

Abstract
Glioblastoma (GBM) is a highly angiogenic malignancy that is resistant to standard therapy; neo-formed vessels of this aggressive malignancy are thought to arise by sprouting of pre-existing brain capillaries. However, the conventional anti-angiogenic therapy, which seemed promising initially, shows transitory and incomplete efficacy. The discovery of vasculogenic mimicry (VM) has offered a new horizon for understanding tumor vascularization. VM is a tumor cell-constituted, matrix-embedded fluid-conducting meshwork that is independent of endothelial cells and is positively correlated with poor prognosis. Therefore, a better understanding of GBM vasculature is needed to optimize anti-angiogenic therapy. This review focuses on the signaling molecules and cascades involved in VM in relation to ongoing glioma research, as well as the clinical translational advances in GBM that have been offered by the development of optimized anti-angiogenesis treatment modalities.
Keywords: Vasculogenic mimicry (VM), Glioblastoma (GBM), Angiogenesis, Cell signaling
Introduction
Glioblastoma (GBM) is the most common and lethal malignant brain tumor in adults. It is an extraordinarily aggressive malignancy characterized by extensive micro-vascular proliferation and is highly resistant to intensive combination therapies. The prognosis for GBM patients is extremely poor despite the use of comprehensive treatment involving gross tumor resection, chemotherapy and/or radiotherapy, with an average life expectancy of 12 to 15 months once diagnosed [1,2].
GBM is one of the most vascularized tumors, and its poor prognosis primarily results from its invasive properties. Indeed, an accepted tenet underlying tumor survival is that a blood supply is required to sustain growth and invasion [3]. The neoplastic angiogenesis research ignited by this important premise was the basis for enthusiastic preclinical trials, which ultimately reaped disappointing clinical results [4]. Thus, investigators accepted that tumor perfusion mechanisms are much more sophisticated than we previously realized [2].
The newly discovered vascular network structure, vasculogenic mimicry (VM), was first described and defined by Maniotis et al. for malignant melanoma in 1999 [5]. The discovery of VM simultaneously sparked intensive controversy [6] and brought a new vision to tumor therapy. VM is defined by a fluid-conducting, matrix-embedded meshwork that is independent of endothelial cells (ECs), but instead is formed by certain types of tumor cells through their acquirement of plasticity to mimic endothelial function [7]. Since the discovery of VM, cumulative studies have contributed new insights into the underlying molecular pathways supporting its existence in a variety of non-melanoma aggressive tumors [8-13], including GBM [14]. CD34 or CD31 and PAS dual-staining have been applied to visualize the EC-free, matrix-rich morphological pattern of VM [5,8-14]; and immunohistochemistry and microarray analysis have been applied to identify its undifferentiated, embryonic-like phenotype [5,7,14]. Two distinctive VM types--tubular type [15] and patterned matrix type [14,16]--have been described. From the extensive literature across this vast field, we now appreciate that the simplistic model of sprouting angiogenesis is far too limited to describe the complex tumor vasculature. There are several other paradigms reported in addition to VM (Figure 1), including vascular co-option, vascular intussusception, bone marrow-derived vasculogenesis and cancer stem-like cell-derived vasculogenesis [3]. For glioma, the phenomenon of VM is associated with high grades of tumor invasiveness and poor prognosis [1,17]. As an alternative to traditional anti-angiogenic therapy, the identification of molecules and signaling pathways relating to VM may offer potential therapeutic targets to improve treatment [18]. In this review, we summarize the current progress in understanding the molecular mechanisms revealed by ongoing glioma VM research and discuss potential VM-targeting strategies for the future development of GBM therapies.
Figure 1.
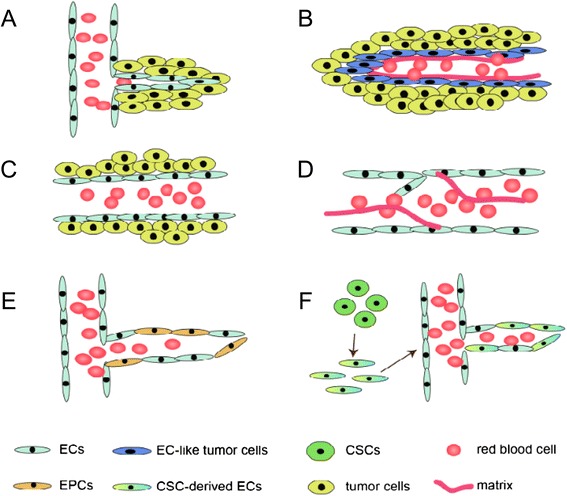
Mechanisms of tumor vascularization. The different mechanisms of tumor vascularization are depicted in the figure. These include: (A) sprouting angiogenesis: sprouting of pre-existing brain capillaries through proliferation and migration of local ECs; (B) vasculogenic mimicry: tumor cell-constituted, matrix-embedded fluid-conducting meshwork; (C) vascular co-option: tumor cells grow along pre-existing blood vessels; (D) vascular intussusception: internal division of the preexisting capillary plexus; (E) bone marrow-derived vasculogenesis: recruitment of circulating endothelial precursors (EPCs) to form new vessels; (F) cancer stem-like cell-derived vasculogenesis: transdifferentiation of CSCs into ECs to form new vessels.
Functional characterization of VM
Tumors depend on an adequate blood supply for growth and metastasis [2,3]; therefore, intratumoral microvessel density is assessed as a biomarker for tumor progression and is considered a valuable prognostic indicator [19]. However, clinical trials have shown that some tumors with low intratumoral microvessel density have poor prognosis, and conventional anti-angiogenic results are not consistently predictive. The inconsistency between theory and practice suggests that alternative vascular mechanisms exist, which was the basis for proposing VM as a new topic of research [2].
The presence of characteristic VM structures in tumor tissues is associated with poor clinical outcome, suggesting that VM contributes functionally to tumor progression. Blood circulation demonstrated by confocal indocyanine green angiography revealed that VM patterns are not part of the EC-lined vascular system. Fluid enters these channels through leakage and circulates within the VM network rather than accumulating in a pool [20].
In addition, microarray analysis illustrated that tumor cells involved in the VM structure convert to a dedifferentiated, embryonic-like phenotype, which appears to be multipotent to act as either ECs or common tumor cells. Moreover, the co-localization of tumor cells and ECs lining the vascular endothelium (referred to as mosaic vessels [16]) indicates a direct or indirect physical connection between endothelial vessels and VM networks.
Biological phenomena of Glioma stem-like cells (GSCs) related to VM
Tumor cells capable of VM formation exhibit high plasticity indicative of a multipotent phenotype that resembles embryonic stem cells. This phenomenon is confirmed for cancer stem cells (CSCs). A simple recapitulation of the CSC theory is as follows: genetically dysregulated tumor cells are embryonic-like, therefore “plastic”, and therefore capable of expressing vascular-like phenotypes [16]. Though specific criteria for the identification of CSCs have not been established, screening and enrichment of CSCs prospectively in a series of malignancies has identified stem cell biomarkers such as CD133 and Nestin, which are also expressed in GSCs [21,22]. CSCs can be enriched by the use of anti-CD133 antibodies or through the generation of neurospheres in a certain culture condition, mostly serum-free media containing epidermal growth factor and basic fibroblast growth factor [23]; and GSC screening undergoes similar procedures. As for the origins of GSCs, in addition to the multistep mutations of normal stem cell genomes, GSCs might also be derived partly due to dedifferentiation via the process of epithelial-mesenchymal-transition (EMT), a process about regaining dedifferentiated phenotypes and mesenchymal features [24]. Therefore, GSCs may be considered a bridge between EMT and VM formation.
Similar to normal neural stem cells, GSCs possess the capacity of self-renewal and multi-lineage differentiation; however, aberrant gene regulation confers their tumorigenicity. GSCs participate in VM formation by interacting with the vascular niche to shape the proper tumor microenvironment and by differentiating into EC-like tumor cells to constitute VM structure. The cells derived from GSCs present multi-phenotype features of different lineages, such as neural cells and mural cells, thus effectively mimicking the microcirculatory system. The tumor vasculature of GSCs in an incomplete stage is thought to be formed by VM channel [23], consistent with the three stages of tumor blood supply pattern proposed by Zhang et al. [25], namely VM channels, mosaic vessels and the endothelium-dependent vessels (Figure 2).
Figure 2.
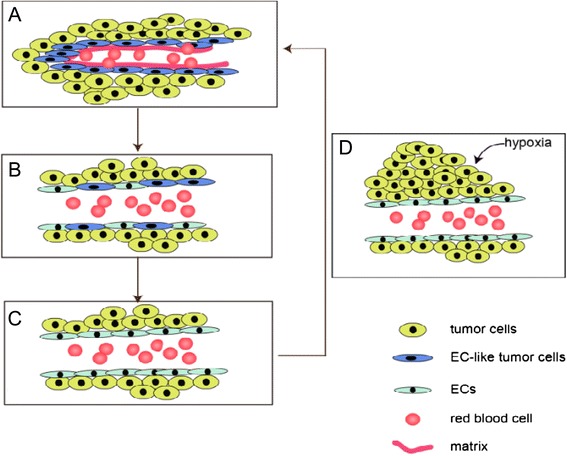
The three stages of tumor blood supply pattern. The possible connection between VM channels and the endothelium-dependent vessels: different stages of a continuous process--(A) VM channels, (B) mosaic vessels and (C) the endothelium-dependent vessels. Figure 2(D)--tumor proliferation acts as a trigger point to anther tumor blood supply cycle.
Signaling cascades involved in VM
There are two main aspects that underlie the formation of VM, i.e. highly plastic tumor cells and the tumor microenvironment. The tumor cells lining the vasculature display an undifferentiated embryonic-like biological and molecular phenotype [26], suggesting the involvement of CSCs. CSCs are a subpopulation of tumor cells that possess the capacity of self-renewal, multi-lineage differentiation, tumor initiation and resistance to chemo- or radio-therapy [27]. Additionally, the tumor microenvironment is a critical factor that conventional tumor cell-targeted therapy fails to take into account. Changes in the extracellular microenvironment may persist after removal or destruction of an aggressive tumor, thus resulting in the recurrence or continuance of certain tumors [28]. The over-expression of VEGFR-2 [21,26,29], EphA2 [30], VE-cadherin [31], MMPs and TGF-β [32], among other molecules, have been demonstrated to be closely associated with vasculature formation. Hypoxia, which can be induced by Bevacizumab therapy [33], represents an additionally factor that might partially activate remodeling of the extracellular microenvironment as a result of oxygen-glucose deprivation.
The multipotent phenotype underlying VM is supported by a complex network of potential signaling pathways, and an increasing number of studies have been conducted to illustrate the fundamental mechanisms in order to establish new treatment regimens. We present some of the signaling pathways attributed to VM formation in GBM (Figure 3, Table 1) according to their association with the embryonic/stem cell phenotype (section Stem cell pathway of GBM), the glioma microenvironment (section Glioma microenvironment-related signaling pathways) and hypoxic conditions (section Hypoxia-related mechanism of VM).
Figure 3.
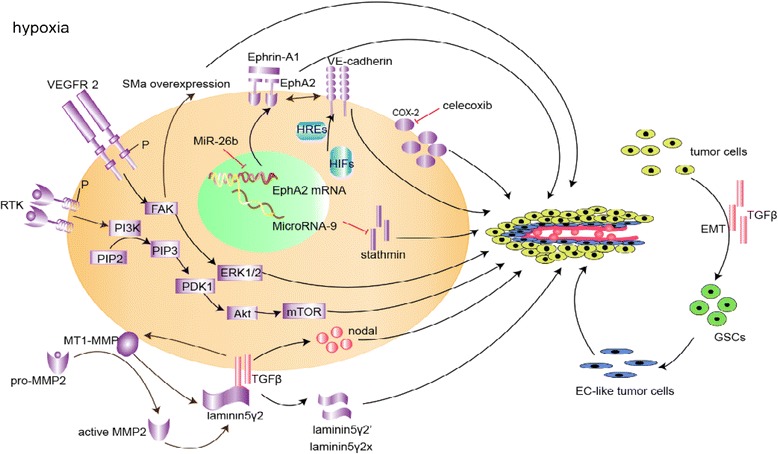
Schematic model of signaling pathways implicated in glioma vasculogenic mimicry (VM). The whole VM process proceeds under the condition of hypoxia. Only signaling molecules which have been specifically modulated using small inhibitory RNAs, blocking antibodies or small molecule inhibitors are depicted -- demonstrating their ability to directly or indirectly affect VM, and are categorized as microenvironment (purple), embryonic/stem cell (red), and hypoxia signaling pathways (blue). There are no specific boundary lines between the three parts and overlap between major VM signaling pathways demonstrated coordinated work of these pathways.
Table 1.
Brief summary of the potential molecular targets for optimized anti-angiogenesis therapy
| Project | Potential molecular targets | Related process | Tumor types/cell lines | method | PMID |
|---|---|---|---|---|---|
| Signaling pathway | |||||
| TGFβ Signaling pathway | TGFβ | EMT-embryonic phenotype regain; | U251 | Transfection and RNAi | 22104964 |
| tumor microenvironment remodulation | SHG44 | 24370825 | |||
| VEGFR-2/Flk-1 pathway | VEGFR-2 | Self-renewal, tumorigenicity and tubular formation | U87 derived GSCs, | Transfection and RNAi; | 23536763 |
| GBM samples | Immunohistochemistry, | ||||
| Xenograft models | |||||
| U87 | RNAi; | 22654102 | |||
| GSDC | xenotransplantation | ||||
| VE-cadherin/CDH5 and EphA2 pathways | VE-cadherin | hypoxia induced microenvironment remodulation; | Glioma samples;GSCs | RT-PCR, western blot, immunohistochemistry; | 23645533 |
| RNAi and xenograft models | |||||
| EphA2 and MiR-26b | Cell proliferation, invasion and tubular formation | Glioma samples; | RT-PCR; | 21264258 | |
| U251 and C6 | Transfection and RNAi | ||||
| RTK/PI3K/Akt/mTOR pathway | RTK/PI3K/mTOR | Cell proliferation, apoptosis, microenvironment remodulation. | U251 and T98G | Pharmacologic inhibitor, RNAi | 24418474 |
| MMP-laminin5γ2 chain pathway | MMPs | a shared downstream signaling pathway | U251 | Transfection and RNAi | 22104964 |
| SHG44 | |||||
| others | COX-2 | Promote cell survival, proliferation, and angiogenesis and prohibit apoptosis | Glioma specimen | Immunohistochemistry, Kaplan–Meier survival analysis and log-rank tests | 21533525 |
| MiR-9 | Cell proliferation and apoptosis | U87MG | Transfection and RNAi | 24043603 | |
| U251 | |||||
| SHG44 |
Stem cell pathway of GBM
Tumor cells capable of VM formation exhibit a high plasticity indicative of a multipotent phenotype resembling embryonic stem cells. The in vitro tubular formation capacity and unique gene expression signature of highly invasive melanoma cells suggests that tumor cell VM activity is associated with distinctive genetic dysregulation [5]. Microarray analysis of VM-positive tissues revealed increased expression of genes associated with an undifferentiated embryonic-like phenotype [7]. GSCs participate in VM formation in several ways: they interact with the vascular niche to shape the proper tumor microenvironment [26,32,34] and differentiate into EC-like tumor cells to constitute VM structure [21,22]. Several molecules associated with anaplastic properties of tumor cells are over-expressed in CSCs and are associated with VM formation, including transforming growth factorβ (TGFβ) [32], Nodal [34], and vascular endothelial growth factor receptor-2 (VEGFR-2) [26]. TGFβ and Nodal are members of the TGFβ superfamily and are discussed in section The TGFβ signaling pathway below, while VEGFR-2 is discussed in section The vascular endothelial growth factor receptor-2 (VEGFR-2)/Flk-1 pathway on the basis of its more well-established role in affecting the tumor microenvironment.
The TGFβ signaling pathway
The TGFβ superfamily of growth factors is a group of ubiquitous multifunctional cytokines that regulate a plethora of cellular activities, including proliferation, differentiation, migration and survival. Both TGFβ and Nodal are critical members of this family [35]. TGFβ is named after its function in inducing EMT [34], a process whereby fully differentiated epithelial cells divert to a dedifferentiated state and acquire mesenchymal features. TGFβ regulates two different aspects of VM formation: the maintenance of the undifferentiated embryonic state of stem cell via EMT [36] and the induction of tubular formation through modulation of the tumor microenvironment. Studies of breast cancer and melanoma show that TGFβ stimulates tubular formation by regulating MMP expression in epithelial tumors [37,38]. Thus, TGFβ enhances or reduces the activity of the MMP-laminin 5γ2 chain signaling pathway (discussed further in section The MMP-laminin 5γ2 chain signaling pathway). Similar regulatory mechanisms have been detected in GBM [32].
Ling, et al. demonstrated that TGFβ levels correspond with glioma malignancy in vitro. Plasmid transfection of TGFβ mRNA activates the dose-dependent expression of VM-related molecules in glioma cell lines as assessed by RT-PCR, and the activity of MMP2 and MMP9 as assessed by gelatin zymography. Furthermore, inhibition of TGFβ results in a decline in both the quantity and activity of MT1-MMP, which in turn reduces MMP2 activation, thereby impairing tubular formation. EphA2 was not modulated in response to TGFβ regulation in this study, and VE-cadherin (CDH5), which can be induced by potent down-regulation of TGFβ through EMT, was detectably absent [32]. This is inconsistent with the role for VE-cadherin in VM reported by Mao, et al. [31]. A possible explanation for this discrepancy involves the use of glioma cell lines versus GSCs. The putative roles for VE-cadherin and EphA2 in regulating the glioma microenvironment are discussed in further detail below (section The VE-cadherin (CDH5) and EphA2 pathways).
Nodal, a potent embryonic morphogen in the TGFβ superfamily, is a biomarker that normally loses expression during the differentiate process, but becomes aberrantly re-expressed in highly aggressive tumors. Nodal contributes to tumor progression and plasticity through a variety of mechanisms [39]. Though the role for Nodal has not precisely been demonstrated for GBM, the activation of Nodal expression by TGFβ has been shown to promote glioma cell growth [34]. Given the above information, we can presume that both the activation of EMT and the TGFβ-induced expression of MMPs and Nodal play a role in VM formation.
Glioma microenvironment-related signaling pathways
Pathways known to modulate the glioma microenvironment include the vascular endothelial growth factor receptor-2 (VEGFR-2)/Flk-1 pathway (section The vascular endothelial growth factor receptor-2 (VEGFR-2)/Flk-1 pathway), VE-cadherin (CDH5) and EphA2 pathways (section The VE-cadherin (CDH5) and EphA2 pathways), the RTK/PI3K/Akt/mTOR signaling pathway (section The RTK/PI3K/Akt/mTOR signaling pathway), and the MMP-laminin 5γ2 chain signaling pathway (section The MMP-laminin 5γ2 chain signaling pathway). The potential role of other related molecules in determining the glioma microenvironment in VM is discussed in section Other molecules that regulate the glioma microenvironment in VM.
The vascular endothelial growth factor receptor-2 (VEGFR-2)/Flk-1 pathway
VEGFR-2 has emerged as an essential angiogenic mediator of signaling cascades induced by vascular endothelial growth factor (VEGF). High levels of both VEGFR-2 and VEGF are co-expressed during angiogenesis. The VEGF-VEGFR pathway and its downstream molecular cascades are thought to activate both traditional angiogenesis and VM in some aggressive tumors [18,40]. VEGFR-2, a receptor tyrosine kinase, is the earliest differentiation marker for ECs. Its expression in adults is normally restricted primarily to ECs, but is up-regulated transiently during angiogenesis [29]. VEGFR-2 has been reported to be over-expressed in GBM by GSCs at both the mRNA and protein level. Once activated by VEGF, this signaling pathway mediates chemotaxis, self-renewal, tumorigenicity, tubule formation and over-expression of critical VM markers [26]. VEGFR-2 phosphorylation at specific tyrosine residues in an intracellular kinase domain in its C-terminus subsequently activates down-stream intracellular signaling molecular cascades, including the focal adhesion kinase (FAK) and mitogen-activated protein kinase (MAPK)- extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) cascades, and smooth muscle actin (SMa) expression. Activation of the FAK and MAPK-ERK1/2 signaling pathways then mediate cell proliferation, migration, and tubule formation [26]. However, these events do not require VEGF stimulation in GBM-derived tumor cell lines [26,29], which might partially be explained by the constitutive phosphorylation of VEGFR-2 in GSCs [26]. VEGFR-2 also may be activated indirectly by other factors in the tumor microenvironment, either through binding to membrane-associated integrins or by coordinate induction of integrins, leading to increased VEGFR-2 activation [26]. Follow-up studies using animal tumor models also validate the indispensable role of VEGFR-2, which is independent of VEGF and regulates mural-like tumor cell-associated VM in GBM [21,26].
The VE-cadherin (CDH5) and EphA2 pathways
VE-cadherin, a member of the cadherin-family, is a transmembrane glycoprotein that is thought to be specifically expressed in ECs. VE-cadherin promotes homotypic cell-cell interactions and was one of the first molecules identified as an important player in VM for melanoma [41]. The Eph family is the largest family of receptor tyrosine kinases (RTKs) and regulates cell proliferation, migration and angiogenesis. Similar to the findings for melanoma [42], both VE-cadherin and EphA2 are expressed at higher levels in VM-positive glioma than VM-negative glioma, and the expression of these genes correlates with the glioma grade and is known to be required for VM network formation [30,31,43]. Low expression levels in glioma cell lines that are incapable of VM formation further indicates a role for these molecules in VM [31,32]. Additionally, knockdown of these genes with short hairpin RNA causes a predominant decrease in VM formation [31,42]. For hypoxia-induced VM, VE-cadherin is up-regulated by hypoxia-inducible factor (HIF) 2α and 1α through direct binding to the VE-cadherin promoter [31]. Furthermore, EphA2 over-expression couples with reduced miR-26b expression, properly speaking, miR-26b down-regulates the levels of endogenous EphA2 protein by binding to a specific microRNA response element in its 3’UTR [30]. In 2006, Hess et al. demonstrated that in VM-positive melanoma, VE-cadherin co-localizes with EphA2 at areas of cell-cell contact, and these two molecules interact either directly or indirectly during the process of VM formation [42]. VE-cadherin regulates EphA2 activity by mediating its auto-phosphorylation through interaction with its membrane bound ligand, Ephrin-A1. Phosphorylated EphA2 subsequently regulates the phosphoinositide 3-kinase (PI3K) and FAK [44,45] pathways, thus activating the PI3K-MMP-laminin 5γ2 chain signaling pathways and related intracellular signaling cascades. Whether a similar pathway of regulation occurs in GBM awaits further verification.
The RTK/PI3K/Akt/mTOR signaling pathway
The mutant RTK/PI3K/Akt/mTOR pathway is the most frequently deregulated signaling cascade in GBM and regulates various cellular processes such as proliferation, growth, apoptosis, and cytoskeletal rearrangement [46]. This pathway has been accepted as a novel genetic target for acquired glioma resistance [45,47,48].
RTKs are the largest group of the enzyme linked receptor families, which possess an N-terminal extracellular ligand-binding domain, a single anchoring transmembrane-helix, and a cytosolic C-terminal region that contains the catalytic domain [46]. In glioma, EGFR is one of the most prominent members [46,47]. PI3K is a cytoplasmic lipid kinase consisting of a regulatory subunit, p85, and a catalytic subunit, p110. Combination of p85 and the RTK results in the activation of catalytic subunit (p110), which then catalyzes the phosphorylation of PI 3,4-bisphosphate (PIP2) into 3,4,5-triphosphate (PIP3) [46]. PIP3 in turn activates phosphoinositide-dependent kinase-1 (PDK1), which subsequently activates Akt by phosphorylating specific amino acid sites. Activated Akt regulates a series of downstream molecules such as mTOR, which controls a variety of cellular functions [46]. Deficiency of PTEN and related regulators in glioma, which serve as negative regulators of PIP2 and PIP3, promotes tumor invasiveness [49]. Conversely, targeting of the mutant RTK/PI3K/Akt/mTOR signaling pathway leads to consistent reduction in the invasion and migration by VM channels in U251 glioma cells [49]. In concert with these results, VM-related molecules such as EphA2 and MMPs also show a corresponding reduction [49]. These findings suggest that the aberrant RTK/PI3K/Akt/mTOR pathway plays a role in GBM VM formation.
The MMP-laminin 5γ2 chain signaling pathway
The laminin 5γ2 chain is the main component of the basal membrane, which can be degraded by MMPs. The over-expression of MMPs in several malignant tumors such as aggressive melanoma [50] and GBM [17,32] can promote tumor invasion and migration. In GBM cell lines [32] and GBM samples [17], MMPs showed a positive correlation with VM formation.
The MMP-laminin5γ2 chain pathway has been suggested as a common downstream signaling pathway of several molecular cascades. The MMP-laminin 5γ2 chain and other signaling regulators associated with VM (TGFβ [32], VE-cadherin, EphA2, PI3K [42] etc.) are coordinately over-expressed in a number of malignancies including GBM [32] and aggressive melanoma [50]. MMP transcription is regulated by upstream regulators such as TGFβ [32]. When activated by upstream molecules, MT1-MMP converts proMMP2 to active MMP2. Both MT1-MMP and MMP2 promote the cleavage of the laminin 5γ2 chain into promigratory γ2′and γ2x fragments, which in turn stimulate migration, invasion, and VM formation [32]. MT1-MMP siRNA promotes a sharp decrease in the cleavage of the laminin 5γ2 chain and VM formation [32]. Thus, the MMP-laminin5γ2 chain pathway may serve as the final executor of VM formation for several molecular cascades.
Other molecules that regulate the glioma microenvironment in VM
Cyclo-oxygenase-2 (COX-2), an inducible isoform of the prostaglandin synthesis enzyme cyclooxygenase, shows a positive association with VM channel formation [51]. Pathophysiological factors induce the expression of this enzyme, which is not constitutively expressed in physiological states. COX-2 has been shown to promote cell survival, proliferation, and angiogenesis and prohibit apoptosis, all of which are involved in tumor progression [51]. Recent reports have revealed that highly invasive human glioma cell lines exhibit higher COX-2 expression with vascular channels formation when cultured on three-dimensional Matrigel, whereas non-invasive cell lines do not exhibit this biological phenomenon. These results are supported by studies of human glioma specimens [17]. Inhibition of COX-2 with Celecoxib or specific siRNAs caused a noteworthy reduction in VM formation, suggesting that COX-2 functions in the formation of VM structures.
Other molecules potentially associated with VM formation in GBM include microRNA-9 (miR-9) and Galectin-1 (Gal-1). MiR-9, a tissue-specific microRNA in the central nervous system, inhibits VM formation of glioma cell lines by suppressing Stathmin expression [52]. Gal-1 is regulated by a brain-expressed X-linked gene and is reported to be associated with VM in vitro and in vivo in an oligodendroglioma model [53]; therefore, a similar role for Gal-1 might characterize GBM. Though the exact mechanisms of these molecules are not well elucidated, it is likely that these so-called VM-related pathways co-operate with other pathways in the remodeling of the VM extracellular microenvironment.
Hypoxia-related mechanism of VM
Hypoxia may potentially be the earliest inducer of VM and influences VM throughout the process. Hypoxia is more easily detectable in more invasive and rapidly proliferating tumors. Furthermore, the role for hypoxia in VM is supported by findings that in vitro hypoxic conditions can lead to VM formation [31]. Induction of VM formation in the absence of oxygen may explain the poor outcome of conventional anti-angiogenic therapy in aggressive melanoma [2]. As a hallmark of most tumors, hypoxia participates in different pathways to maintain a stem cell-like phenotype, regulate cellular differentiation, and promote tumor invasion, metastasis, resistance to apoptosis, angiogenesis and VM [18].
During VM formation in GBM, HIF2α and 1α regulate VE-cadherin expression by directly interacting with hypoxia responsive elements (HREs) in its promoter [31]; however, in aggressive melanoma, though hypoxia induces VM via VE-cadherin, VE-cadherin is up-regulated by Bcl-2 rather than HIFs [54]. Moreover, during the process of GSC generation and transdifferentiation towards tubular formation in vitro, hypoxia offers an indispensable condition for GSCs to differentiate into a group of specialized cells that express specific biomarkers [55], many of which are thought to be promoted by HIFs [54]. Though much work has been done on VM formation, the substantial cellular and molecular mechanisms underlying the effects of hypoxia in the pathogenesis of VM in GBM remain largely elusive.
Clinical-translational advances in the treatment of GBM and future prospects of VM as a therapeutic target
As highlighted by the multitude of signaling pathways discussed above, comprehensive treatments are indispensable for the control of highly aggressive tumors. Consequently, the understanding of how mono-therapies coordinate to optimize therapeutic outcomes may help to further the understanding of GBM. To achieve this goal, mounting efforts have focused on identifying the related mechanisms. The highly plastic phenotype, sufficient blood perfusion and an adequate nutrient supply are critical for sustaining continual progression of neoplasms. However, the regulatory mechanisms that create these conditions may provide the key to control the process: the tumor cells, along with their stem cells and pericytes, construct their habitat with the assistance of autocrine or paracrine signals, which in turn influence the tumor properties. To obtain the maximum neoplastic effect, a tumor must implement all of the above strategies for the positive reinforcement of growth. Hence, the available tumor therapies, on one hand, are aimed at targeting tumor cells and their progenitor CSCs; and on the other hand, are aimed at simultaneously suppressing the remodeling of the microenvironment and related molecules. For optimal design of therapeutics, the removal or disruption of tumor-related cells, the elimination of essential oxygen and energy supply and the intervention of underlying signaling cascades should all be taken into consideration.
Aside from tumor cell-targeting therapy, the mechanisms of angiogenesis and vascularization are perplexing enough to elicit detailed interpretation. Because the conventional anti-angiogenesis strategies exhibited disappointing results in cumulative clinical trials, several other paradigms about tumor blood supply have emerged, including VM, which has drawn intense controversy since it was first described in 1999. Since then, evidence has begun to accumulate to validate the existence and significance of this novel circulatory structure [1,17,56,57], and meta-analysis has validated the association between VM and poor prognosis [58]. A variety of staining methods from physicochemical dyeing to immunofluorescence techniques have been utilized to reveal the histological features and origins of VM; multidisciplinary approaches have been applied for understanding the aberrant gene transcription and protein translation associated with VM. On the basis of these studies, the functions of a number of genes have been validated in VM, and several molecular pathways have been demonstrated that could provide potential targets for therapy.
Among the potential therapies for VM, anti-angiogenesis by the VEGF mono-antibody Bevacizumab showed minimal efficacy and enhanced tumor invasiveness triggered by hypoxia induction, which may be partially due to VM [33]; and the potent angiogenesis inhibitor Endostar did not affect GBM VM formation [59]. While therapies aimed at classical anti-angiogenesis have shown limited effects, the VM associated mechanisms offered new insights. A handful of preclinical studies suggest that specific compounds affecting components of the previously described vascular, embryonic or hypoxia pathways in tumor cells can inhibit VM formation in xenograft tumor models. VEGFR-2 kinase inhibitors SU1498 and AZD2171 have been shown to reduce VM channel formation in GBM cell lines in vitro and in vivo, concurrent with a reduction in chemotaxis, proliferation and tumorigenicity [26]. Likewise, the MMP inhibitor chemically modified tetracycline [28] and the TGF inhibitor isoxanthohumol [60] downregulate VE-cadherin, EphA2, laminin5γ2 and MMPs and impair VM formation. Lidamycin suppresses tubular structure in a dose-dependent manner, potentially through an apoptosis-related mechanism [61]; while targeting of the RTK/PI3K/Akt pathway enhances the cytotoxic effect of radiation and temozolomide in malignant glioma cells [49].
The increasing abundance of targeted techniques, such as gene knockdown technology, delineates a tangible realization of the potential for counteracting tumor-related mechanisms, which should eventually lead to effective treatments for GBM and other human diseases. In addition, newly devised targeted drug delivery systems circumvent multidrug resistance and demonstrate an enhanced chemotherapeutic efficacy [62,63] . The use of strategies targeting multiple signaling pathways in a combinatorial manner may lead to increased therapeutic efficiency; and studies on VM as a novel and distinct circulatory system will certainly contribute significantly to the future development of anti-tumor treatment regimens.
Acknowledgement
This work was supported by the National Natural Science Foundation of China (No. 81201991), the Basic Research Program of Shanxi Province of China (No. 2012021035–5), the Program for the Outstanding Innovative Teams of Higher Learning Institutions of Shanxi, and the Scientific Research Subject of Shanxi Provincial Health Department (No. 201301066).
Abbreviations
- GBM
Glioblastoma
- VM
Vasculogenic mimicry
- ECs
Endothelial cells
- PAS
Periodic acid-schiff
- GSCs
Glioma stem-like cells
- CSCs
Cancer stem cells
- EMT
Epithelial-mesenchymal-transition
- TGFβ
Transforming growth factor β
- VEGFR-2/Flk-1
Vascular endothelial growth factor receptor-2
- MMPs
Matrix metalloproteinases
- MT1-MMP
Membrane-type 1
- PDK1
Phosphoinositide-dependent kinase-1
- FAK
Focal adhesion kinase
- MAPK
Mitogen-activated protein kinase
- ERK1/2
Extracellular signal-regulated protein kinases 1 and 2
- RTKs
Receptor tyrosine kinases
- EGFR
Epidermal growth factor receptor
- PI3K
Phosphoinositide 3-kinase
- HIF
Hypoxia-inducible factor
- COX-2
Cyclo-oxygenase-2
- HREs
Hypoxia responsive elements
- miR-9
microRNA-9
- Gal-1
Galectin-1
Footnotes
Jin-ming Mao and Jing Liu contributed equally to this work.
Competing interests
All authors disclose no financial and personal relationships with other people or organizations that could inappropriately influence (bias) our work.
Authors’ contributions
Geng Guo and Xing-gang Mao designed the study, Jin-ming Mao and Jing Liu drafted the manuscript. All authors have contributed to revising the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Jin-ming Mao, Email: 1195360990@qq.com.
Jing Liu, Email: jingliu0813@163.com.
Geng Guo, Email: guogeng973@163.com.
Xing-gang Mao, Email: xgmao@hotmail.com.
Chang-xin Li, Email: 476900031@qq.com.
References
- 1.Wang SY, Ke YQ, Lu GH, Song ZH, Yu L, Xiao S, et al. Vasculogenic mimicry is a prognostic factor for postoperative survival in patients with glioblastoma. J Neurooncol. 2013;112(3):339–45. doi: 10.1007/s11060-013-1077-7. [DOI] [PubMed] [Google Scholar]
- 2.Soda Y, Myskiw C, Rommel A, Verma IM. Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. J Mol Med (Berl) 2013;91(4):439–48. doi: 10.1007/s00109-013-1019-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plate KH, Scholz A, Dumont DJ. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012;124(6):763–75. doi: 10.1007/s00401-012-1066-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–52. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fausto N. Vasculogenic mimicry in tumors. Fact or artifact? Am J Pathol. 2000;156(2):359. doi: 10.1016/S0002-9440(10)64738-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–21. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 8.Shirakawa K, Wakasugi H, Heike Y, Watanabe I, Yamada S, Saito K, et al. Vasculogenic mimicry and pseudo-comedo formation in breast cancer. Int J Cancer. 2002;99(6):821–8. doi: 10.1002/ijc.10423. [DOI] [PubMed] [Google Scholar]
- 9.Sood AK, Fletcher MS, Zahn CM, Gruman LM, Coffin JE, Seftor EA, et al. The clinical significance of tumor cell-lined vasculature in ovarian carcinoma: implications for anti-vasculogenic therapy. Cancer Biol Ther. 2002;1(6):661–4. doi: 10.4161/cbt.316. [DOI] [PubMed] [Google Scholar]
- 10.Fan YZ, Sun W, Zhang WZ, Ge CY. Vasculogenic mimicry in human primary gallbladder carcinoma and clinical significance thereof. Zhonghua Yi Xue Za Zhi. 2007;87(3):145–9. [PubMed] [Google Scholar]
- 11.Guzman G, Cotler SJ, Lin AY, Maniotis AJ, Folberg R. A pilot study of vasculogenic mimicry immunohistochemical expression in hepatocellular carcinoma. Arch Pathol Lab Med. 2007;131(12):1776–81. doi: 10.5858/2007-131-1776-apsovm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao H, Gu XM. Study on vasculogenic mimicry in malignant esophageal stromal tumors. World J Gastroenterol. 2008;14(15):2430–3. doi: 10.3748/wjg.14.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vartanian AA, Stepanova EV, Gutorov SL, Solomko E, Grigorieva IN, Sokolova IN, et al. Prognostic significance of periodic acid-Schiff-positive patterns in clear cell renal cell carcinoma. Can J Urol. 2009;16(4):4726–32. [PubMed] [Google Scholar]
- 14.Yue WY, Chen ZP. Does vasculogenic mimicry exist in astrocytoma? J Histochem Cytochem. 2005;53(8):997–1002. doi: 10.1369/jhc.4A6521.2005. [DOI] [PubMed] [Google Scholar]
- 15.El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, et al. A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain. 2010;133(Pt 4):973–82. doi: 10.1093/brain/awq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004;112(7–8):508–25. doi: 10.1111/j.1600-0463.2004.apm11207-0810.x. [DOI] [PubMed] [Google Scholar]
- 17.Liu XM, Zhang QP, Mu YG, Zhang XH, Sai K, Pang JC, et al. Clinical significance of vasculogenic mimicry in human gliomas. J Neurooncol. 2011;105(2):173–9. doi: 10.1007/s11060-011-0578-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18(10):2726–32. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weidner N. Tumoural vascularity as a prognostic factor in cancer patients: the evidence continues to grow. J Pathol. 1998;184(2):119–22. doi: 10.1002/(SICI)1096-9896(199802)184:2<119::AID-PATH17>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 20.Frenkel S, Barzel I, Levy J, Lin AY, Bartsch DU, Majumdar D, et al. Demonstrating circulation in vasculogenic mimicry patterns of uveal melanoma by confocal indocyanine green angiography. Eye (Lond) 2008;22(7):948–52. doi: 10.1038/sj.eye.6702783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scully S, Francescone R, Faibish M, Bentley B, Taylor SL, Oh D, et al. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J Neurosci. 2012;32(37):12950–60. doi: 10.1523/JNEUROSCI.2017-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 23.Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468(7325):824–8. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 24.Fan YL, Zheng M, Tang YL, Liang XH. A new perspective of vasculogenic mimicry: EMT and cancer stem cells (Review) Oncol Lett. 2013;6(5):1174–80. doi: 10.3892/ol.2013.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Guo H, Zhang D, Zhang W, Zhao X, Ren Z, et al. Microcirculation patterns in different stages of melanoma growth. Oncol Rep. 2006;15(1):15–20. [PubMed] [Google Scholar]
- 26.Yao X, Ping Y, Liu Y, Chen K, Yoshimura T, Liu M, et al. Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) Plays a Key Role in Vasculogenic Mimicry Formation, Neovascularization and Tumor Initiation by Glioma Stem-like Cells. PLoS One. 2013;8(3):e57188. doi: 10.1371/journal.pone.0057188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi L, Zhou ZH, Ping YF, Chen JH, Yao XH, Feng H, et al. Isolation and characterization of stem cell-like precursor cells from primary human anaplastic oligoastrocytoma. Mod Pathol. 2007;20(10):1061–8. doi: 10.1038/modpathol.3800942. [DOI] [PubMed] [Google Scholar]
- 28.Seftor RE, Seftor EA, Kirschmann DA, Hendrix MJ. Targeting the tumor microenvironment with chemically modified tetracyclines: inhibition of laminin 5 gamma2 chain promigratory fragments and vasculogenic mimicry. Mol Cancer Ther. 2002;1(13):1173–9. [PubMed] [Google Scholar]
- 29.Francescone R, Scully S, Bentley B, Yan W, Taylor SL, Oh D, et al. Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 protein activation. J Biol Chem. 2012;287(29):24821–31. doi: 10.1074/jbc.M111.334540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang Y, et al. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PLoS One. 2011;6(1):e16264. doi: 10.1371/journal.pone.0016264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao XG, Xue XY, Wang L, Zhang X, Yan M, Tu YY, et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro Oncol. 2013;15(7):865–79. doi: 10.1093/neuonc/not029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling G, Wang S, Song Z, Sun X, Liu Y, Jiang X, et al. Transforming growth factor-beta is required for vasculogenic mimicry formation in glioma cell line U251MG. Cancer Biol Ther. 2011;12(11):978–88. doi: 10.4161/cbt.12.11.18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu B, Guo L, Ma J, Lv Y. Antiangiogenesis therapy might have the unintended effect of promoting tumor metastasis by increasing an alternative circulatory system. Med Hypotheses. 2010;74(2):360–1. doi: 10.1016/j.mehy.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 34.Sun J, Liu SZ, Lin Y, Cao XP, Liu JM. TGF-beta promotes glioma cell growth via activating Nodal expression through Smad and ERK1/2 pathways. Biochem Biophys Res Commun. 2014;443(3):1066–72. doi: 10.1016/j.bbrc.2013.12.097. [DOI] [PubMed] [Google Scholar]
- 35.Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21(2):123–32. doi: 10.1111/j.1755-148X.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- 36.Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 2008;21(1):39–55. doi: 10.1111/j.1755-148X.2007.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim ES, Sohn YW, Moon A. TGF-beta-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF)2 in human breast epithelial cells. Cancer Lett. 2007;252(1):147–56. doi: 10.1016/j.canlet.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Kuo YC, Su CH, Liu CY, Chen TH, Chen CP, Wang HS. Transforming growth factor-beta induces CD44 cleavage that promotes migration of MDA-MB-435 s cells through the up-regulation of membrane type 1-matrix metalloproteinase. Int J Cancer. 2009;124(11):2568–76. doi: 10.1002/ijc.24263. [DOI] [PubMed] [Google Scholar]
- 39.Topczewska JM, Postovit LM, Margaryan NV, Sam A, Hess AR, Wheaton WW, et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med. 2006;12(8):925–32. doi: 10.1038/nm1448. [DOI] [PubMed] [Google Scholar]
- 40.Paulis YW, Soetekouw PM, Verheul HM, Tjan-Heijnen VC, Griffioen AW. Signalling pathways in vasculogenic mimicry. Biochim Biophys Acta. 2010;1806(1):18–28. doi: 10.1016/j.bbcan.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci U S A. 2001;98(14):8018–23. doi: 10.1073/pnas.131209798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hess AR, Seftor EA, Gruman LM, Kinch MS, Seftor RE, Hendrix MJ. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: implications for vasculogenic mimicry. Cancer Biol Ther. 2006;5(2):228–33. doi: 10.4161/cbt.5.2.2510. [DOI] [PubMed] [Google Scholar]
- 43.Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3(10):541–51. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- 44.Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117(Pt 10):2037–49. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- 45.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170(5):1445–53. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV, et al. Aberrant signaling pathways in glioma. Cancers (Basel) 2011;3(3):3242–78. doi: 10.3390/cancers3033242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan QW, Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr Top Microbiol Immunol. 2010;347:279–96. doi: 10.1007/82_2010_67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sami A, Karsy M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: novel therapeutic agents and advances in understanding. Tumour Biol. 2013;34(4):1991–2002. doi: 10.1007/s13277-013-0800-5. [DOI] [PubMed] [Google Scholar]
- 49.Choi EJ, Cho BJ, Lee DJ, Hwang YH, Chun SH, Kim HH, et al. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer. 2014;14(1):17. doi: 10.1186/1471-2407-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seftor RE, Seftor EA, Koshikawa N, Meltzer PS, Gardner LM, Bilban M, et al. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001;61(17):6322–7. [PubMed] [Google Scholar]
- 51.Fan YZ, Sun W. Molecular regulation of vasculogenic mimicry in tumors and potential tumor-target therapy. World J Gastrointest Surg. 2010;2(4):117–27. doi: 10.4240/wjgs.v2.i4.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song Y, Mu L, Han X, Li Q, Dong B, Li H, et al. MicroRNA-9 inhibits vasculogenic mimicry of glioma cell lines by suppressing Stathmin expression. J Neurooncol. 2013;115(3):381–90. doi: 10.1007/s11060-013-1245-9. [DOI] [PubMed] [Google Scholar]
- 53.Le Mercier M, Fortin S, Mathieu V, Roland I, Spiegl-Kreinecker S, Haibe-Kains B, et al. Galectin 1 proangiogenic and promigratory effects in the Hs683 oligodendroglioma model are partly mediated through the control of BEX2 expression. Neoplasia. 2009;11(5):485–96. doi: 10.1593/neo.81526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao N, Sun BC, Sun T, Ma YM, Zhao XL, Liu ZY, et al. Hypoxia-induced vasculogenic mimicry formation via VE-cadherin regulation by Bcl-2. Med Oncol. 2012;29(5):3599–607. doi: 10.1007/s12032-012-0245-5. [DOI] [PubMed] [Google Scholar]
- 55.Dong J, Zhang Q, Huang Q, Chen H, Shen Y, Fei X, et al. Glioma stem cells involved in tumor tissue remodeling in a xenograft model. J Neurosurg. 2010;113(2):249–60. doi: 10.3171/2010.2.JNS09335. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, Jing Z, Luo C, Zhuang M, Xia J, Chen Z, et al. Vasculogenic mimicry-potential target for glioblastoma therapy: an in vitro and in vivo study. Med Oncol. 2012;29(1):324–31. doi: 10.1007/s12032-010-9765-z. [DOI] [PubMed] [Google Scholar]
- 57.Chen YS, Chen ZP. Vasculogenic mimicry: a novel target for glioma therapy. Chin J Cancer. 2013;33(2):74–9. doi: 10.5732/cjc.012.10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao Z, Bao M, Miele L, Sarkar FH, Wang Z, Zhou Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: a systemic review and meta-analysis. Eur J Cancer. 2013;49(18):3914–23. doi: 10.1016/j.ejca.2013.07.148. [DOI] [PubMed] [Google Scholar]
- 59.Liu Z, Li Y, Zhao W, Ma Y, Yang X. Demonstration of vasculogenic mimicry in astrocytomas and effects of Endostar on U251 cells. Pathol Res Pract. 2011;207(10):645–51. doi: 10.1016/j.prp.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 60.Serwe A, Rudolph K, Anke T, Erkel G. Inhibition of TGF-beta signaling, vasculogenic mimicry and proinflammatory gene expression by isoxanthohumol. Invest New Drugs. 2012;30(3):898–915. doi: 10.1007/s10637-011-9643-3. [DOI] [PubMed] [Google Scholar]
- 61.Li XQ, Zhang SH, Ouyang ZG, Zhen YS. Inhibitory effect of lidamycin upon vasculogenic mimicry and its induction of apoptosis in glioma cells. Zhonghua Yi Xue Za Zhi. 2009;89(25):1736–40. [PubMed] [Google Scholar]
- 62.Li XY, Zhao Y, Sun MG, Shi JF, Ju RJ, Zhang CX, et al. Multifunctional liposomes loaded with paclitaxel and artemether for treatment of invasive brain glioma. Biomaterials. 2014;35(21):5591–604. doi: 10.1016/j.biomaterials.2014.03.049. [DOI] [PubMed] [Google Scholar]
- 63.Ju RJ, Mu LM, Lu WL. Targeting drug delivery systems for circumventing multidrug resistance of cancers. Ther Deliv. 2013;4(6):667–71. doi: 10.4155/tde.13.39. [DOI] [PubMed] [Google Scholar]