

Abstract
Programmed capillary regression and remodeling are essential developmental processes. However, the cellular and molecular mechanisms that regulate vessel regression are only beginning to be understood. Here, using in vivo, dynamic, confocal imaging of mouse transgenic reporters as well as static confocal and electron microscopy, we studied the embryonic development and postnatal regression of the transient mouse pupillary membrane (PM) vasculature. This approach allowed us to directly observe the precise temporal sequence of cellular events preceding and during the elimination of the PM from the mouse eye. Imaging of Tcf/Lef-H2B::GFP Wnt-reporter mice uncovered that, unlike the hyaloid vasculature of the posterior eye, a PM endothelial cell (EC) Wnt/β-catenin response is unlikely to be part of the regression mechanism. Live imaging of EC and macrophage dynamics revealed highly active Csf1r-GFP+ macrophages making direct contact with the Flk1-myr::mCherry+ vessel surface and with membrane protrusions or filopodia extending from the ECs. Flk1-myr::mCherry+ EC membrane particles were observed on and around ECs as well as within macrophages. Electron microscopy studies confirmed that they were in phagosomes within macrophages, indicating that the macrophages engulfed the membrane particles. Interestingly, EC plasma membrane uptake by PM macrophages did not correlate with apoptosis and was found shortly after vessel formation at mid-gestation stages in the embryo; long before vessel regression begins during postnatal development. Additionally, genetic ablation of macrophages showed that EC membrane particles were still shed in the absence of macrophages suggesting that macrophages do not induce the formation or release of EC microparticles. These studies have uncovered a novel event during programmed capillary regression in which resident macrophages scavenge endothelial cell microparticles released from the PM vessels. This finding suggests that there may be an initial disruption in vessel homeostasis embryonically as the PM forms that may underlie its ultimate regression postnatally.
Keywords: eye, pupillary membrane, endothelial cells, macrophages
INTRODUCTION
During mammalian eye development, the immature lens, retina and vitreous are supported by transient capillary beds that ultimately regress, postnatally in rodents and prenatally in humans, to allow light to pass through the eye unimpeded (Ito and Yoshioka, 1999; Zhu et al., 2000). This transient network is comprised of the pupillary membrane (PM) on the anterior surface of the lens and the hyaloid vasculature (HV) behind the lens, which includes the hyaloid artery (HA), tunica vasculosa lentis (TVL) and the vasa hyaloidea propria (VHP) (Ito and Yoshioka, 1999). When regression of the hyaloid network fails to occur, conditions such as persistent hyperplastic primary vitreous (PHPV) and persistent pupillary membrane (PPM) result and can lead to vision deficiencies. Therefore, elucidation of the cellular and molecular mechanisms driving HV and PM regression will lead to better treatments of PHPV and PPM. Of great interest to developmental biologists, HV and PM regression is an exquisitely precise developmental event in which vessel segments are selectively eliminated from an otherwise healthy tissue. Thus, the mechanisms that underlie these events are likely integrated with other vital developmental pathways and a better understanding of these mechanisms could impact anti-neoangiogenic strategies to treat diseases such as cancer.
Recent decades of research on the mechanisms regulating PM and HV regression have uncovered an essential role for resident macrophages (Diez-Roux and Lang, 1997; Lang et al., 1994; Lang and Bishop, 1993; Lobov et al., 2005). From the analyses of transgenic mice that express diphtheria toxin specifically in macrophages, and later in macrophage-deficient PU.1 null mice, it was shown that both the PM and HV vascular beds persist well into postnatal stages (Lang and Bishop, 1993; Lobov et al., 2005). In the case of the HV, further experiments showed that macrophage-mediated Wnt7b signaling is an essential molecular trigger of endothelial cell (EC) apoptosis and consequential vessel regression (Lobov et al., 2005). HV macrophages express Wnt7b during postnatal regression and genetic down-regulation of Wnt7b signaling results in the same persistent HV phenotype as the macrophage-deficient PU.1 null mice (Lobov et al., 2005). However, despite an obvious requirement for macrophages, PM vascular regression has not been reported to require Wnt7b activity (Lobov et al., 2005). Thus, it is currently not clear whether the mechanism driving regression of the PM is similar to that of the HV.
Here, we report an unbiased approach using live imaging of the mouse PM to delineate cellular and molecular signaling events that take place during PM regression. Specifically, we employed five different, previously validated transgenic fluorescent reporters (Table 1) to label individual cell populations that comprise the PM. Flk1-myr::mCherry and Flk1-H2B::YFP transgenes were used to label EC plasma membranes and nuclei, respectively (Fraser et al., 2005; Larina et al., 2009; Poche et al., 2009). To visualize the resident macrophages, we used the Csf1r-GFP transgenic line and vascular pericytes were marked with an NG2-dsRed transgene (Sasmono et al., 2003; Zhu et al., 2008). In order to monitor Wnt signaling in vivo, we have utilized a Tcf/Lef-H2B::GFP Wnt reporter transgenic mouse in which cells responding to Wnt signaling have a bright, nuclear GFP (Ferrer-Vaquer et al.). Our imaging experiments have led us to revise the timeline of PM development and regression and have uncovered several unexpected phenomena. Specifically, we failed to detect a Wnt/β-catenin response within PM endothelial cells during the regression period. However, we did identify physical interactions between endothelial cells and macrophages that support the role of macrophages in PM regression. We observed macrophages engulfing membrane particles emanating from PM endothelial cells although these events were not associated with cell death or were the macrophages necessary for the release of EC particles. Our findings support a model where there is an early disruption in vessel homeostasis that impedes the ability of the PM vessel network to expand concomitantly with the growing lens and key breaks in vessel segments lead to vessels being cleared from the lens surface.
Table 1.
Transgenic fluorescent reporter mice used in this study.
| Flk1-myr::mCherry | Endothelial cell membranes | Larina et al. (2009) |
| Flk1-H2B::YFP | Endothelial cell nuclei | Fraser et al. (2005) |
| Csf1r-GFP | Macrophages | Sasmono et al. (2003) |
| NG2-dsRed | Pericytes | Zhu et al. (2008) |
| Tcf/Lef-H2B::GFP | Wnt-responding cells | Ferrer-Vaquer et al. (2010) |
METHODS
Mouse Strains
Flk1-myr::mCherry tg/tg and Flk1-H2B::YFP tg/tg mice were maintained on a mixed FVB/CD1 background and all breeding pairs were genotyped for the absence of the retinal degeneration (rd) allele using previously published conditions (Gimenez and Montoliu, 2001). Csf1r-GFP tg/tg (Sasmono et al., 2003), NG2-dsRed +/tg (Zhu et al., 2008) and Tcf/Lef-H2B::GFP +/t g (Ferrer-Vaquer et al.) mice were maintained on a C57BL/6, B6SJLF1 and mixed B6129SF1/ICR background, respectively. All mice were genotyped for the presence of their respective transgenes by screening embryonic and neonatal litters or adult tail snips under a fluorescence microscope. The PU.1+/− and PU.1−/− mice were genotyped as previously described (Henkel et al., 1996; McKercher et al., 1996).
Pupillary membrane and tunica vasculosa lentis whole mounts and immunofluorescence
Enucleated eyes were fixed with 4% paraformaldehyde (PFA) for 1 hour at 4° C. For pupillary membrane (PM) whole mounts, the anterior portion of the eye (including the cornea, iris, ciliary margin and attached lens) was isolated. Using a small spring scissors, the ciliary margin was further trimmed away leaving behind the cornea, iris and lens. #5 forceps were then used to carefully tease away the lens leaving behind the cornea and iris with most of the attached PM capillaries in tact. From there, the tissue was floated onto a glass slide and the cornea layer separated from the iris/PM layer and discarded. The PM with attached iris border was left in a drop of 1X PBS for subsequent immunofluorescence staining on the slide. For tunica vasculosa lentis (TVL) whole mounts, the posterior part of the eye just behind the lens was removed to expose the posterior lens covered with TVL capillaries. These samples were immunolabeled in 1.5 mL Eppendorf tubes. For immunofluorescence, the samples were washed 3 times in 1X PBS containing 0.1% Triton X (10 minutes each wash) and blocked in 2% normal serum (in 1X PBS) for 1 hour at room temperature. These samples were then incubated with primary antibodies (diluted in 2% serum) overnight at 4° C. The following day, the samples were washed 3 times in 1X PBS (10 minutes each), incubated in secondary antibodies for 1 hour at room temperature, washed three times in 1X PBS and stained with DAPI (1:500) for 15 minutes. At this point, the PM slide preparations were mounted under a coverslip using Fluoromount-G (SouthernBiotech) mounting media and the TVLs were mounted on a 35 mm glass bottom dish (MatTek) and overlaid with a coverslip. The samples were imaged immediately using a Zeiss LSM 510 META confocal microscope and a Plan-Apochromat 20X/0.8, C-Apochromat 40X/1.2, or Plan-Apochromat 63X/1.4 objective. Primary antibodies used: anti-dsRed (1:200) (Clontech #632496), anti-Caspase 3 (1:200) (BD Pharmingen #559565), anti-NG2 (1:200) (Millipore #AB5320). Secondary antibodies used: goat, anti-rabbit Alexa Fluor 488, goat, anti-rabbit Alexa Fluor 555 and goat, anti-rabbit Alexa Fluor 647 (1:400) (Molecular Probes).
Immunogold labeling and transmission electron microscopy
Eyes from P3 Flk1-myr::mCherry+/tg mice were enucleated and fixed in 0.1M Sodium Cacodylate (pH 7.2) containing 4% paraformaldehyde for 30 minutes at room temperature. All subsequent steps were performed at room temperature. Next, the posterior eyecup was removed using a small spring scissors leaving behind the anterior eye with the lens attached and this was fixed for an additional hour. This was followed by three ten-minute (3 X 10 mins.) washes in 1X PBS. Next, the samples were blocked in 2% normal goat serum (in 1X PBS) for 1 hour and then incubated for 3 hours with an anti-dsRed antibody, (Clontech #632496), diluted 1:100 in 2% normal goat serum. After incubation with the primary antibody, the samples were then washed in 1X PBS (3 X 10 mins.) and incubated for 1 hour with FluoroNanogold™-anti-rabbit Fab’-Alexa Fluor® 488 secondary antibodies (Nanoprobes, cat. #7204) diluted 1:250 in 2% normal goat serum. Following another round of 1X PBS washes (3 X 10 mins.), the samples were post-fixed with 2% glutaraldehyde (in 1X PBS) for 30 minutes and washed again in 1X PBS (3 X 10 mins.). At this point, the samples were washed in deionized water (3 X 10 mins.) and then subjected to the HQ SILVER™ (Nanoprobe #2012) silver enhancement system following manufacturer instructions. Following 5-minute incubation with the silver enhance solution, the samples were washed in 0.1M Sodium Cacodylate buffer (pH 7.2) (3 X 10 mins.). The samples were then fixed in 0.1% osmium, dehydrated through an acetone series, infiltrated and embedded in Embed-812 (Electron Microscopy Sciences, Hatfield, PA), sectioned (80-100 nm) and stained with lead and uranyl acetate. Sections were viewed on an FEI Tecnai 12 transmission electron microscope operating at 80 keV.
Flk1-H2B::YFP + pupillary membrane image processing and nuclear counting
Eyes from Flk1-H2B::YFP+/tg mice (E12.5 to P10) were enucleated and immediately placed on a 35 mm glass bottom petri dish (MatTek) with the corneal surface facing the glass. The eyes were then imaged en face using a Zeiss LSM 510 META confocal microscope and Plan-Apochromat 20X/0.8 objective. Using LSM Image Examiner software and either the rim of the ciliary body in the embryos or the postnatal iris as a guide, the PM within each image was cropped and its area measured. This was followed by manual counting of every Flk1-H2B::YFP+ cell within the entire PM. Statistically significant differences (t-test) between varying time points were determined for the total number of cells per PM and the number of cells per PM area.
Vital imaging of the pupillary membrane
P0 to P10 fluorescent reporter mice were anesthetized in an induction chamber with 2.5% isoflurane with an oxygen flow rate of 1 L/min. for 5 mins. Once anesthesia was induced, the pup was transferred to the stage of a dissection microscope and isoflurane was continuously administered via a nose cone. To prepare the eye for live imaging, 70% ethanol was swabbed over the eyelid, which was then removed using a #5 forceps and a small spring scissors. The exposed eye was immediately covered in lubricating eye drops (GenTeal, Alcon) to prevent drying. For the imaging session, the pup was positioned on a custom made imaging platform, which contained a #1 cover glass bottom well. The well was filled with additional lubricant gel and the pup placed on the platform with its head sitting on the rim of the well and the exposed eye sitting within the gel-filled well. The pup was then immobilized to the imaging platform using 0.5 mm wide strips of laboratory tape placed across the head and abdomen and gently secured to the imaging platform. The pup and platform were then positioned on the stage of a Zeiss LSM 5 LIVE fast scanning confocal microscope equipped with a heated environmental chamber set to 37° C. The nose cone was re-attached to the immobilized pup and 2.5% isoflurane with an oxygen flow rate of 1 L/min. for 5 mins. was administered for the duration of the imaging session. Time-lapse images were acquired using a LCI Plan-Neofluar 25X/0.8 objective every 3 minutes for up to 9 hours. Once the imaging session was complete, the pup was sacrificed by CO2 asphyxiation followed by decapitation. All supplemental movies have a playback rate of 20 frames per second.
RESULTS
Pupillary membrane endothelial cells do not appear to respond to canonical Wnt/β− catenin signaling
Based on previously published work describing an essential role for macrophage-secreted Wnt7b signaling in HV regression (Lobov et al., 2005), we were interested in determining whether we could capture dynamic Wnt signaling events using the PM as an in vivo model system. To achieve this, we generated Flk1-myr::mCherry; Tcf/Lef-H2B::GFP mice in which the EC plasma membranes are labeled with mCherry and the Wnt-responding cell nuclei are labeled with GFP (Ferrer-Vaquer et al.; Poche et al., 2009). As a control, we first confirmed that the Wnt reporter faithfully recapitulates the pattern of Wnt signaling previously described for the HV (Lobov et al., 2005). Confocal microscopy of TVL and HA whole mounts from E18.5, postnatal day 3 (P3) and postnatal day 7 (P7) mice revealed the expected strong Wnt reporter GFP expression within mCherry+ EC nuclei (Figure 1A-C and data not shown). Interestingly, confocal analysis of the PM at the same developmental stages showed a complete lack of Wnt-reporter activity within the mCherry+ PM vessels (arrows in Figure 1D-F). Instead, Tcf/Lef-H2B::GFP expression was observed in a subset of cells within the lens epithelium (Figure 1D-F) just adjacent to the PM vessels. Thus, it appears unlikely that PM ECs respond canonically to Wnt ligands secreted by macrophages, indicating that another molecular mechanism of PM capillary regression must exist.
Figure 1. Pupillary membrane endothelial cells are not responsive to canonical Wnt signaling.
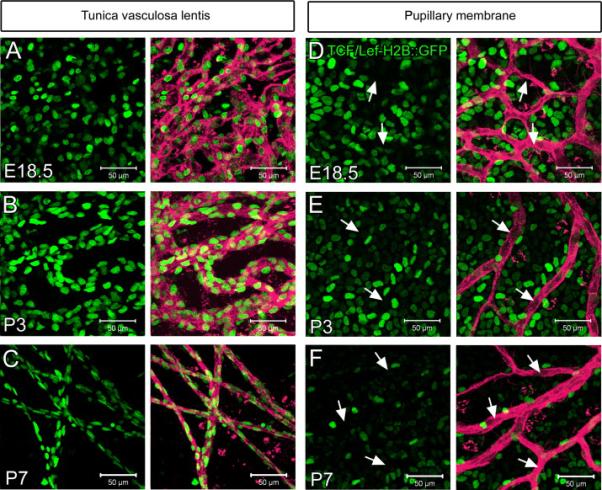
At E18.5, P3 and P7 the tunica vasculosa lentis exhibits Tcf/Lef-H2B::GFP+ reporter expression within Flk1-myr::mCherry+ endothelial cells (A-C). The pupillary membrane Flk1-myr::mCherry+ capillaries are negative for Tcf/Lef-H2B::GFP expression (D-F, arrows). N>3.
Vital imaging of the postnatal pupillary membrane uncovered a role for resident macrophages in scavenging of endothelial cell membrane particles
Resident macrophages clearly play a role in PM regression since a persistent PM phenotype has been observed in the macrophage-deficient and depleted mice (Lang and Bishop, 1993; Lobov et al., 2005). To further define how macrophages contribute to PM regression, we generated Flk1-myr::mCherry; Csf1r-GFP double transgenic mice to monitor the cell-cell interactions between the PM endothelial cells and macrophages. Static imaging of freshly isolated P5 eyes revealed several interesting features. First, every GFP+ macrophage within the PM contained Cherry+ inclusions suggesting that the macrophages had either engulfed dead ECs or had taken up EC membrane particles (Figure 2 A and data not shown). This phenomenon was not observed in other vessels beds that do not undergo regression within the P5 brain, skin, kidney and muscle (Supplementary Figure 1). We also observed discrete, Cherry+ cytoplasmic protrusions extending from the PM ECs and making direct contact with macrophages (Figure 2A and arrowheads in B). In addition to EC projections, Cherry+ EC puncta were also detected on the surface of and adjacent to PM vessels indicating that ECs might be shedding plasma membrane into the extracellular space in the form of microparticles and/or exosomes (arrows in Figure 2B).
Figure 2. Csf1r-GFP+ PM macrophages make contact with Flk1-myr::mCherry+ endothelial cells and engulf endothelial cell plasma membrane.
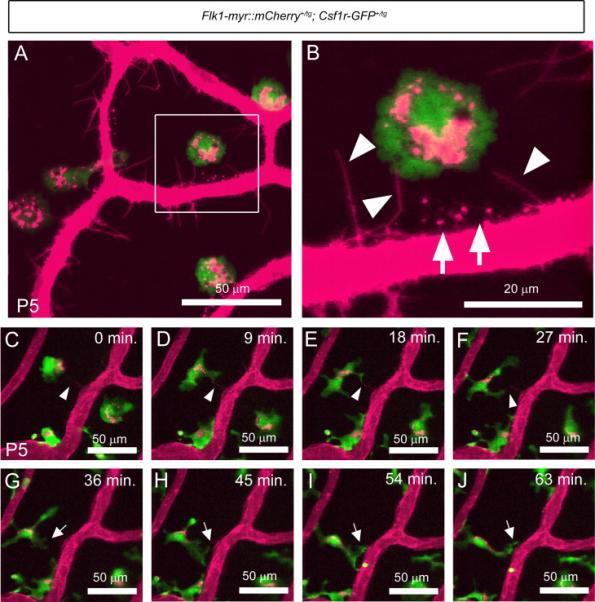
Static imaging at P5, showed Cherry+ plasma membrane particles localized to GFP+ macrophages (A). Higher magnification of the boxed area in panel A shows the PM endothelium as sending out cytoplasmic extensions which project toward the macrophages (arrowheads in B) as well as Cherry+ puncta localized outside of the vasculature (arrows in B). Live imaging of the PM at P5 confirmed that macrophage filopodia make intermittent contact with the Cherry+ cytoplasmic extensions (arrowheads in C to F). Macrophage filopodia also make contact with the walls of the PM vasculature and appear to “grab” plasma membrane particles from the vessel surface (arrows in G to J). N>5.
To further clarify our observations of Flk1-myr::mCherry+/tg; Csf1r-GFP+/tg static images, we performed vital imaging of the postnatal PM within the intact eye. Live imaging of the PM at P3, a time when it is undergoing regression, revealed that the macrophages were extremely dynamic and showed direct contact with mCherry labeled endothelial cells. In low magnification movies of the PM, we observed most GFP+ macrophages extending and retracting lamellipodia/filopodia, contacting and probing the mCherry+ PM vessel surface (Supplemental Movie 1). Higher magnification time lapse imaging of the PM at P5 confirmed that macrophage lamellipodia make intermittent contact with cytoplasmic extensions of the ECs (follow the arrowheads in Figure 2C-F and Supplemental Movie 2). We also observed lamellipodia making contact with the vessel surface and exhibit a “grabbing” behavior toward the EC plasma membrane suggestive of a phagocytic cup (follow the arrows in Figure 2G-J and Supplemental Movie 2).
Transmission electron microscopy further supports a role for PM macrophages in scavenging endothelial cell membrane particles
For additional characterization of the size and location of myr::mCherry+ membrane particles within the PM and adjacent macrophages, we performed immunogold labeling and transmission electron microscopy (TEM). P3 PM ECs were labeled with an anti-dsRed antibody (Clontech) that cross-reacts with mCherry. Immunofluorescent labeling of PM whole mounts confirmed the ability of the antibody to recognize mCherry thereby highlighting Flk1-myr::mCherry expression (Figure 3 A-C). Using immunogold TEM imaging, we labeled the PM ECs with the dsRed antibody that gave the expected punctate, electron-dense signal specifically on the EC surface (Figure 3D-E). We also occasionally observed small, nano-scale vesicular structures (approximately 200-300 nm in diameter) that were labeled with the dsRed antibody and were located outside of the vasculature, similar to those observed by confocal microscopy (arrowheads in Figure 3D-H). The TEM images also revealed that mCherry+ particles observed by confocal microscopy (Figure 3I-K) were indeed localized to phagosomes of the PM resident macrophages (Figure 3L-N). The presence of mCherry particles within macrophage phagosomes clearly shows that PM macrophages scavenge and phagocytose plasma membrane derived from the postnatal PM ECs.
Figure 3. Transmission electron microscopy confirms that macrophages phagocytose endothelial cell plasma membrane.
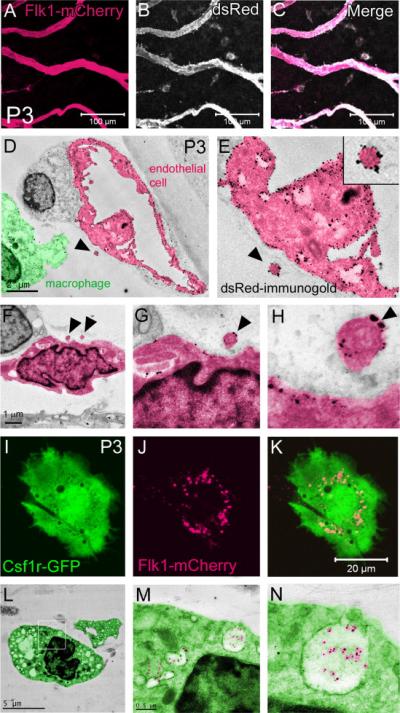
An anti-dsRed antibody effectively recognizes mCherry on the PM endothelial cell plasma membranes (A-C). Immunogold labeling, using the same dsRed antibody, specifically labels the endothelium with gold nanoparticles that are detected by transmission electron microscopy (TEM) (D-E). Discrete, gold+ vesicular structures were observed outside of the endothelium (arrowheads in D-H). Confocal microscopy of a single Csf1-GFP+ PM macrophage containing mCherry+ inclusions (I-K). TEM uncovered gold+ particles localized to macrophage phagosomes confirming that the Flk1-myr::mCherry+ plasma membrane particles are phagocytosed by macrophages L-N). N>5.
Macrophage engulfment of Flk1-myr::mCherry+ cell membrane particles is not due to phagocytosis of apoptotic endothelial cells
Since PM ECs are thought to undergo cell death during early postnatal stages resulting in complete vessel regression by P14 (Ito and Yoshioka, 1999), we next performed static imaging of freshly isolated embryonic (E11.5 to E18.5) eyes from Flk1-myr::mCherry; Csf1r-GFP mice (Figure 4 and data not shown). Here, our expectation was that embryonic macrophages would not contain mCherry+ particles because the developmentally programmed capillary regression has yet to initiate. To our surprise, we found that mCherry+ particles were present in the embryonic PM macrophages. At E11.5, resident macrophages were present within the presumptive PM; however, vessels have yet to invade the area (data not shown). By E12.5, the first PM vessels were visible but mCherry+ inclusions were not seen within the macrophages (Figure 4A-C). Beginning at E14.5, we first observed a few macrophages that were mCherry+ (Figure 4D and arrowheads 4E-F) and by E16.5 all resident macrophages contained mCherry+ particles (data not shown). This persisted to E18.5 and into postnatal stages (Figure 4G and arrowheads in 4H-I).
Figure 4. Macrophage engulfment of endothelial cell membrane particles begins during embryogenesis.
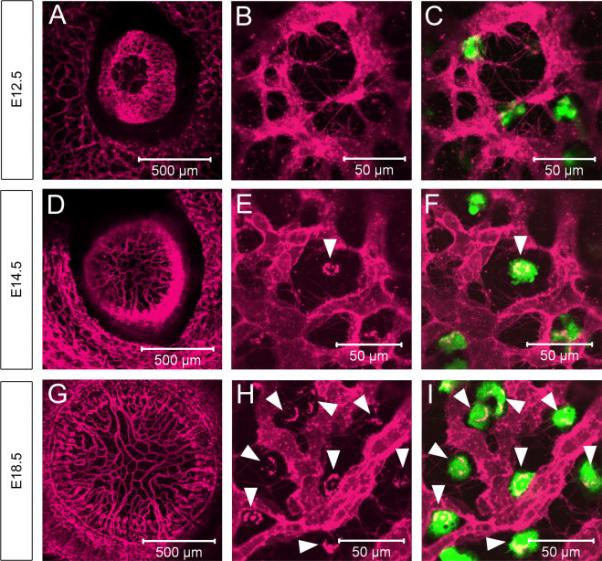
At E12.5, the vasculature has begun to form within the PM, but the resident macrophages have yet to acquire Cherry+ membrane particles (A-C). By E14.5, the PM vasculature is established (D) and a subset of macrophages contains membrane particles (arrowhead in E-F). All of the PM macrophages are positive for membrane particles at E18.5 (G and arrowheads in H-I). N>5.
The above data indicated that macrophage engulfment of EC membrane particles begins shortly after vessel formation and precedes the known timing of PM vessel regression and EC apoptosis. To further determine the relationship between membrane particle engulfment and cell death, we performed two additional experiments. First, PM vessels (E12.5 to E18.5) of Flk1-myr::mCherry+/tg; Flk1-H2B::YFP+/tg mice were analyzed to determine if nuclear fragmentation correlated with PM membrane engulfment. A representative example from the E16.5 PM shows YFP+ EC nuclei within intact mCherry+ vessels. However, we never detected fluorescent signals from YFP+ apoptotic EC nuclear fragments outside of the vascualture or overlapping with the mCherry+ particles within the macrophages (arrowheads in Figure 5A-C), showing that macrophages were not simply engulfing whole dead cells, but rather EC membrane material specifically. We next examined activated Caspase 3+ immunofluorescence in the embryonic PM vasculature and we were not able to detect Caspase 3+ cells within the embryonic PM (data not shown). However, at later postnatal stages (P7 shown), we occasionally observed YFP+ nuclear fragments that clearly correlated with activated Caspase 3+ cells. These data showed that apoptotic cells are present within the postnatal PM and that Flk1-H2B::YFP+ nuclear fragments are a valid readout EC cell death (arrowheads in Figure 5D-F), but the appearance of Flk1-myr::mCherry+ membrane particles within macrophages did not correlate temporally or spatially with endothelial cell apoptosis.
Figure 5. Macrophage engulfment of embryonic endothelial cell membrane particles is not due to apoptosis.
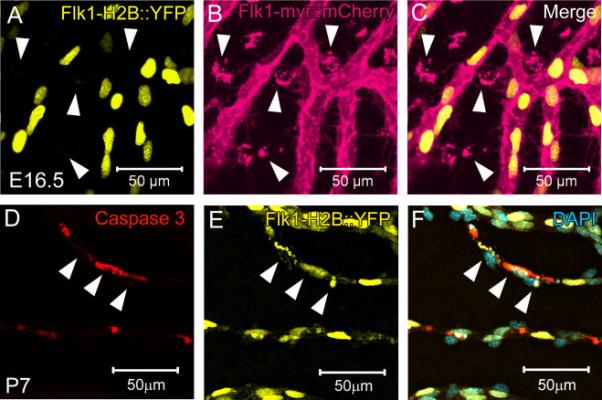
At E16.5, there is no indication of Flk1-H2B::YFP+ apoptotic nuclear fragments or co-localization with Flk1-myr::mCherry+ membrane particles within the macrophages (arrowheads in A-C). By the time EC apoptosis is known to occur (postnatal day 7), activated Caspase 3+ staining is observed as co-localizing with YFP+ nuclear fragments thereby validating Flk1-H2B::YFP+ as a readout for apoptosis (D-F). N>5.
Since it is known that pericytes are often required to maintain vessel homeostasis (Armulik et al.; Augustin et al., 2009), we also investigated whether there was any indication of embryonic pericyte death that might be related to instability of the PM capillary network and EC membrane uptake by macrophages. Here, we performed immunofluorescence on PM whole mounts using an antibody against the N2 proteoglycan, which has been previously reported to be expressed in pericytes in developing capillary beds (Ozerdem et al., 2001; Ruiter et al., 1993). Immunofluorescent labeling of the P3 PM of Flk1-myr::mCherry+/tg mice showed clear investment of NG2+ pericytes within the mCherry+ vasculature (Supplemental Figure 2A-C). To more easily track NG2+ pericytes throughout PM development and regression in situ, the NG2-dsRed transgenic line was used to brightly label NG2+ cells with red fluorescent protein (Zhu et al., 2008). We noticed that at E12.5, a time when the PM has begun to form, NG2+ pericytes are localized to the periphery outside of the PM (Supplemental Figure 2D). This suggests that the pericytes are likely recruited into the PM subsequent to EC invasion. By E14.5, once the PM has formed, pericyte coverage appeared throughout the vascular bed, persisting into early postnatal stages (Supplemental Figure 2E-F). At no point, from E14.5 to P5, did we observe indication of NG2+ cell death (data not shown). However, by P7 NG2-dsRed+ cell fragments, suggestive of dead or dying pericytes, were observed surrounding vessel segments (Supplemental Figure 2G and arrows in H-I) and within resident macrophages (Supplemental Figure 2G and arrowheads in H-I). From these data, there is no indication that pericyte death is correlated with the macrophage engulfment of Flk1-myr::mCherry+ EC membrane particles. While EC membrane particles are detected throughout embryonic and postnatal development, pericyte and EC apoptosis appear concurrently by P7, consistent with published reports of pericyte and EC death in the mouse and rat PM (Diez-Roux et al., 1999; Ito and Yoshioka, 1999).
Pupillary membrane endothelial cell numbers do not begin to decline until postnatal day five
The Flk1-H2B::YFP transgenic line provides superior en face resolution of individual ECs within the PM. Thus, in order to place the appearance of Flk1-myr::mCherry+ membrane particles in the context of EC development and regression specifically, we performed confocal microscopy on freshly isolated E14.5 to P10 Flk1-H2B::YFP+ eyes and generated 3D Z-stack projections. Using either the rim of the ciliary body in the embryos or the postnatal iris as a guide, the region of the PM within these images was cropped and YFP+ cells manually counted (Figure 6A-H). Between E14.5 to E18.5, a statistically significant, two-fold increase in total YFP+ ECs was detected within the PM (Figure 6I). From E18.5 to P5, there was no significant change in total PM EC numbers suggesting that ECs are neither added nor eliminated (Figure 6I). Thus, the PM EC developmental time course can be broken up into three distinct periods – a growth period when the PM is actively developing and adding new ECs (E14.5 to E18.5); a lag period when the numbers of ECs remain constant (E18.5 to P5); and a regression period, when vessel segments are cleared from the PM (P5 to P10). However, comparing total EC numbers per PM area revealed a steady decline in EC density beginning at P0 (Figure 6J). These data indicate that the early growth phase can counteract the increase in PM area to maintain the density in late embryonic development, but once ECs are no longer added or begin to be lost, there is steady decline in EC density as the area of the PM continues to increase.
Figure 6. Time course of pupillary membrane endothelial cell addition and elimination.
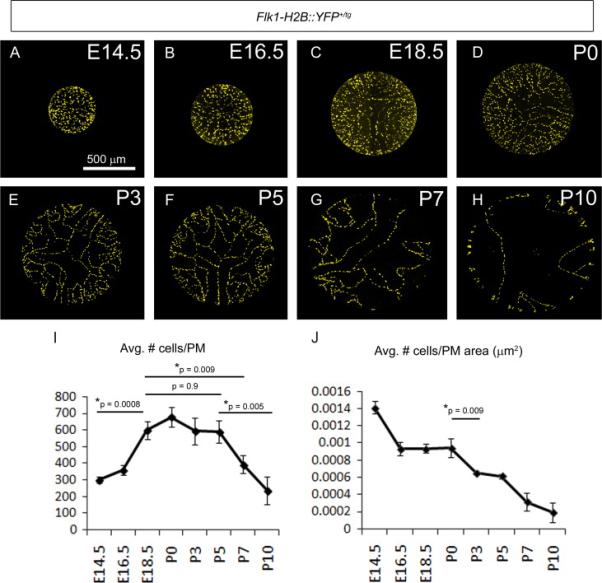
The eyes of embryonic and postnatal Flk1-H2B::YFP+/tg mice were imaged en face, the PM cropped and YFP+ nuclei counted (A-H). Between E14.5 and E18.5, there is a doubling of the EC numbers within the PM after which the EC numbers plateau until P5. Between P5 to P10, PM EC numbers dramatically decline (I). When comparing YFP+ nuclei to PM area, there is a sharp decline in the EC density beginning at P0 that is likely due to the rapid expansion of PM area at a time when EC numbers are constant (J). N=3 per time point.
To directly visualize dynamic events during regression, live imaging was performed of the P0 Flk1-myr::mCherry; Flk1-H2B::YFP PM. During the nine-hour imaging session of the central PM, we noticed the characteristic scavenging movement of mCherry-containing macrophages. Macrophages were observed interacting directly with mCherry labeled vessels as reported previously and several vessel segments showed reduced diameters compared with other vessels within the field of view. However, surprisingly the YFP+ ECs showed no signs of cell migration, death or division as might be expected in a regressing vessel bed (Supplemental movie 3). Instead, ECs were remarkably static, despite the high activity level of macrophages. Thus, ECs do not appear to undergo an active process that could promote remodeling, although vessel thinning was observed. These data clearly show that vessel regression does not involve the mass death or migration of individual ECs but suggests that either vessel thinning from the continued loss of EC membrane material and/or from vessels being stretched as the PM expands is likely to be the mechanism by which vessels are cleared.
EC membrane particles are present even when macrophages are absent in the PU.1 knockout mice
Finally, we sought to determine whether the appearance of EC membrane particles relates specifically to the presence of macrophages with the PM. We crossed the Flk1-myr::mCherry transgene into the PU.1−/− knockout mice that are deficient in macrophages and exhibit a persistent PM (Lobov et al., 2005; McKercher et al., 1996). At P0, the PM vascular plexus in PU.1 mutants contained smaller intervascular spaces as well as larger caliber vessel segments as compared to controls (compare Figure 7A-B). These observations are consistent with the previously described persistent PM phenotype of these mutants (Lang and Bishop, 1993; Lobov et al., 2005). Next, we examined the presence of myr::mCherry+ membrane particles and cytoplasmic protrusions of PU.1 heterozygotes and homozygotes carrying the Flk1-myr::mCherry transgene. Heterozygous control PMs displayed the characteristic large intervascular myr::Cherry+ puncta corresponding to macrophage inclusions (Figure 7C, arrows) while the PU.1−/− mice, which lack macrophages(Lobov et al., 2005), did not display the collections of particles normally found within macrophages (Figure 7D). However, high magnification images of PU.1−/− PMs showed an abundance of myr::mCherry+ particles surrounding the vessels and located within the intervascular spaces, where macrophages are usually found (compare Figure 7C and E to arrows in Figure 7D and F). EC protrusions were also present in PU.1−/− PMs but appeared reduced in number (Figure 7D, arrowhead). These data show that macrophages are not required for the formation of EC membrane protrusions and particle release and clearly indicate that the membrane particles are not a consequence of a macrophage-induced death.
Figure 7. Pupillary membrane protrusions and particle shedding occurs in the absence of macrophages.
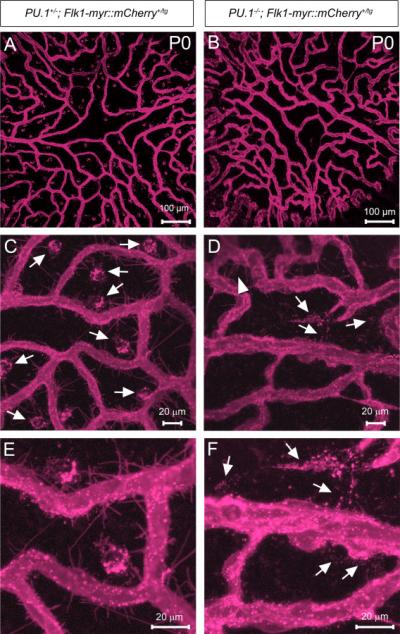
The PM of P0 PU.1+/− and PU.1−/− mice carrying the Flk1-myr::mCherry transgene was imaged en face to examine the expression of mCherry+ EC membrane protrusions and particles (A-B). Compared to the PU.1+/− mice (arrows in C), the PU.1−/− PM is devoid of all large Cherry+ macrophage inclusions due to the loss of macrophages (D). The PU.1−/− PM still exhibits EC protrusions (arrowhead in D) and an apparent increase in intervascular membrane particles (arrows in D and F) as compared to controls (E).
DISCUSSION
Using transgenic fluorescent protein reporters to perform high resolution live and static imaging of cell types within the PM, we have assembled a revised timeline of events leading to PM regression that now includes details of vessel abnormalities even at embryonic stages (Figure 8A). By E12.5, the PM capillary network has developed to cover the anterior lens and there is no sign EC stress. However, by E14.5 we identified EC membrane particle phagocytosis by macrophages which continues throughout embryonic development as the PM grows. The release of EC membrane microparticles has been shown to be a stress response in endothelial cells (Dignat-George and Boulanger; Horstman et al., 2004) so it is possible that the PM vessels experience stress that disrupts homeostasis shortly after formation that may ultimately contribute to their regression. By E18.5, and into postnatal stages, no new ECs are added to the PM despite the continued rapid growth of the eye and especially the lens. As a result, there is progressive stretching of the PM vasculature, due to its adhesion to the growing lens surface and the postnatal iris, and it is likely that key breaks between vessel segments and discrete cell death events releases some of the tension. Ultimately, the PM vessels subjected to these tugging forces of the growing eye (and possibly iris contraction) are pulled away from the lens surface and toward the iris at the PM periphery (Figure 8B-C).
Figure 8. Revised timeline and working model of pupillary membrane regression.
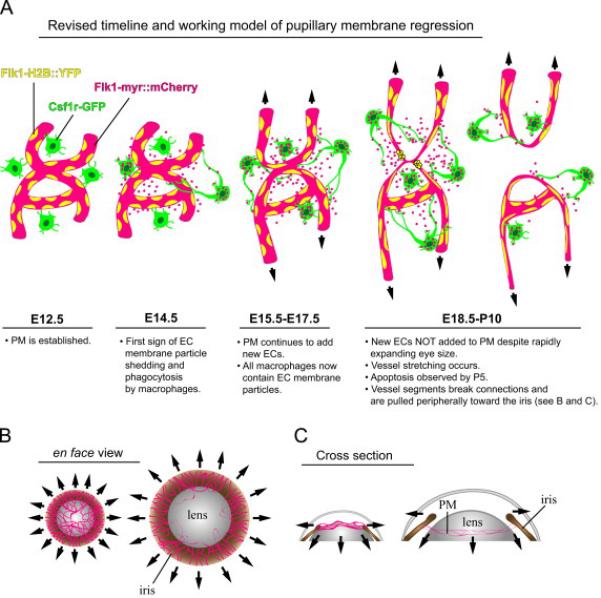
See text for details. Abbreviations: Pupillary membrane (PM), endothelial cell (EC).
In keeping with this model, previous studies have indicated that rat PM regression is initiated by constriction and dilation of the iris preceding macrophage activation (Morizane et al., 2006). By treating the rat eye with mydriatics to prevent iris movement, it was found that PM vessels persisted and the number of apoptotic cells was reduced. These data suggested that repeated constriction and dilation of the iris causes repetitive periods of bending and relaxation of the PM vessels at the iris margin. This, in turn, was thought to cause constant cessation and resumption of blood flow that ultimately induces vascular apoptosis (Morizane et al., 2006). Indeed, previous vital imaging studies of the rat PM also implicated flow stasis as a trigger for apoptosis observed in discrete vessel segments during later stages of regression (Meeson et al., 1996). Further evidence of a role for iris movement in PM regression comes from recent analysis of Fibrillin-2 (Fbn2) knockout mice that exhibit iris coloboma. In these animals, the PM regressed normally near regions of intact iris. However, in areas where the iris was notched, the PM persisted into adulthood (Shi et al., 2013). In addition to the data presented above, we performed live imaging of P9 Flk1-myr::mCherry+/tg; Flk1-H2B::YFP+/tg PMs and observed what appeared to be iris movement causing the stretching of PM vessels (Supplemental Movie 4). Thus, it is certainly possible that iris movement contributes to PM blood flow changes and/or vessel stretching and ultimately regression. However, it is still not obvious exactly how this is connected to the dependence of PM regression on macrophages.
It is clear from models in which macrophages are depleted or missing that macrophages are required for PM regression (Diez-Roux and Lang, 1997; Lang et al., 1994; Lang and Bishop, 1993; Lobov et al., 2005). The complete lack of Tcf/Lef-H2B::GFP expression in the mouse PM ECs lies in stark contrast to the robust levels of Tcf/Lef-H2B::GFP activity we observed in the HV ECs. These data suggest that the molecular mechanism of PM vessel regression is distinct from the canonical Wnt/β-catenin-dependent mechanism driving HV regression. Indeed, others have reported a potential role for Bone morphogenetic protein 4 (BMP4) paracrine signaling from the lens in promoting rat PM vessel apoptosis (Kiyono and Shibuya, 2003). However, a true test of this model, in the form of a lens-specific Bmp4 conditional knockout mouse and the consequence on PM regression, has not been reported. Another significant difference between the capillary regression of the PM versus the HV is that the PM vasculature does not require functional Bax and Bak proapoptotic gene activity for regression to occur whereas the HV of Bax−/−; Bak−/− double mutants persists into adulthood (Hahn et al., 2005). It is not obvious why the PM apparently uses a different apoptotic program than the HV and this raises the question of whether apoptosis on its own is the driving force of PM regression. Also worthy of discussion is the finding that Tcf/Lef-H2B::GFP-positive lens epithelial cells lie adjacent to Tcf/Lef-H2B::GFP-negative PM vessels, suggesting a role for Wnt signaling from the PM to the lens epithelium. Lens epithelial cells are known to require Wnt signaling for proper differentiation as Lrp6 null mice exhibit incompletely formed anterior lens epithelium (Stump et al., 2003). Thus, the source of a Wnt signal could derive from the PM endothelial cells, macrophages or pericytes and may be instructive for proper lens development.
The data presented here reveal a surprising and interesting new role for macrophages in the regression of the PM as facilitators of EC plasma membrane phagocytosis. We observed resident macrophages phagocytosing EC membrane particles prior to the first signs of EC apoptosis and vessels regression. This phenomenon could only be observed due to the fact that the Flk1-myr::mCherry reporter specifically targets the mCherry fluorescent protein to the EC plasma membrane (Larina et al., 2009; Poche et al., 2009). By crossing this line to Csf1r-GFP mice (Sasmono et al., 2003), we were able to capture PM GFP+ macrophages displaying scavenging behavior. These macrophages send out lamellipodia/filopodia that make direct contact with EC cytoplasmic projections as well as the vessel wall and appear to engulf mCherry+ membrane particles directly from the PM capillaries. Using confocal microscopy and TEM, we also found that the mCherry+ EC membrane particles end up in macrophage phagosomes and are present there as early as E14.5. In macrophages, filopodia function as environmental sensors that were recently described as “phagocytic tentacles” which extend and then quickly retract toward the soma upon binding a particle to facilitate its phagocytosis (Kress et al., 2007; Mattila and Lappalainen, 2008). This description appears strikingly similar to our in vivo observations of PM macrophages extending projections to the outer vessel wall and making point-to-point contact with EC cytoplasmic projections likely resulting in the phagocytosis of EC plasma membrane particles. The point-to-point contact between the macrophage filopodia and the EC cytoplasmic projections is also analogous to the description of the mechanism by which macrophages contact E. coli via the binding of the macrophage filopodia to the bacterial fimbriae. This allows the macrophage to “hook” the bacterium which is followed by lamellipodial contact which “shovels” the bacterium into a phagocytic cup (Moller et al.). Whether the PM macrophages use similar mechanisms to engulf EC plasma membrane particles remains to be determined.
It is particularly interesting that EC membrane engulfment begins by E14.5, which is approximately 2 days after the PM forms and 9-10 days before we and others have observed EC death within the mouse PM (Ito and Yoshioka, 1999). This suggests that the trigger for the developmentally programmed PM regression may exist much earlier than thought and regression may begin coincident with the phagocytosis of EC plasma membrane particles by macrophages.
To test whether the macrophages are required for the formation or release of particles, we examined PU.1−/−; Flk1-myr::mCherry+/tg animals and found that the EC membrane particles were still present, showing that macrophages do not induce the particle release. Since macrophages are known to be required ultimately for PM regression (Lang and Bishop, 1993; Lobov et al., 2005), it is possible that the continued removal and sequestration of EC membrane by macrophages could be facilitating regression by several possible mechanisms. First, since we observed that ECs are not added to the PM (Figure 6), it is possible that there is no way to counter the prolonged loss of membrane material from the cells and as the lens continues to grow and stretch, the vessels become thinner and ultimately break. Second, recent studies have shown that microparticles exuded from ECs can also be taken up by ECs and promote angiogenesis (Cantaluppi et al., 2012; Deregibus et al., 2007; Jansen et al., 2013b; Leroyer et al., 2009; Ranghino et al., 2012; van Balkom et al., 2013). Thus, the engulfment of PM microparticles by macrophages may lead to a withdrawal of important angiogenic and/or homeostatic cues leading to a reduction of EC branching or membrane recycling. Third, we did observe a low rate of apoptosis in PM cells that could account for some vessel clearing. Published data indicates that membrane particles derived from ECs can prevent apoptosis when applied to cells directly (Abid Hussein et al., 2007; Perez-Casal et al., 2009). Removal of the particles by macrophages may counter the anti-apoptotic effect, however we observed such a low apoptotic rate, it is unclear if this is significant. Fourth, it was clear from our studies that membrane engulfment precedes not only vessel regression, but also pericyte cell death. The studies we performed using the NG2-dsRed reporter showed that pericytes were present around PM vessels at both embryonic and early postnatal stages but by P7 we detected dying and fragmented pericytes. Thus, it is tempting to speculate that perhaps the steady release of EC membrane and potentially membrane-associated factors necessary for EC-pericyte interactions may be released. This, in turn, might disrupt the association of the pericytes with ECs making the vessels susceptible to regression. Finally, we also have to account for the possibility that shed EC particles contain signaling molecules that might communicate directly with resident macrophages. Indeed, several recent studies have suggested that EC microparticles promote monocyte adhesion to ECs and induce monocyte activation during EC inflammation (Burger et al., 2011; Jansen et al., 2013a; Jy et al., 2004). A true test of these models would require blockage of EC particle release and/or recycling specifically in the PM of embryos and we have not yet identified methods to achieve this.
In conclusion, this study has established that macrophage phagocytosis of shed EC plasma membrane particles occurs within the embryonic PM prior to the onset of capillary regression and apoptosis. This finding raises the intriguing possibility that macrophages directly mediate PM regression by engulfing EC plasma membrane which in turn hinders the ability of PM vessels to incorporate new ECs, recycle EC membrane and/or expand concomitantly with the growing lens and iris leading to progressive vessel stretching and subsequent elimination from the lens surface. Our data also raise the question of whether apoptosis alone is necessary and sufficient for complete PM regression or do a significant number of PM vessel segments retract peripherally to become part of the iris vasculature. However, to investigate this possibility we must be able to image PM regression over several days in the same animal. It is also formally possible that the described phagocytic behavior of macrophages is a response to an unidentified EC stress or dysfunction that begins in embryogenesis. Whatever the case, future PM macrophage/EC live imaging experiments, coupled to genetic loss-of-function and/or pharmacological manipulation, will be enlightening.
Supplementary Material
Highlights.
We report in vivo imaging of the mouse pupillary membrane (PM) vasculature.
PM endothelial cells (ECs) show no evidence of Wnt/β-catenin-mediated cell death.
PM ECs shed membrane particles that are engulfed but not induced by macrophages.
These studies implicate an embryonic, endothelial origin of PM regression.
ACKNOWLEDGEMENTS
We are very grateful for the following funding sources: P30EY007551 (ARB); F32EY019436 (RAP); R21EY020632 (MED). We also thank Kat Hadjantonakis for providing the Tcf/Lef-H2B::GFP mice, Stanley Appel and David Beers for proving the PU.1+/− mutant mice, David Hume for providing the Csf1r-GFP mice and Richard Lang to helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abid Hussein MN, Boing AN, Sturk A, Hau CM, Nieuwland R. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thrombosis and haemostasis. 2007;98:1096–1107. doi: 10.1160/th05-04-0231. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/ Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol. 2011;31:1898–1907. doi: 10.1161/ATVBAHA.110.222703. [DOI] [PubMed] [Google Scholar]
- Cantaluppi V, Biancone L, Figliolini F, Beltramo S, Medica D, Deregibus MC, Galimi F, Romagnoli R, Salizzoni M, Tetta C, Segoloni GP, Camussi G. Microvesicles derived from endothelial progenitor cells enhance neoangiogenesis of human pancreatic islets. Cell transplantation. 2012;21:1305–1320. doi: 10.3727/096368911X627534. [DOI] [PubMed] [Google Scholar]
- Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, Bruno S, Bussolati B, Camussi G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007;110:2440–2448. doi: 10.1182/blood-2007-03-078709. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Argilla M, Makarenkova H, Ko K, Lang RA. Macrophages kill capillary cells in G1 phase of the cell cycle during programmed vascular regression. Development. 1999;126:2141–2147. doi: 10.1242/dev.126.10.2141. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Lang RA. Macrophages induce apoptosis in normal cells in vivo. Development. 1997;124:3633–3638. doi: 10.1242/dev.124.18.3633. [DOI] [PubMed] [Google Scholar]
- Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. 31:27–33. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev Biol. 10:121. doi: 10.1186/1471-213X-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ST, Hadjantonakis AK, Sahr KE, Willey S, Kelly OG, Jones EA, Dickinson ME, Baron MH. Using a histone yellow fluorescent protein fusion for tagging and tracking endothelial cells in ES cells and mice. Genesis. 2005;42:162–171. doi: 10.1002/gene.20139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez E, Montoliu L. A simple polymerase chain reaction assay for genotyping the retinal degeneration mutation (Pdeb(rd1)) in FVB/N-derived transgenic mice. Lab Anim. 2001;35:153–156. doi: 10.1258/0023677011911525. [DOI] [PubMed] [Google Scholar]
- Hahn P, Lindsten T, Tolentino M, Thompson CB, Bennett J, Dunaief JL. Persistent fetal ocular vasculature in mice deficient in bax and bak. Arch Ophthalmol. 2005;123:797–802. doi: 10.1001/archopht.123.6.797. [DOI] [PubMed] [Google Scholar]
- Henkel GW, McKercher SR, Yamamoto H, Anderson KL, Oshima RG, Maki RA. PU.1 but not ets-2 is essential for macrophage development from embryonic stem cells. Blood. 1996;88:2917–2926. [PubMed] [Google Scholar]
- Horstman LL, Jy W, Jimenez JJ, Ahn YS. Endothelial microparticles as markers of endothelial dysfunction. Front Biosci. 2004;9:1118–1135. doi: 10.2741/1270. [DOI] [PubMed] [Google Scholar]
- Ito M, Yoshioka M. Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat Embryol (Berl) 1999;200:403–411. doi: 10.1007/s004290050289. [DOI] [PubMed] [Google Scholar]
- Jansen F, Yang X, Franklin BS, Hoelscher M, Schmitz T, Bedorf J, Nickenig G, Werner N. High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovascular research. 2013a;98:94–106. doi: 10.1093/cvr/cvt013. [DOI] [PubMed] [Google Scholar]
- Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, Wenzel D, Vosen S, Franklin BS, Fleischmann BK, Nickenig G, Werner N. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. 2013b;128:2026–2038. doi: 10.1161/CIRCULATIONAHA.113.001720. [DOI] [PubMed] [Google Scholar]
- Jy W, Minagar A, Jimenez JJ, Sheremata WA, Mauro LM, Horstman LL, Bidot C, Ahn YS. Endothelial microparticles (EMP) bind and activate monocytes: elevated EMP-monocyte conjugates in multiple sclerosis. Front Biosci. 2004;9:3137–3144. doi: 10.2741/1466. [DOI] [PubMed] [Google Scholar]
- Kiyono M, Shibuya M. Bone morphogenetic protein 4 mediates apoptosis of capillary endothelial cells during rat pupillary membrane regression. Mol Cell Biol. 2003;23:4627–4636. doi: 10.1128/MCB.23.13.4627-4636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress H, Stelzer EH, Holzer D, Buss F, Griffiths G, Rohrbach A. Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc Natl Acad Sci U S A. 2007;104:11633–11638. doi: 10.1073/pnas.0702449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R, Lustig M, Francois F, Sellinger M, Plesken H. Apoptosis during macrophage-dependent ocular tissue remodelling. Development. 1994;120:3395–3403. doi: 10.1242/dev.120.12.3395. [DOI] [PubMed] [Google Scholar]
- Lang RA, Bishop JM. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell. 1993;74:453–462. doi: 10.1016/0092-8674(93)80047-i. [DOI] [PubMed] [Google Scholar]
- Larina IV, Shen W, Kelly OG, Hadjantonakis AK, Baron MH, Dickinson ME. A membrane associated mCherry fluorescent reporter line for studying vascular remodeling and cardiac function during murine embryonic development. Anat Rec (Hoboken) 2009;292:333–341. doi: 10.1002/ar.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroyer AS, Ebrahimian TG, Cochain C, Recalde A, Blanc-Brude O, Mees B, Vilar J, Tedgui A, Levy BI, Chimini G, Boulanger CM, Silvestre JS. Microparticles from ischemic muscle promotes postnatal vasculogenesis. Circulation. 2009;119:2808–2817. doi: 10.1161/CIRCULATIONAHA.108.816710. [DOI] [PubMed] [Google Scholar]
- Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, Kurup S, Glass DA, Patel MS, Shu W, Morrisey EE, McMahon AP, Karsenty G, Lang RA. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–421. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. Embo J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- Meeson A, Palmer M, Calfon M, Lang R. A relationship between apoptosis and flow during programmed capillary regression is revealed by vital analysis. Development. 1996;122:3929–3938. doi: 10.1242/dev.122.12.3929. [DOI] [PubMed] [Google Scholar]
- Moller J, Luhmann T, Chabria M, Hall H, Vogel V. Macrophages lift off surface-bound bacteria using a filopodium-lamellipodium hook-and-shovel mechanism. Sci Rep. 3:2884. doi: 10.1038/srep02884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane Y, Mohri S, Kosaka J, Tone S, Kiyooka T, Miyasaka T, Shimizu J, Ogasawara Y, Shiraga F, Minatogawa Y, Sasaki J, Ohtsuki H, Kajiya F. Iris movement mediates vascular apoptosis during rat pupillary membrane regression. Am J Physiol Regul Integr Comp Physiol. 2006;290:R819–825. doi: 10.1152/ajpregu.00602.2005. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. 2001;222:218–227. doi: 10.1002/dvdy.1200. [DOI] [PubMed] [Google Scholar]
- Perez-Casal M, Downey C, Cutillas-Moreno B, Zuzel M, Fukudome K, Toh CH. Microparticle-associated endothelial protein C receptor and the induction of cytoprotective and anti-inflammatory effects. Haematologica. 2009;94:387–394. doi: 10.3324/haematol.13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poche RA, Larina IV, Scott ML, Saik JE, West JL, Dickinson ME. The Flk1-myr::mCherry mouse as a useful reporter to characterize multiple aspects of ocular blood vessel development and disease. Dev Dyn. 2009;238:2318–2326. doi: 10.1002/dvdy.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranghino A, Cantaluppi V, Grange C, Vitillo L, Fop F, Biancone L, Deregibus MC, Tetta C, Segoloni GP, Camussi G. Endothelial progenitor cell-derived microvesicles improve neovascularization in a murine model of hindlimb ischemia. International journal of immunopathology and pharmacology. 2012;25:75–85. doi: 10.1177/039463201202500110. [DOI] [PubMed] [Google Scholar]
- Ruiter DJ, Schlingemann RO, Westphal JR, Denijn M, Rietveld FJ, De Waal RM. Angiogenesis in wound healing and tumor metastasis. Behring Inst Mitt. 1993:258–272. [PubMed] [Google Scholar]
- Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- Shi Y, Tu Y, Mecham RP, Bassnett S. Ocular phenotype of fbn2-null mice. Invest Ophthalmol Vis Sci. 54:7163–7173. doi: 10.1167/iovs.13-12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Tu Y, Mecham RP, Bassnett S. Ocular phenotype of Fbn2-null mice. Invest Ophthalmol Vis Sci. 2013;54:7163–7173. doi: 10.1167/iovs.13-12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump RJ, Ang S, Chen Y, von Bahr T, Lovicu FJ, Pinson K, de Iongh RU, Yamaguchi TP, Sassoon DA, McAvoy JW. A role for Wnt/beta-catenin signaling in lens epithelial differentiation. Developmental biology. 2003;259:48–61. doi: 10.1016/s0012-1606(03)00179-9. [DOI] [PubMed] [Google Scholar]
- van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM, van Eijndhoven MA, Pegtel DM, Stoorvogel W, Wurdinger T, Verhaar MC. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood. 2013;121:3997–4006. S3991–3915. doi: 10.1182/blood-2013-02-478925. [DOI] [PubMed] [Google Scholar]
- Zhu M, Madigan MC, van Driel D, Maslim J, Billson FA, Provis JM, Penfold PL. The human hyaloid system: cell death and vascular regression. Exp Eye Res. 2000;70:767–776. doi: 10.1006/exer.2000.0844. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.