J = 13.8, 11.3, 3.2 Hz, 1H), 1.32 (d, J = 6.2 Hz, 3H). 13C NMR (62.5 MHz, CDCl3) δ 165.4, 165.3,133.6,133.04, 132.98, 129.8, 129.7, 129.6, 129.4, 128.2, 117.3, 95.4, 70.3, 68.0, 66.6, 29.5, 17.7.
4.1.33. Synthesis of (2R,3R,5R,6S)-2-(but-3-en-1-yloxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (33)
The title compound was prepared in a manner similar to that of 16. Started with 400 mg (1.129 mmol) dibenzoyl ascarylose (1), 0.16 mL (1.796 mmol) alcohol 30, 130 mg 3Å molecular sieves, and 0.61 mL (4.838 mmol) BF3·OEt2 in 12 mL CH2Cl2. Purified to obtain 429.8 mg (93%) of a colorless oil. [α]D25= 8.3, c 0.4; HRMS (m/z): [M+Na] calcd. for C24H26O6Na 433.1622, found 433.1629; 1H NMR (250 MHz, Chloroform-d) δ 8.12 (d, J = 7.0 Hz, 2H), 8.03 (d, J = 7.0 Hz, 2H), 7.54 (dt, J = 7.1, 3.6 Hz, 2H), 7.43 (td, J = 7.6, 4.5 Hz, 4H), 5.87 (ddt, J = 17.0, 10.2, 6.7 Hz, 1H), 5.31 – 5.15 (m, 3H), 5.10 (dd, J = 10.2, 1.9 Hz, 1H), 4.87 (s, 1H), 4.11 (dd, J = 9.8, 6.0 Hz, 1H), 3.81 (dt, J = 9.6, 6.8 Hz, 1H), 3.58 (dt, J = 9.7, 6.6 Hz, 1H), 2.41 (q, J = 6.9 Hz, 3H), 2.22 (ddd, J = 13.9, 11.3, 3.1 Hz, 1H), 1.31 (d, J = 6.2 Hz, 3H). 13C NMR (62.5 MHz, CDCl3) δ 165.4, 165.3, 134.6, 133.00, 132.94, 129.7, 129.6, 129.4, 128.2, 116.6, 96.1, 70.3, 66.9, 66.6, 33.8, 29.5, 17.7.
4.1.34. Synthesis of (2R,3R,5R,6S)-2-(hex-5-en-1-yloxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (34)
The title compound was prepared in a manner similar to that of 16. Started with 300 mg (0.842 mmol) dibenzoyl ascarylose (1), 0.16 mL (1.332 mmol) alcohol 31, 90 mg 3Å molecular sieves, and 0.46 mL (3.6 mmol) BF3·OEt2 in 9 mL CH2Cl2. Purified to obtain 262.6 mg (71%) of a colorless oil. [α]D25= 7.7, c 0.48; HRMS (m/z): [M+Na] calcd. For C26H30O6Na 461.1935, found 461.1945; 1H NMR (400 MHz, Chloroform-d) δ 8.11 (d, J = 7.2 Hz, 2H), 8.04 (d, J = 7.2 Hz, 2H), 7.64 – 7.52 (m, 2H), 7.51 – 7.42 (m, 4H), 5.84 (m, 1H), 5.24 – 5.12 (m, 2H), 5.10 – 4.94 (m, 2H), 4.83 (s, 1H), 4.07 (dq, J = 9.8, 6.3 Hz, 1H), 3.77 (dt, J = 9.7, 6.6 Hz, 1H), 3.52 (dt, J = 9.7, 6.4 Hz, 1H), 2.42 (dt, J = 13.6, 3.9 Hz, 1H), 2.21 (ddd, J = 13.9, 11.4, 3.2 Hz, 1H), 2.17 – 2.07 (m, 2H), 1.68 (dq, J = 9.2, 6.5, 5.6 Hz, 2H), 1.59 – 1.46 (m, 2H), 1.30 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.94, 165.85, 138.8, 133.5, 133.4, 130.08, 130.05, 129.8, 128.6, 114.9, 96.6, 70.8, 67.9, 67.0, 33.7, 30.0, 29.2, 25.7, 18.1.
4.1.35. Synthesis of (2R,3R,5R,6S)-2-(((E)-4-methoxy-4-oxobut-2-en-1-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (35)
See ref. 2d for representative procedure. Started with 262.8 mg (0.663 mmol) 32, 0.30 mL (3.3 mmol) methyl acrylate, and 56.3 mg (66.3 μmol) Grubbs second generation catalyst in 22.2 mL CH2Cl2. Purified to obtain 112.4 mg (37%) of a colorless oil. [α]D25= 4.5, c 0.5; HRMS (m/z): [M+Na] calcd. For C25H26O8Na 477.1531, found 477.1520; 1H NMR (250 MHz, Chloroform-d) δ 8.10 (d, J = 7.3 Hz, 2H), 8.03 (d, J = 7.3 Hz, 2H), 7.64 – 7.51 (m, 2H), 7.51 – 7.37 (m, 4H), 7.02 (dt, J = 15.7, 4.3 Hz, 1H), 6.16 (dt, J = 15.7, 2.0 Hz, 1H), 5.32 – 5.11 (m, 2H), 4.89 (s, 1H), 4.45 (ddd, J = 16.0, 4.3, 2.1 Hz, 1H), 4.24 (ddd, J = 16.1, 4.6, 2.1 Hz, 1H), 4.13 – 3.98 (m, 1H), 3.76 (s, 3H), 2.46 (dt, J = 13.6, 4.0 Hz, 1H), 2.23 (ddd, J = 14.0, 11.3, 3.2 Hz, 1H), 1.29 (d, J = 6.2 Hz, 3H). 13C NMR (62.5 MHz, CDCl3) δ 166.4, 165.50, 165.46, 143.3, 133.2, 133.1, 129.8, 129.6, 129.5, 128.4, 121.0, 96.0, 70.2, 70.1, 67.0, 65.7, 51.6, 29.6, 17.7.
4.1.36. Synthesis of (2R,3R,5R,6S)-2-(((E)-5-methoxy-5-oxopent-3-en-1-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (36)
See ref. 2d for representative procedure. Started with 429.8 mg (1.047 mmol) 33, 0.47 mL (5.2 mmol) methyl acrylate, and 88.9 mg (104 μmol) Grubbs second generation catalyst in 36.6 mL CH2Cl2. Purified to obtain 258.8 mg (53%) of a colorless oil. [α]D25= 11.2, c 0.2; HRMS (m/z): [M+Na] calcd. For C26H28O8Na 491.1676, found 491.1672; 1H NMR (250 MHz, Chloroform-d) δ 8.09 (d, J = 7.3 Hz, 2H), 8.02 (d, J = 7.3 Hz, 2H), 7.62 – 7.49 (m, 2H), 7.49 – 7.35 (m, 4H), 7.00 (dt, J = 15.7, 7.0 Hz, 1H), 5.94 (m, 1H), 5.28 – 5.08 (m, 2H), 4.84 (s, 1H), 4.04 (m, 1H), 3.86 (dt, J = 9.8, 6.5 Hz, 1H), 3.71 (s, 3H), 3.63 (dt, J = 9.8, 6.2 Hz, 1H), 2.62 – 2.47 (m, 2H), 2.41 (dt, J = 13.5, 4.0 Hz, 1H), 2.17 (ddd, J = 13.9, 11.4, 3.2 Hz, 1H), 1.28 (d, J = 6.2 Hz, 3H). 13C NMR (62.5 MHz, CDCl3) δ 166.5, 165.5, 165.4, 145.2, 133.1, 133.0, 129.73, 129.67, 129.6, 129.4, 128.3, 122.8, 96.2, 70.2, 66.8, 65.6, 51.3, 32.2, 29.5, 17.7.
4.1.37. Synthesis of (2R,3R,5R,6S)-2-(((E)-7-methoxy-7-oxohept-5-en-1-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (37)
See ref. 2d for representative procedure. Started with 100 mg (0.228 mmol) 34. Evaporation of the volatiles afforded 165.2 mg of ruby oil. Silica gel column chromatography (12 g silica gel, gradient run from 50% hexanes in CH2Cl2 to 100% CH2Cl2) afforded 60.6 mg (53%) of colorless oil. [α]D25= 4.1, c 0.48; HRMS (m/z): [M+Na] calcd. For C27H32O6Na 519.1989, found 519.1996. 1H NMR (400 MHz, Chloroform-d) δ 8.11 (d, J = 8.4, 1.6 Hz, 2H), 8.05 (d, 2H), 7.64 – 7.54 (m, 2H), 7.47 (ddd, J = 8.8, 7.1, 5.2 Hz, 4H), 7.01 (dt, J = 15.7, 7.0 Hz, 1H), 5.88 (dt, J = 15.6, 1.6 Hz, 1H), 5.25 – 5.13 (m, 2H), 4.83 (s, 1H), 4.06 (dq, J = 9.9, 6.2 Hz, 1H), 3.78 (dt, J = 9.8, 6.3 Hz, 1H), 3.73 (s, 3H), 3.52 (dt, J = 9.8, 6.1 Hz, 1H), 2.48 – 2.37 (m, 1H), 2.29 (qd, J = 7.1, 1.6 Hz, 2H), 2.21 (ddd, J = 13.5, 11.3, 3.1 Hz, 1H), 1.74 – 1.56 (m, 4H), 1.30 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.0 165.7, 165.6, 149.0, 133.24, 133.16, 130.0, 129.84, 129.79, 129.6, 128.4, 121.3, 96.4, 70.5, 67.4, 66.8, 51.4, 31.9, 29.7, 28.9, 24.7, 17.9.
4.1.38. Synthesis of (2R,3R,5R,6S)-2-(4-methoxy-4-oxobutoxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (38)
The title compound was prepared in a manner similar to that of 23. Started with 89.7 mg (0.197 mmol) 35, 9.3 mg 10% palladium on activated carbon in 9 mL ethyl acetate. Purified to obtain 85.7 mg (95%) colorless oil. [α]D25= 5.8, c 0.2; HRMS (m/z): [M+Na] calcd. For C25H28O8Na 479.1676, found 479.1700; 1H NMR (250 MHz, Chloroform-d) δ 8.10 (d, J = 7.0, 1.4 Hz, 2H), 8.04 (d, J = 7.1, 1.4 Hz, 2H), 7.65 – 7.51 (m, 2H), 7.51 – 7.38 (m, 4H), 5.27 – 5.09 (m, 2H), 4.82 (s, 1H), 4.14 – 3.97 (m, 1H), 3.82 (dt, J = 9.8, 6.2 Hz, 1H), 3.70 (s, 3H), 3.54 (dt, J = 9.7, 6.1 Hz, 1H), 2.55 – 2.34 (m, 3H), 2.19 (ddd, J = 13.8, 11.3, 3.2 Hz, 1H), 1.99 (h, J = 6.5 Hz, 2H), 1.29 (d, J = 6.2 Hz, 3H). 13C NMR (62.5 MHz, CDCl3) δ 173.8, 165.6, 165.5, 133.14, 133.07, 129.8, 129.73, 129.68, 129.5, 128.3, 96.3, 70.4, 67.2, 66.7, 51.4, 33.6, 29.6, 28.8, 21.7, 17.8.
4.1.39. Synthesis of (2R,3R,5R,6S)-2-((5-methoxy-5-oxopentyl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (39)
The title compound was prepared in a manner similar to that of 23. Started with 131.1 mg (0.280 mmol) 36, 13.5 mg palladium on activated carbon 10% palladium in 13.44 mL ethyl acetate. Purified to obtain 120.1 mg (91%) colorless oil. [α]D25= 12.4, c 0.3; HRMS (m/z): [M+Na] calcd. For C26H30O8Na 493.1833, found 493.1826 1H NMR (250 MHz, Chloroform-d) δ 8.10 (d, 2H), 8.03 (d, J = 7.0, 1.6 Hz, 2H), 7.64 – 7.51 (m, 2H), 7.44 (ddd, J = 8.8, 6.9, 3.6 Hz, 4H), 5.27 – 5.09 (m, 2H), 4.82 (s, 1H), 4.14 – 3.99 (m, 1H), 3.78 (dt, J = 9.4, 6.0 Hz, 1H), 3.67 (s, 3H), 3.51 (dt, J = 9.7, 5.7 Hz, 1H), 2.50 – 2.31 (m, 3H), 2.20 (ddd, J = 13.9, 11.3, 3.1 Hz, 1H), 1.75 (ddt, J = 12.3, 7.9, 4.7 Hz, 4H), 1.29 (d, J = 6.2 Hz, 3H). 13C NMR (62.5 MHz, CDCl3) δ 173.8, 165.6, 165.5, 133.14, 133.07, 129.8, 129.73, 129.68, 129.5, 128.3, 96.3, 70.4, 67.2, 66.7, 51.4, 33.6, 29.6, 28.8, 21.7, 17.8.
4.1.40. Synthesis of (2R,3R,5R,6S)-2-((7-methoxy-7-oxoheptyl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate of (40)
The title compound was prepared in a manner similar to that of 23. Started with 103.4 mg (0.208 mmol) 37, 10.6 mg 10% palladium on activated carbon in 10.6 mL ethyl acetate. Purified to obtain 84.4 mg (81%) colorless oil. [α]D25= 2.8, c 0.3; HRMS (m/z): [M+Na] calcd. For C28H34O8Na 521.2146, found 521.2163; 1H NMR (400 MHz, Chloroform-d) δ 8.11 (d, J = 7.2 Hz, 2H), 8.04 (d, J = 7.2 Hz, 2H), 7.64 – 7.54 (m, 2H), 7.52 – 7.42 (m, 4H), 5.24 – 5.12 (m, 2H), 4.82 (s, 1H), 4.06 (dq, J = 9.8, 6.3 Hz, 1H), 3.76 (dt, J = 9.7, 6.7 Hz, 1H), 3.67 (s, 3H), 3.50 (dt, J = 9.7, 6.5 Hz, 1H), 2.41 (dt, J = 13.7, 4.0 Hz, 1H), 2.34 (t, J = 7.5 Hz, 2H), 2.21 (ddd, J = 13.8, 11.4, 3.2 Hz, 1H), 1.67 (m, 4H), 1.41 (tdd, J = 10.6, 7.3, 4.2 Hz, 4H), 1.30 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.5, 166.0, 165.9, 133.5, 133.4, 130.2, 130.09, 130.06, 129.8, 128.7, 96.6 70.8, 68.0, 67.0, 51.7, 34.2, 30.0, 29.5, 29.2, 26.1, 25.1, 18.1.
4.1.41. Synthesis of (E)-4-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)but-2-enoic acid (asc-ωΔC4)
See ref. 4b for representative procedure. Started with 44.3 mg (0.097 mmol) 35 and 7.8 mL (1N) LiOH in 7.8 mL of tert-Butanol. Purified to obtain 7.0 mg (31%) colorless oil. [α]D25= -100.2, c 0.1; HRMS (m/z): [M+Na] calcd. For C10H16O6Na 255.0839, found 255.0841; 1H NMR (400 MHz, Methanol-d4) δ 6.89 (dt, J = 15.6, 4.5 Hz, 1H), 6.04 (d, J = 15.6, 2.1 Hz, 1H), 4.57 (s, 1H), 4.34 (ddd, J = 16.0, 4.3, 2.1 Hz, 1H), 4.16 (ddd, J = 16.0, 4.7, 2.0 Hz, 1H), 3.83 (q, J = 2.6 Hz, 1H), 3.54 (dq, J = 9.9, 5.0, 4.2 Hz, 2H), 3.34 (s, 1H), 1.98 (dt, J = 13.0, 3.6 Hz, 1H), 1.80 (ddd, J = 13.4, 10.6, 3.1 Hz, 1H), 1.23 (d, J = 5.6 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 170.8, 144.6, 123.9, 100.2, 71.4, 69.4, 68.4, 66.7, 36.2, 18.2.
4.1.42. Synthesis of (E)-5-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)pent-2-enoic acid (asc-ωΔC5)
See ref. 4b for representative procedure. Started with 102.1 mg (0.218 mmol) 36 and 17.9 mL (1N) LiOH in 17.9 mL of tert-Butanol. Purified to obtain 23 mg (43%) colorless oil. [α]D25= -53.8, c 0.3; HRMS (m/z): [M+Na] calcd. For C11H18O6Na 269.0996, found 269.1003;1H NMR (400 MHz, Methanol-d4) δ 6.96 (dt, J = 15.7, 6.9 Hz, 1H), 5.88 (d, J = 15.7 Hz, 1H), 4.52 (s, 1H), 3.86 – 3.72 (m, 2H), 3.54 (ddt, J = 14.9, 10.5, 5.3 Hz, 3H), 3.35 – 3.25 (m, 1H), 2.50 (q, J = 6.5 Hz, 2H), 1.94 (dt, J = 13.0, 3.8 Hz, 1H), 1.75 (ddd, J = 13.2, 10.5, 3.2 Hz, 1H), 1.22 (d, J = 5.7 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 170.1, 147.7, 124.4, 100.7, 71.3, 69.5, 68.4, 66.9, 36.1, 33.6, 18.2.
4.1.43. Synthesis of (E)-7-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)hept-2-enoic acid (asc-ωΔC7)
See ref. 4b for representative procedure. Started with 42 mg (0.082 mmol) 37 and 7.4 mL (1N) LiOH in 7.4 mL of tert-Butanol. Purified to obtain 8.5 mg (36%) colorless oil. [α]D25= -49.0, c 0.2; HRMS (m/z): [M-H]- calcd. For C13H22O6Na 273.1344, found 273.1343; 1H NMR (400 MHz, Methanol-d4) δ 6.92 (dt, J = 15.6, 7.0 Hz, 1H), 5.81 (d, J = 15.6 Hz, 1H), 4.49 (s, 1H), 3.76 (td, J = 3.1, 1.5 Hz, 1H), 3.70 (dt, J = 9.9, 6.1 Hz, 1H), 3.60 – 3.38 (m, 3H), 2.25 (qd, J = 7.0, 1.6 Hz, 2H), 1.99 – 1.89 (m, 1H), 1.76 (ddd, J = 13.1, 10.9, 3.1 Hz, 1H), 1.60 (tdd, J = 11.9, 8.5, 5.3 Hz, 4H), 1.22 (d, J = 5.9 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 170.6, 150.5, 100.6, 71.1, 69.6, 68.5, 68.1, 49.6, 36.2, 33.0, 30.3, 26.2, 18.3.
4.1.44. Synthesis of 4-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)butanoic acid (asc-ωC4)
See ref. 4b for representative procedure. Started with 48.6 mg (0.106 mmol) 38 and 8.5 mL (1N) LiOH in 8.5 mL of tert-butanol. Purified to obtain 7 mg (28%) colorless oil. [α]D25= -88.5, c 0.2; HRMS (m/z): [M+Na] calcd. For C11H20O6Na 257.0996, found 257.1004; 1H NMR (400 MHz, Methanol-d4) δ 4.49 (s, 1H), 3.76 (tt, J = 3.9, 1.9 Hz, 1H), 3.74 – 3.68 (m, 1H), 3.60 – 3.39 (m, 3H), 2.37 (t, J = 7.3 Hz, 2H), 1.99 – 1.82 (m, 3H), 1.76 (ddd, J = 13.2, 10.9, 3.1 Hz, 1H), 1.21 (d, J = 5.9 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 177.8, 100.6, 71.1, 69.5, 68.5, 67.5, 36.1, 32.3, 26.4, 18.2.
4.1.45. Synthesis of 5-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)pentanoic acid (asc-ωC5)
See ref. 4b for representative procedure. Started with 40.3 mg (0.095 mmol) 39 and 7.8 mL (1N) LiOH in 7.8 mL of tert-Butanol. Purified to obtain 5.8 mg (23%) colorless oil. [α]D25= -64.8, c 0.3; HRMS (m/z): [M+Na] calcd. For C11H20O6Na 271.1152, found 271.1143; 1H NMR (400 MHz, Methanol-d4) δ 4.50 (s, 1H), 3.77 (q, J = 2.6 Hz, 1H), 3.72 (dd, J = 9.8, 6.2 Hz, 1H), 3.62 – 3.38 (m, 3H), 2.32 (t, J = 6.7 Hz, 2H), 1.95 (dt, J = 13.0, 3.8 Hz, 1H), 1.83 – 1.76 (m, 1H), 1.76 – 1.56 (m, 5H), 1.23 (d, J = 5.9 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 177.8, 100.4, 70.9, 69.4, 68.3, 67.9, 36.0, 34.9, 30.1, 23.1, 18.1.
4.1.46. Synthesis of 7-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)heptanoic acid (asc-ωC7)
See ref. 4b for representative procedure. Started with 53 mg (0.11 mmol) 40 and 9.4 mL (1N) LiOH in 9.4 mL of tert-Butanol. Purified to obtain 12.7 mg (43%) colorless oil. [α]D25= -63.0, c 0.2; HRMS (m/z): [M-H]- calcd. For C13H24O6Na 275.1500, found 275.1497; 1H NMR (400 MHz, Methanol-d4) δ 4.48 (s, 1H), 3.75 (td, J = 3.1, 1.4 Hz, 1H), 3.68 (dt, J = 9.7, 6.6 Hz, 1H), 3.60 – 3.45 (m, 2H), 3.41 (dt, J = 9.6, 6.2 Hz, 1H), 2.28 (t, J = 7.4 Hz, 2H), 1.99 – 1.89 (m, 1H), 1.76 (ddd, J = 13.3, 10.9, 3.1 Hz, 1H), 1.61 (ddt, J = 10.3, 7.3, 4.5 Hz, 4H), 1.47 – 1.32 (m, 4H), 1.22 (d, J = 5.9 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 178.0, 100.6, 71.0, 69.6, 68.5, 68.4, 36.2, 35.2, 30.7, 30.2, 27.2, 26.3, 18.3.
4.1.47. Representative cross metathesis procedure: synthesis of (2R,3R,5R,6S)-2-(((2R,7R,E)-7-hydroxyoct-4-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (41)
This procedure is representative of all cross metathesis procedures not involving methyl acrylate. To a solution of 235.2 mg (0.5541 mmol) alkene 15 and 0.57 mL (5.5 mmol) alcohol 11 in 17 mL CH2Cl2 at reflux was added 47.0 mg (0.0553 mmol) Grubbs 2nd generation ruthenium catalyst at once. The resulting solution was allowed to stir at reflux for 4 h and allowed to cool to 25°C. The reaction mixture was concentrated to 381.2 mg of brown oil. Silica gel column chromatography (silica gel, gradient run from 10% ethyl acetate in hexanes to 35% ethyl acetate in hexanes) afforded 223.6 mg (84%) of a yellow oil. [α]D25 = 24.5, c 0.965 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C28H34O7Na 505.2197, found 505.2210; 1H NMR (250 MHz, CDCl3): δ 8.11 (d, J = 7.1 Hz, 2H), 8.05 (d, J = 7.0 Hz, 2H), 7.57 (m, 2H), 7.46 (m, 4H), 5.59 (m, 2H), 5.16 (m, 2H), 4.95 (s, 1H), 4.14 (dq, J = 9.9, 6.2 Hz, 1H), 3.86 (m, 2H), 2.27 (m, 6H), 1.28 (d, J = 6.2 Hz, 3H), 1.20 (d, J =6.3 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H).
4.1.48. Synthesis of (2R,3R,5R,6S)-2-(((2R,8R,E)-8-hydroxynon-5-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (42)
The title compound was prepared in a manner similar to that of 43. Started with 1.1364 g (2.5915 mmol) alkene 16, 2.67 mL (25.9 mmol) alcohol 11, and 220.1 mg (0.2589 mmol) Grubbs 2nd generation ruthenium catalyst in 80 mL CH2Cl2. Purified to obtain 1.0211 g (79%) of a yellow oil. [α]D25 = -6.4, c 11.47 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C29H36O7Na 519.2353, found 519.2356; 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.4 Hz, 2H), 8.03 (d, J = 7.5 Hz, 2H), 7.54 (m, 2H), 7.43 (m, 4H), 5.53 (m, 2H), 5.18 (m, 2H), 4.95 (s, 1H), 4.13 (dq, J = 12.1, 6.3 Hz, 1H), 3.83 (m, 2H), 2.42 (dt, J = 13.0, 4.1 Hz, 1H), 2.18 (m, 5H), 1.71 (m, 1H), 1.58 (m, 1H), 1.28 (d, J = 6.2 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H), 1.17 (d, J = 6.3 Hz, 3H).
4.1.49. Synthesis of (2R,3R,5R,6S)-2-(((2R,9R,E)-9-hydroxydec-5-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (43)
The title compound was prepared in a manner similar to that of 43. Started with 18.8 mg (0.0429 mmol) alkene 16, 52 μL (0.43 mmol) alcohol 12, and 3.7 mg (0.0044 mmol) Grubbs 2nd generation ruthenium catalyst in 1.3 mL CH2Cl2. Purified to obtain 20.4 mg (93%) of a yellow oil. [α]D25 = -23.7, c 0.76(CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C30H38O7Na 533.2510, found 533.2522; 1H NMR (250 MHz, CDCl3): δ 8.12 (d, J = 7.2 Hz, 2H), 8.05 (d, J = 7.0 Hz, 2H), 7.59 (m, 2H), 7.47 (m, 4H), 5.53 (m, 2H), 5.16 (m, 2H), 4.96 (s, 1H), 4.12 (dq, J = 12.7, 6.2 Hz, 1H), 3.85 (m, 2H), 2.42 (dt, J = 13.4, 4.0 Hz, 1H), 2.21 (m, 5H), 1.55 (m, 5H), 1.28 (d, J = 6.2 Hz, 3H), 1.20 (m, 6H).
4.1.50. Synthesis of (2R,3R,5R,6S)-2-(((2R,7R)-7-hydroxyoctan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (44)
The title compound was prepared in a manner similar to that of 23. Started with 168.7 mg (0.3496 mmol) unsaturated alcohol 41 and 25.8 mg 10 % palladium on activated carbon in 23 mL EtOAc. Obtained 160.1 mg (95%) of a colorless oil that required no further purification. [α]D25 = -27, c 0.18 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C28H36O7Na 507.2353, found 507.2348; 1H NMR (400 MHz, CDCl3): δ 8.11 (m, 2H), 8.05 (m, 2H), 7.58 (m, 2H), 7.46 (td, J = 7.7, 3.5 Hz, 4H), 5.17 (m, 2H), 4.95 (s, 1H), 4.12 (dq, J = 9.8, 6.2 Hz, 1H), 3.85 (m, 2H), 2.42 (dt, J = 13.6, 4.0 Hz, 1H), 2.21 (ddd, J = 13.9, 11.4, 3.2 Hz, 1H), 1.50 (m, 8H), 1.28 (d, J = 6.2 Hz, 3H), 1.21 (d, J = 6.2 Hz, 3H); 1.19 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 166.0; 165.9; 133.5; 133.4; 130.2; 130.1; 129.8; 128.7; 94.0; 72.8; 71.5; 70.9; 68.3; 67.2; 39.5; 37.3; 29.9; 26.0 (2C); 23.8; 19.4; 18.1.
4.1.51. Synthesis of (2R,3R,5R,6S)-2-(((2R,8R)-8-hydroxynonan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (45)
The title compound was prepared in a manner similar to that of 46. Started with 786.6 mg (1.584 mmol) unsaturated alcohol 42 and 94.2 mg 10 % palladium on activated carbon in 88 mL EtOAc. Obtained 743.4 mg (94%) of a colorless oil that required no further purification. [α]D25 = 7.04, c 1.81 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C29H38O7Na 521.2510, found 521.2506; 1H NMR (250 MHz, CDCl3) δ 8.11 (m, 2H), 8.04 (m, 2H), 7.58 (m, 2H), 7.42 (m, 4H), 5.18 (m, 2H), 4.95 (s, 1H), 4.12 (m, 1H), 3.84 (m, 2H), 2.42 (dt, J = 14.2, 3.9 Hz, 1H), 2.24 (m, 1H), 1.49 (d, J = 46.5 Hz, 10H), 1.28 (d, J = 6.3 Hz, 3H), 1.21 (d, J = 2.6 Hz, 3H), 1.18 (d, J = 2.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 166.0; 165.9; 133.41; 133.36; 130.2; 130.0; 129.8; 128.6; 94.0; 72.8; 71.4; 70.9; 68.3; 67.1; 39.5; 37.3; 29.9; 29.8; 25.9 (2C); 23.7; 19.3; 18.1.
4.1.52. Synthesis of (2R,3R,5R,6S)-2-(((2R,9R)-9-hydroxydecan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (46)
The title compound was prepared in a manner similar to that of 23. Started with 376.7 mg (0.7382 mmol) unsaturated alcohol 43 and 44.9 mg 10 % palladium on activated carbon in 41 mL EtOAc. Obtained 375.4 mg (99%) of a colorless oil that required no further purification. [α]D25 = -36, c 0.12 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C30H40O7Na 535.2666, found 535.2654; 1H NMR (250 MHz, CDCl3): δ 8.11 (d, J = 7.7 Hz, 2H), 8.04 (d, J = 7.7 Hz, 2H), 7.59 (m, 2H), 7.47 (m, 4H), 5.18 (m, 2H), 4.95 (s, 1H), 4.12 (dq, J = 12.0, 6.1 Hz, 1H), 3.83 (m, 2H), 2.42 (dt, J = 13.6, 4.1 Hz, 1H), 2.19 (m, 1H), 1.46 (m, 13H), 1.28 (d, J = 6.2 Hz, 3H), 1.19 (d, J = 6.0 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 166.0; 165.9; 133.43; 133.39; 130.2; 130.1; 129.8; 128.6; 94.0; 72.9; 71.5; 70.9; 68.3; 67.2; 39.6; 37.3; 29.9; 29.84; 29.77; 26.0; 25.9; 23.7; 19.4; 18.1.
4.1.53. Representative procedure for PCC oxidation: synthesis of (2S,3R,5R,6R)-2-methyl-6-(((R)-7-oxooctan-2-yl)oxy)tetrahydro-2H-pyran-3,5-diyl dibenzoate (47)
To a suspension of 129.7 mg (0.6017 mmol) pyridinium chlorochromate and 111.2 mg 4Å molecular sieves in 21 mL CH2Cl2 at 0°C was added 72.2 mg (0.149 mmol) saturated alcohol 44 at once. The resulting suspension was allowed to stir at 0°C for 2 h. The reaction mixture was concentrated to 89.2 mg of dark red oil. Silica gel column chromatography (30.24 g silica gel, gradient run from 7.5% diethyl ether in CH2Cl2 to 12% diethyl ether in CH2Cl2) afforded 71.3 mg (99%) of a colorless oil. [α]D25 = -66, c 0.065 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C28H34O7Na 505.2197, found 505.2181; 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.7 Hz, 2H), 8.05 (d, J = 7.4 Hz, 2H), 7.57 (t, J = 7.5 Hz, 2H), 7.45 (m, 4H), 5.16 (m, 2H), 4.94 (s, 1H), 4.10 (dq, J = 12.3, 6.4 Hz, 1H), 3.83 (m, 1H), 2.47 (t, J = 7.4 Hz, 2H), 2.41 (dt, J = 13.7, 3.9 Hz, 1H), 2.21 (m, 1H), 2.15 (s, 3H), 1.56 (m, 6H), 1.28 (d, J = 6.3 Hz, 3H), 1.18 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 209.1; 166.0; 165.9; 133.5; 133.4; 130.2; 130.1; 129.8; 128.7; 94.0; 72.7; 71.4; 70.9; 67.2; 43.8; 37.1; 30.2; 29.9; 25.5; 24.0; 19.3; 18.1.
4.1.54. Synthesis of (2S,3R,5R,6R)-2-methyl-6-(((R)-8-oxononan-2-yl)oxy)tetrahydro-2H-pyran-3,5-diyl dibenzoate (48)
The title compound was prepared in a manner similar to that of 47. Started with 605.7 mg (2.810 mmol) pyridinium chlorochromate, 328.3 mg (0.6584 mmol) saturated alcohol 45, and 513.8 mg 4Å molecular sieves in 120 mL CH2Cl2. Purified to obtain 313.6 mg (96%) of a colorless oil. [α]D25 = 3.33, c 3.83 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C29H36O7Na 519.2353, found 519.2363; 1H NMR (400 MHz, CDCl3): δ 8.11 (m, 2H), 8.05 (m, 2H), 7.58 (td, J = 7.3, 2.2 Hz, 2H), 7.46 (td, J = 7.4, 2.9 Hz, 4H), 5.16 (m, 2H), 4.95 (s, 1H), 4.11 (m, 1H), 3.84 (m, 1H), 2.44 (m, 3H), 2.15 (m, 4H), 1.50 (m, 8H), 1.29 (d, J = 6.1 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 209.2; 166.0; 165.9; 133.41; 133.37; 130.2; 130.0; 129.8; 128.6; 93.9; 72.7; 71.4; 70.8; 67.2; 43.9; 37.1; 30.1; 29.9; 29.4; 25.8; 23.9; 19.3; 18.1.
4.1.55. Synthesis of (2S,3R,5R,6R)-2-methyl-6-(((R)-9-oxodecan-2-yl)oxy)tetrahydro-2H-pyran-3,5-diyl dibenzoate (49)
The title compound was prepared in a manner similar to that of 47. Started with 123.4 mg (0.5725 mmol) pyridinium chlorochromate, 58.6 mg (0.114 mmol) saturated alcohol 46, and 93.9 mg 4Å molecular sieves in 20 mL CH2Cl2. Purified to obtain 52.0 mg (89%) of a colorless oil. [α]D25 = 26.6, c 0.775 (CH2Cl2); HRMS (m/z): [M+Na]+calcd. for C30H38O7Na 533.2510, found 533.2525; 1H NMR (250 MHz, CDCl3) δ 8.12 (d, J = 7.0 Hz, 2H), 8.05 (d, J = 7.1 Hz, 2H), 7.57 (m, 2H), 7.46 (m, 4H), 5.17 (m, 2H), 4.95 (s, 1H), 4.12 (dq, J = 12.6, 6.6 Hz, 1H), 3.84 (m, 1H), 2.43 (m, 3H), 2.14 (m, 4H), 1.54 (m, 10H), 1.29 (d, J = 6.2 Hz, 3H), 1.19 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 209.0; 165.7; 165.6; 133.11; 133.07; 129.9; 129.7; 129.5; 128.3; 93.6; 72.5; 72.4; 71.1; 70.5; 66.8; 43.6; 36.9; 36.8; 29.7; 29.6; 29.3; 29.0; 25.5; 23.6; 19.2; 17.8.
4.1.56. Representative procedure for methanolysis with K2CO3: synthesis of (2R,3R,5R,6S)-2-(((2R,7R)-7-hydroxyoctan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diol (asc-C8-OH)
To a suspension of 10.8 mg (0.0223 mmol) saturated alcohol 44 in 1.5 mL MeOH was added 1.8 mg (0.013 mmol) K2CO3 at once. The resulting solution was allowed to stir at reflux for 5 h and then allowed to cool to 25°C. The reaction mixture was concentrated to 17.3 mg of colorless oil. HPLC purification afforded 2.7 mg (44%) of a colorless oil. [α]D25 = -22, c 0.14 (CH3OH); HRMS (m/z): [M+Na]+calcd. for C14H28O5Na 299.1829, found 299.1838; 1H NMR (400 MHz, CD3OD): δ 4.64 (s, 1H), 3.74 (m, 5H), 3.51 (ddd, J = 11.4, 9.3, 4.5 Hz, 1H), 1.94 (dddd, J = 13.0, 4.4, 3.2, 1.0 Hz, 1H), 1.76 (ddd, J = 13.1, 11.3, 3.1 Hz, 1H), 1.44 (m, 8H), 1.21 (m, 3H), 1.14 (dd, J = 6.3, 1.9 Hz, 3H), 1.12 (d, J = 6.0 Hz, 3H); 13C NMR (400 MHz, CDCl3): δ 96.1; 71.6; 70.0; 69.5; 68.34; 68.28; 39.5; 37.2; 35.4; 26.0; 25.9; 24.0; 19.2; 17.9.
4.1.57. Synthesis of (2R,3R,5R,6S)-2-(((2R,8R)-8-hydroxynonan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diol (asc-C9-OH)
The title compound was prepared in a manner similar to that of asc-C10-OH. Started with 344.5 mg (0.6909 mmol) alcohol 45 and 49.0 mL sat. 1 M LiOH in 49.0 mL MeOH. Purified to obtain 178.5 mg (89%) of a colorless oil. [α]D25 = -49.1, c 2.53 (CH3OH); HRMS (m/z): [M+Na]+calcd. for C15H30O5Na 313.1985, found 313.1979; 1H NMR (400 MHz, CD3OD): δ 4.63 (s, 1H), 3.69 (m, 4H), 3.51 (ddd, J = 11.4, 9.4, 4.6 Hz, 1H), 1.94 (dt, J = 13.1, 3.9 Hz, 1H), 1.76 (ddd, J = 13.2, 11.3, 3.0 Hz, 1H), 1.42 (m, 10H), 1.21 (d, J = 6.1 Hz, 3H), 1.12 (dd, J = 10.3, 6.1 Hz, 6H); 13C NMR (100 MHz, CD3OD): δ 97.7; 72.7; 71.3; 70.1; 68.7; 68.5; 40.3; 38.5; 36.1; 30.9; 27.03; 27.0; 23.6; 19.5; 18.3.
4.1.58. Representative procedure for hydrolysis with LiOH/MeOH: synthesis of (2R,3R,5R,6S)-2-(((2R,9R)-9-hydroxydecan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diol (asc-C10-OH)
To a suspension of 141.3 mg (0.2756 mmol) saturated alcohol 46 in 20 mL MeOH was added 20 mL sat. 1M LiOH at once. The resulting solution was allowed to stir at 23°C for 24 h. 10 mL 20% iPrOH in CH2Cl2 was added to the reaction mixture, and the layers were separated. The aqueous layer was extracted with an additional 5x 10 mL 20% iPrOH in CH2Cl2. Solid NaCl was then added until saturation was obtained, and the aqueous layer was further extracted with 3 × 10 mL 20% iPrOH in CH2Cl2. The combined organic layers were dried over Na2SO4 and filtered. Evaporation of solvent afforded 117.2 mg of an oil. Silica gel column chromatography (46.72 g silica gel, gradient run from 5% iPrOH in CH2Cl2 to 20% iPrOH in CH2Cl2) afforded 78.9 mg (94%) of a colorless oil. [α]D25 = -35.3, c 1.43 (CH3OH); HRMS (m/z): [M+Na]+calcd. for C16H32O5Na 327.2142, found 327.2133; 1H NMR (400 MHz, CD3OD): δ 4.64 (s, 1H), 3.69 (m, 4H), 3.52 (ddd, J = 11.3, 9.3, 4.6 Hz, 1H), 1.95 (dt, J = 13.1, 3.8 Hz, 1H), 1.76 (m, 1H), 1.44 (m, 12H), 1.21 (d, J = 6.2 Hz, 3H), 1.13 (dd, J = 9.9, 6.1 Hz, 6H); 13C NMR (400 MHz, CD3OD): δ 97.7; 72.6; 71.3; 70.1; 68.7; 68.4; 40.3; 38.5; 36.1; 30.9; 30.8; 27.0; 26.9; 23.6; 19.5; 18.2.
4.1.59. Synthesis of (R)-7-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)octan-2-one (asc-C8-MK)
The title compound was prepared in a manner similar to that of asc-C10-OH. Started with 49.7 mg (0.103 mmol) ketone 47 and 7.1 mL 1 M LiOH in 7.1 mL MeOH. Purified to obtain 21.7 mg (77%) of a colorless oil. [α]D25 = -71.0, c 1.15 (CH3OH); HRMS (m/z): [M+Na]+calcd. for C14H26O5Na 297.1672, found 297.1670; 1H NMR (400 MHz, CD3OD): δ 4.67 (s, 1H), 3.67 (m, 4H), 2.53 (t, J = 7.3 Hz, 2H), 2.16 (s, 3H), 1.98 (dddd, J = 13.0, 4.4, 3.2, 1.1 Hz, 1H), 1.79 (ddd, J = 13.0, 11.2, 3.1 Hz, 1H), 1.53 (m, 6H), 1.24 (d, J = 6.1 Hz, 3H), 1.15 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 209.8; 96.0; 71.1; 70.0; 69.4; 68.2; 43.8; 37.1; 35.4; 30.2; 25.4; 23.9; 19.1; 17.9.
4.1.60. Synthesis of (R)-8-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)nonan-2-one (asc-C9-MK)
The title compound was prepared in a manner similar to that of asc-C10-OH. Started with 234.1 mg (0.4714 mmol) ketone 48 and 33.3 mL 1 M LiOH in 33.3 mL MeOH. Purified to obtain 103.4 mg (76%) of a colorless oil. [α]D25 = -40.0, c 3.11 (CH3OH); HRMS (m/z): [M+Na]+calcd. for C15H28O5Na 311.1829, found 311.1830; 1H NMR (250 MHz, CD3OD): δ 4.63 (s, 1H), 3.62 (m, 4H), 2.48 (t, J = 7.3 Hz, 2H), 2.12 (s, 3H), 1.94 (dddd, J = 13.1, 4.4, 3.3, 1.0 Hz, 1H), 1.75 (ddd, J = 12.9, 11.1, 3.0 Hz, 1H), 1.44 (m, 8H), 1.20 (d, J = 6.0 Hz, 3H), 1.11 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 212.1; 97.5; 72.4; 71.2; 69.9; 68.3; 44.3; 38.2; 36.0; 30.2; 29.8; 26.7; 24.8; 19.3; 18.1.
4.1.61. Synthesis of (R)-9-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)decan-2-one (asc-C10-MK)
The title compound was prepared in a manner similar to that of asc-C10-OH. Started with 36.9 mg (0.0723 mmol) ketone 49 and 5.3 mL sat. 1 M LiOH in 5.3 mL MeOH. Purified to obtain 18.3 mg (84%) of a colorless oil. [α]D25 = -42, c 0.28 (CH3OH); HRMS (m/z): [M+Na]+calcd. for C28H34O7Na 325.1985, found 325.1987; 1H NMR (250 MHz, CD3OD): δ 4.63 (s, 1H), 3.62 (m, 4H), 2.47 (t, J = 7.3 Hz, 2H), 2.12 (s, 3H), 1.94 (dt, J = 13.1, 4.0 Hz, 1H), 1.75 (ddd, J = 13.0, 10.8, 2.9 Hz, 1H), 1.46 (m, 10H), 1.20 (d, J = 6.0 Hz, 3H), 1.11 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 212.2; 97.6; 72.5; 71.2; 70.0; 68.3; 44.3; 38.3; 36.0; 30.5; 30.2; 29.8; 26.7; 24.9; 19.3; 18.1.
4.1.62. Synthesis of (2R,3R,5R,6S)-2-(((2R,5R)-5-hydroxyhexan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl dibenzoate (51)
The title compound was prepared in a manner similar to that of 16. Began with 400.0 mg dibenzoyl ascarylose (1), column chromatography (50 g silica gel, gradient run from 20% ethyl acetate in hexanes to 100% ethyl acetate) yielded 297.5 mg (58%) of a colorless oil. [α]D25 = 3.1, c 1.46 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C26H32O7Na 479.2040, found 479.2046; 1H NMR (250 MHz, MeOD) δ 8.08 (d, J = 7.3 Hz, 2H), 8.00 (d, J = 7.7 Hz, 2H), 7.60 (t, J = 6.6 Hz, 2H), 7.56 – 7.40 (m, 4H), 5.21 – 5.03 (m, 2H), 4.96 (s, 1H), 4.16 (dq, J = 12.5, 6.4 Hz, 1H), 3.93 – 3.70 (m, 2H), 2.39 (dt, J = 13.4, 4.0 Hz, 1H), 2.19 (ddd, J = 13.9, 11.3, 3.1 Hz, 1H), 1.90 – 1.39 (m, 4H), 1.25 (d, J = 6.2 Hz, 3H), 1.20 (d, J = 6.1 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 165.75, 165.62, 133.21, 133.14, 129.89, 129.80, 129.75, 129.56, 128.39, 93.65, 72.49, 71.15, 70.56, 67.81, 67.02, 35.05, 33.01, 29.65, 23.57, 19.04, 17.81.
4.1.63. Synthesis of (2R,3R,5R,6S)-2-(((2R,5R)-5-hydroxyhexan-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diol (asc-C6-OH)
See ref. 4b for representative procedure. Began with 48.0 mg 51, purification yielded 20.9 mg (80%) of a colorless oil. [α]D25 = -58.4, c 0.495 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C12H24O5Na 271.1516, found 271.1510; 1H NMR (250 MHz, MeOD) δ 4.64 (s, 1H), 3.87 – 3.68 (m, 3H), 3.68 – 3.42 (m, 2H), 1.94 (dt, J = 13.0, 3.8 Hz, 1H), 1.75 (ddd, J = 13.2, 10.9, 3.0 Hz, 1H), 1.69 – 1.39 (m, 4H), 1.21 (d, J = 6.0 Hz, 3H), 1.16 (d, J = 6.2 Hz, 3H), 1.13 (d, J = 6.0 Hz, 3H); 13C NMR (63 MHz, MeOD) δ 97.47, 72.37, 71.17, 69.91, 68.29, 68.25, 36.07, 35.91, 34.39, 23.55, 19.33, 18.07.
4.1.64. Synthesis of (2S,3R,5R,6R)-2-methyl-6-(((R)-5-oxohexan-2-yl)oxy)tetrahydro-2H-pyran-3,5-diyl dibenzoate (52)
The title compound was prepared in a manner similar to that of 47. Began with 220.0 mg 76, column chromatography (8 g silica gel, 5% Et2O in CH2Cl2) yielded 210.0 mg (96%) of a colorless oil. [α]D25 = 16.8, c 0.495 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C26H30O7Na 477.1884, found 477.1905; 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.4 Hz, 2H), 8.03 (d, J = 7.5 Hz, 2H), 7.58 – 7.52 (m, 2H), 7.47 – 7.40 (m, 4H), 5.21 – 5.10 (m, 2H), 4.93 (s, 1H), 4.06 (dq, J = 9.7, 6.3 Hz, 1H), 3.87 (h, J = 5.8 Hz, 1H), 2.58 (t, J = 7.4 Hz, 2H), 2.41 (dt, J = 13.3, 3.8 Hz, 1H), 2.21 – 2.13 (m, 4H), 1.85 (q, J = 7.6 Hz, 2H), 1.28 (d, J = 6.2 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 208.11, 165.55, 165.44, 133.10, 133.04, 129.76, 129.66, 129.61, 129.43, 128.28, 93.40, 71.29, 70.92, 70.39, 66.96, 39.46, 30.67, 29.75, 29.51, 18.72, 17.71.
4.1.65. Synthesis of (R)-5-(((2R,3R,5R,6S)-3,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)hexan-2-one (asc-C6-MK)
See ref. 4b for procedure. Began with 210.0 mg 52, HPLC purification yielded 88.9 mg (78%) of a colorless oil. [α]D25 = -138.5, c 0.895 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C12H22O5Na 269.1359, found 269.1355; 1H NMR (400 MHz, MeOD) δ 4.63 (s, 1H), 3.78 (ddd, J = 8.0, 6.3, 4.7 Hz, 1H), 3.70 (td, J = 3.1, 1.5 Hz, 1H), 3.59 – 3.47 (m, 2H), 2.61 (t, J = 7.4 Hz, 2H), 2.15 (s, 3H), 1.94 (dt, J = 12.9, 3.6 Hz, 1H), 1.81 – 1.66 (m, 3H), 1.21 (d, J = 5.6 Hz, 3H), 1.12 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, MeOD) δ 211.77, 97.39, 71.63, 71.40, 69.94, 68.38, 40.58, 36.04, 32.20, 30.02, 19.22, 18.20.
4.1.66. Representative procedure for diacylation with indole-3-carbonyl chloride (9): synthesis of (2S,3R,5R,6R)-2-methyl-6-(((R)-5-oxohexan-2-yl)oxy)tetrahydro-2H-pyran-3,5-diyl bis(1H-indole-3-carboxylate) (53)
To a solution of 43.5 mg (0.1766 mmol) methyl ketone 2 and 250 μL (1.413 mmol) N,N-diisopropylethylamine in 12.5 mL THF at 0 °C was added a solution of 190.3 mg (1.060 mmol) acid chloride 9 in 1.5 mL THF dropwise over the course of 3 minutes. The resulting solution was allowed to warm to 23 °C and stir for 6 h. During this time, close monitoring of the reaction by TLC showed formation and disappearance of an intermediate, likely the monoacylated product, followed by appearance of the desired product. 15 mL of H2O was then added at once and the mixture was allowed to stir for an additional 30 minutes. 15 mL EtOAc was then added and the layers were separated. The aqueous layer was extracted with an additional 3 × 15 mL EtOAc. The combined organic layers were dried over Na2SO4, filtered and concentrated to a light orange oil. HPLC purification afforded 83.6 mg (89%) of a white solid. [α]D25 = 37.0, c 1.44 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C30H32N2O7Na 555.2102, found 555.2126; 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1H), 9.51 (s, 1H), 8.24 (d, J = 7.9 Hz, 1H), 8.14 (d, J = 7.3 Hz, 1H), 7.88 (d, J = 3.0 Hz, 1H), 7.85 (d, J = 3.1 Hz, 1H), 7.40 – 7.34 (m, 2H), 7.27 – 7.15 (m, 4H), 5.32 (td, J = 10.4, 4.5 Hz, 1H), 5.21 (s, 1H), 5.02 (s, 1H), 4.13 (dq, J = 12.2, 6.0 Hz, 1H), 3.91 (h, J = 6.0 Hz, 1H), 2.63 (t, J = 7.4 Hz, 2H), 2.51 (dt, J = 13.3, 3.7 Hz, 1H), 2.29 – 2.23 (m, 1H), 2.21 (s, 3H), 1.89 (q, J = 7.3 Hz, 2H), 1.34 (d, J = 6.3 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 209.09, 164.73, 164.39, 136.26, 136.23, 131.96, 131.82, 125.76, 125.67, 123.16, 123.04, 122.10, 122.05, 121.17, 121.06, 111.87, 111.77, 107.96, 107.72, 93.98, 71.30, 69.94, 69.62, 67.51, 39.67, 30.84, 30.11, 29.90, 18.82, 17.98.
4.1.67. Synthesis of (2S,3R,5R,6R)-5-hydroxy-2-methyl-6-(((R)-5-oxohexan-2-yl)oxy)tetrahydro-2H-pyran-3-yl 1H-indole-3-carboxylate (IC-asc-C6-MK)
To a solution of 10.2 mg (0.0193 mmol) 53 in 3.0 mL i-PrOH at 23 °C was added 3.0 mL 1M LiOH at once. The resulting mixture was heated to 45 °C and allowed to stir for 6.5 h. The reaction was then permitted to cool to 23 °C at which time 5 mL 20% i-PrOH in CH2Cl2 was added and the layers were separated. The aqueous layer was extracted with an additional 2 × 5 mL 20% i-Propanol in CH2Cl2. Solid NaCl was then added to the aqueous layer until saturation was obtained, and it was further extracted with 2 × 5 mL 20% iPrOH in CH2Cl2. The combined organic layers were dried over Na2SO4, filtered and concentrated to yield 15.4 mg of an oil. HPLC purification afforded 2.6 mg (35%) of a colorless oil. [α]D25 = -12.8, c 0.13 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C21H27NO6Na 412.1731, found 412.1735; 1H NMR (250 MHz, CDCl3) δ 8.61 (s, 1H), 8.18 – 8.12 (m, 1H), 7.95 (d, J = 3.1 Hz, 1H), 7.46 – 7.40 (m, 1H), 7.33 – 7.28 (m, 2H), 5.18 – 5.06 (m, 1H), 4.77 (s, 1H), 4.08 – 3.98 (m, 1H), 3.92 – 3.84 (m, 2H), 2.62 (t, J = 7.5 Hz, 2H), 2.34 – 2.17 (m, 4H), 2.13 – 1.99 (m, 1H), 1.93 – 1.79 (m, 2H), 1.29 (d, J = 6.3 Hz, 3H), 1.19 (d, J = 6.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 208.52, 162.30, 136.09, 131.27, 125.75, 123.37, 122.21, 121.49, 111.58, 108.83, 96.22, 70.98, 69.38, 68.79, 68.01, 39.75, 32.31, 30.94, 29.99, 18.96, 17.76.
4.1.68. Synthesis of (2S,3R,5R,6R)-2-methyl-6-((R)-oct-7-en-2-yloxy)tetrahydro-2H-pyran-3,5-diol (54)
See ref. 4b for procedure. Began with 20.6 mg 18, HPLC purification yielded 10.7 mg (69%) of a colorless oil. [α]D25 = 76.8, c 0.09 (MeOH); HRMS (m/z): [M+Na]+ calcd. for C14H26O4Na 281.1723, found 281.1732; 1H NMR (250 MHz, MeOD) δ 5.81 (ddt, J = 17.0, 10.2, 6.8 Hz, 1H), 5.06 – 4.88 (m, 2H), 4.63 (s, 1H), 3.81 – 3.67 (m, 2H), 3.67 – 3.44 (m, 2H), 2.06 (q, J = 5.9, 5.4 Hz, 2H), 1.94 (dt, J = 13.0, 4.0 Hz, 1H), 1.75 (ddd, J = 13.3, 10.9, 3.0 Hz, 1H), 1.59 – 1.33 (m, 6H), 1.20 (d, J = 6.0 Hz, 3H), 1.11 (d, J = 6.1 Hz, 3H); 13C NMR (63 MHz, MeOD) δ 139.94, 114.87, 97.57, 72.47, 71.15, 69.95, 68.30, 38.21, 35.94, 34.83, 30.02, 26.35, 19.37, 18.10.
4.1.69. Synthesis of (2S,3R,5R,6R)-2-methyl-6-((R)-oct-7-en-2-yloxy)tetrahydro-2H-pyran-3,5-diyl bis(1H-indole-3-carboxylate) (55)
The title compound was prepared in a manner similar to that of 53. Began with 43.8 mg 54, HPLC purification yielded 40.0 mg (43%) of a colorless oil. [α]D25 = 43.0, c 0.40 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C32H36N2O6Na 567.2466, found 567.2474; 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H), 9.16 (s, 1H), 8.25 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 7.1 Hz, 1H), 7.91 – 7.88 (m, 1H), 7.88 – 7.83 (m, 1H), 7.41 – 7.32 (m, 2H), 7.28 – 7.16 (m, 4H), 5.85 (ddt, J = 16.9, 10.2, 6.5 Hz, 1H), 5.33 (td, J = 10.6, 4.6 Hz, 1H), 5.22 (s, 1H), 5.06 – 4.93 (m, 3H), 4.20 (dq, J = 10.8, 5.5, 5.0 Hz, 1H), 3.88 (h, J = 5.9 Hz, 1H), 2.51 (dt, J = 13.6, 4.0 Hz, 1H), 2.34 – 2.22 (m, 1H), 2.15 – 2.07 (m, 2H), 1.58 – 1.40 (m, 6H), 1.35 (d, J = 5.9 Hz, 3H), 1.19 (d, J = 5.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 164.65, 164.35, 139.01, 136.29, 136.27, 131.85, 131.57, 126.02, 125.83, 123.44, 123.28, 122.34, 122.32, 121.60, 121.48, 114.59, 111.82, 111.71, 108.71, 108.40, 94.34, 72.54, 70.24, 69.84, 67.46, 37.19, 33.97, 30.33, 28.98, 25.49, 19.39, 18.17.
4.1.70. Synthesis of (2R,3R,5R,6S)-2-(((R,E)-9-methoxy-9-oxonon-7-en-2-yl)oxy)-6-methyltetrahydro-2H-pyran-3,5-diyl bis(1H-indole-3-carboxylate) (56)
See ref. 2d for procedure. Began with 40.0 mg 55, column chromatography (5 g silica gel, CH2Cl2, then a gradient from 5% ethyl acetate in CH2Cl2 to 15% ethyl acetate in CH2Cl2) yielded 42.2 mg (95%) of a white solid. [α]D25 = 53.0, c 0.245 (CH2Cl2); HRMS (m/z): [M+Na]+ calcd. for C34H38N2O8Na 625.2520, found 625.2544; 1H NMR (400 MHz, CDCl3) δ 9.39 (s, 1H), 9.35 (s, 1H), 8.26 (d, J = 7.9 Hz, 1H), 8.17 – 8.12 (m, 1H), 7.94 (d, J = 2.7 Hz, 1H), 7.89 (d, J = 2.8 Hz, 1H), 7.43 – 7.36 (m, 2H), 7.30 – 7.19 (m, 4H), 7.03 (dt, J = 15.6, 6.9 Hz, 1H), 5.88 (d, J = 15.6 Hz, 1H), 5.32 (ddd, J = 14.4, 9.4, 4.0 Hz, 1H), 5.25 – 5.21 (m, 1H), 5.05 (s, 1H), 4.17 (dq, J = 12.2, 6.0 Hz, 1H), 3.91 – 3.85 (m, 1H), 3.72 (s, 3H), 2.56 – 2.49 (m, 1H), 2.32 – 2.24 (m, 3H), 1.72 – 1.63 (m, 1H), 1.59 – 1.49 (m, 4H), 1.49 – 1.42 (m, 1H), 1.35 (d, J = 6.2 Hz, 3H), 1.20 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 167.33, 164.65, 164.33, 149.63, 136.22, 136.18, 131.86, 131.77, 125.79, 125.70, 123.17, 123.03, 122.10, 122.05, 121.26, 121.11, 120.91, 111.82, 111.69, 108.10, 107.88, 94.10, 72.25, 70.01, 69.63, 67.35, 51.44, 36.87, 32.21, 30.12, 27.95, 25.36, 19.15, 18.01.
4.1.71. Synthesis of (R,E)-8-(((2R,3R,5R,6S)-5-((1H-indole-3-carbonyl)oxy)-3-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)non-2-enoic acid (IC-asc-ΔC9)
To a solution of 10.2 mg (0.0169 mmol) methyl ester 56 in 2.5 mL t-Butanol at 23 °C was added 2.5 mL 1M LiOH at once. The resulting mixture was heated to 50 °C and allowed to stir for 11 h. The reaction was then permitted to cool to 23 °C at which time the pH was adjusted to 3 via dropwise addition of 1M HCl. 5 mL 20% 2-propanol in CH2Cl2 was then added and the layers were separated. The aqueous layer was extracted with an additional 2 × 5 mL 20% i-Propanol in CH2Cl2. Solid NaCl was then added until saturation was obtained, and the aqueous layer was further extracted with 2 × 5 mL 20% iPrOH in CH2Cl2. The combined organic layers were dried over Na2SO4, filtered and concentrated to yield 17.3 mg of an oil. HPLC purification afforded 4.9 mg (65%) of a colorless oil. HRMS (m/z): [M+Na]+ calcd. for C30H32N2O7Na 444.2028, found 444.2031. 1H NMR (400 MHz, Methanol-d4) δ 8.03 – 7.99 (m, 1H), 7.96 (s, 1H), 7.46 – 7.41 (m, 1H), 7.23 – 7.14 (m, 2H), 6.84 (dt, J = 15.4, 6.9 Hz, 1H), 5.83 (d, J= 15.6 Hz, 1H), 5.11 (td, J = 10.5, 4.7 Hz, 1H), 4.74 (s, 1H), 4.04 (dd, J = 9.6, 6.2 Hz, 1H), 3.89 – 3.81 (m, 1H), 3.79 (q, J = 2.9 Hz, 1H), 2.29 – 2.17 (m, 3H), 2.00 (ddd, J = 13.4, 11.2, 3.1 Hz, 1H), 1.70 – 1.58 (m, 1H), 1.58 – 1.42 (m, 5H), 1.23 (d, J = 6.3 Hz, 3H), 1.16 (d, J = 6.0 Hz, 3H).
4.2. LC-MS/MS
LC-MS/MS analysis was performed using an Accela UHPLC system equipped with a Kinetex 2.6 μm C18 100 Å column (100 × 2.10 mm) connected to a Thermo TSQ quantum ultra triple quadrupole mass spectrometer. A water/acetonitrile (0.1% acetic acid) solvent gradient was used with a 0.25 mL/min flow rate, starting at 2% acetonitrile for 2 min, ramping from 2-50% acetonitrile over 34 min, ramping from 50-100% acetonitrile over 1 min, holding at 100% acetonitrile for 4 min, ramping from 100-2% acetonitrile over 1 min, then holding at 2% acetonitrile for 4 min. LC-MS/MS was performed using argon collision gas and operating in negative ion, heated (H)-ESI, precursor scanning mode, selecting for a product ion of m/z 73.0. The following parameters were used: spray voltage of 3000 V, vaporizer temperature of 300 °C, sheath gas pressure of 40 arb, auxiliary gas pressure of 20 arb, capillary temperature of 350 °C, skimmer offset of 5 V, collision pressure of 0.8 mtorr, collision energy of 25 eV, and scan time 0.4 s.
4.3. Dauer formation assay
The dauer formation assay was performed as described in ref. 2h. Briefly, synthetic ascarosides or vehicle (ethanol) were added to 100 μL of water at 30 times the assay concentration. This 100 μL was then mixed into 3 mL of NGM-agar (made with Noble agar [BD Biosciences] and without peptone) at 60 °C in a 35 × 10 mm plate, and the agar was allowed to set. A stock of heat-killed E. coli (OP50) was made by diluting OP50 to 8 mg / mL with S basal and heating to 95 °C for 30 min, with vortexing every 5 min. The plates and OP50 stock were stored overnight at 4 °C. 20 μL of the OP50 stock was added to each plate, and plates were allowed to dry in a hood for 15 min. 5 adult worms were placed on each plate, allowed to lay eggs for 3-4 h, and removed. These adult worms were synchronized for the egg lay by performing an egg lay every third day for at least 3 generations. During these 3 generations, it was critical that the worms were not exposed to starvation stress. The assay plates were incubated at 25 °C for 68-72 h, and dauers were identified based on size, shape, and lack of pharyngeal pumping. Data was included only from assay plates that had between 50 and 100 worms. Concentrations tested (220 nM and 6000 nM) were chosen because 220 nM is in the range of the EC50 for many of the dauer pheromone ascarosides and 6000 nM should detect activity from even minimally active ascarosides. In order to avoid day-to-day variability in the assay results, all 29 ascarosides were tested on the same day (in duplicate) in each experiment. The data in Table 1 represent the average of two independent experiments.
Supplementary Material
Acknowledgments
This work was supported with funds provided by Louisiana State University and was partially funded by a subcontract to R.A.B. from Human Frontier Science Program grant RGP0042. Funds for a summer REU for G.M.B. were provided by Howard Hughes Medical Institute, and funds for a summer REU for A.C.G.V.C. were provided by the National Science Foundation. The authors thank Dr. Thomas Weldeghiorghis and Dr. Dale Treleaven (Louisiana State University) for assistance with NMR spectroscopy, Ms. Connie David and Dr. Azeem Hassan (Louisiana State University) for assistance with HR-MS, and Dr. Tim Garrett (University of Florida) for assistance with LC-MS/MS. Our LC-MS/MS work on the triple quadrupole mass spectrometer was supported in part by the National Institutes of Health and National Center for Research Resources CTSA grant 1UL 1RR029890.
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at___ These data include 1H and 13C NMR spectra and LC-MS/MS analysis.
References
- 1.Coghlan A. WormBook. 2005 Sep 7;:1–15. doi: 10.1895/wormbook.1.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) von Reuss SH, Bose N, Srinivasan J, Yim JJ, Judkins JC, Sternberg PW, Schroeder FC. J Am Chem Soc. 2012;134:1817–1824. doi: 10.1021/ja210202y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Srinivasan J, von Reuss SH, Bose N, Zaslaver A, Mahanti P, Ho MC, O’Doherty O, Edison AS, Sternberg PW, Schroeder FC. PloS Biol. 2012;10:1–14. doi: 10.1371/journal.pbio.1001237. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pungaliya C, Srinivasan J, Fox BW, Malik RU, Ludewig AH, Sternberg PW, Schroeder FC. Proc Natl Acad Sci USA. 2009;106:7708–7713. doi: 10.1073/pnas.0811918106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Butcher RA, Ragains JR, Li W, Ruvkun G, Clardy J, Mak HY. Proc Natl Acad Sci USA. 2009;106:1875–1879. doi: 10.1073/pnas.0810338106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Butcher RA, Ragains JR, Clardy J. Org Lett. 2009;11:3100–3103. doi: 10.1021/ol901011c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Srinivasan J, Kaplan F, Ajredini R, Zachariah C, Alborn HT, Teal PEA, Malik RU, Edison AS, Sternberg PW, Schroeder FC. Nature. 2008;454:1115–1119. doi: 10.1038/nature07168. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Butcher RA, Ragains JR, Kim E, Clardy J. Proc Natl Acad Sci USA. 2008;105:14288–14292. doi: 10.1073/pnas.0806676105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Butcher RA, Fujita M, Schroeder FC, Clardy J. Nat Chem Bio. 2007;3:420–422. doi: 10.1038/nchembio.2007.3. [DOI] [PubMed] [Google Scholar]; (i) Jeong P-Y, Jung M, Yim Y-H, Kim H, Park M, Hong E, Lee W, Kim YH, Kim K, Paik Y-K. Nature. 2005;433:541–545. doi: 10.1038/nature03201. [DOI] [PubMed] [Google Scholar]
- 3.(a) Park D, O’Doherty I, Somvanshi RK, Bethke A, Schroeder FC, Kumar U, Riddle DL. Proc Natl Acad Sci USA. 2012;109:9917–9922. doi: 10.1073/pnas.1202216109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McGrath PT, Xu Y, Ailion M, Garrison JL, Butcher RA, Bargmann CI. Nature. 2011;477:321–325. doi: 10.1038/nature10378. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yamada K, Hirotsu T, Matsuki M, Butcher RA, Tomioka M, Ishihara T, Clardy J, Kunimoto H, Lino Y. Science. 2010;329:1647–1650. doi: 10.1126/science.1192020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim K, Sato K, Shibuya M, Zeiger DM, Butcher RA, Ragains JR, Clardy J, Touhara K, Sengupta P. Science. 2009;326:994–998. doi: 10.1126/science.1176331. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Macosko EZ, Pokala N, Feinberg EH, Chalasani SH, Butcher RA, Clardy J, Bargmann CI. Nature. 2009;458:1171–1176. doi: 10.1038/nature07886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Choe A, von Reuss SH, Kogan D, Gasser RB, Platzer EG, Schroeder FC, Sternberg PW. Current Biology. 2012;22:772–780. doi: 10.1016/j.cub.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Noguez JH, Conner ES, Zhou Y, Ciche TA, Ragains JR, Butcher RA. ACS Chem Biol. 2012;7:961–966. doi: 10.1021/cb300056q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bose N, Ogawa A, von Reuss SH, Yim JJ, Ragsdale EJ, Sommer RJ, Schroeder FC. Angew Chem Int Ed. 2012;51:12438–12443. doi: 10.1002/anie.201206797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colley DG, LoVerde PT, Savioli L. Science. 2001;293:1437–1438. doi: 10.1126/science.1060733. [DOI] [PubMed] [Google Scholar]
- 6.Cassada RC, Russell RL. Dev Biol. 1975;46:326–342. doi: 10.1016/0012-1606(75)90109-8. [DOI] [PubMed] [Google Scholar]
- 7.Hallem EA, Dillman AR, Hong AV, Zhang Y, Yano JM, DeMarco SF, Sternberg PW. Current Biology. 2011;21:377–383. doi: 10.1016/j.cub.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas JH, Robertson HR. BMC Biol. 2008;6:42. doi: 10.1186/1741-7007-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagiwara D, Miyake H, Murano K, Morimoto H, Murai M. J Med Chem. 1993;36:2266–2278. doi: 10.1021/jm00068a003. [DOI] [PubMed] [Google Scholar]
- 10.(a) Felix M-A, Duveau F. BMC Biol. 2012;10:59. doi: 10.1186/1741-7007-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Felix M-A, Duveau F. WormBook. 2006 Jan 9;:1–14. [Google Scholar]
- 11.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.