

Abstract
Background
SNPs are the most abundant polymorphism type, and have been explored in many crop genomic studies, including rice and maize. SNP discovery in allotetraploid cotton genomes has lagged behind that of other crops due to their complexity and polyploidy. In this study, genome-wide SNPs are detected systematically using next-generation sequencing and efficient SNP genotyping methods, and used to construct a linkage map and characterize the structural variations in polyploid cotton genomes.
Results
We construct an ultra-dense inter-specific genetic map comprising 4,999,048 SNP loci distributed unevenly in 26 allotetraploid cotton linkage groups and covering 4,042 cM. The map is used to order tetraploid cotton genome scaffolds for accurate assembly of G. hirsutum acc. TM-1. Recombination rates and hotspots are identified across the cotton genome by comparing the assembled draft sequence and the genetic map. Using this map, genome rearrangements and centromeric regions are identified in tetraploid cotton by combining information from the publicly-available G. raimondii genome with fluorescent in situ hybridization analysis.
Conclusions
We report the genotype-by-sequencing method used to identify millions of SNPs between G. hirsutum and G. barbadense. We construct and use an ultra-dense SNP map to correct sequence mis-assemblies, merge scaffolds into pseudomolecules corresponding to chromosomes, detect genome rearrangements, and identify centromeric regions in allotetraploid cottons. We find that the centromeric retro-element sequence of tetraploid cotton derived from the D subgenome progenitor might have invaded the A subgenome centromeres after allotetrapolyploid formation. This study serves as a valuable genomic resource for genetic research and breeding of cotton.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-015-0678-1) contains supplementary material, which is available to authorized users.
Background
Single nucleotide polymorphisms (SNPs) are the most abundant and universal sequence variation across whole genomes, making them attractive markers for the genetic analysis of plants. It is estimated that one SNP occurs every 100 to 300 bp in any genome [1], showing a frequency one magnitude higher than that of simple sequence repeats (SSRs) and other molecular markers [2]. Thus SNP markers are ideal molecular markers for the construction of saturated genetic maps, cloning genes, and quantitative trait loci (QTLs) that control important agronomical traits, marker-assisted crop breeding [3–6], investigating genetic diversity and population structure [7], genome-wide association studies (GWAS) [8], and even to serve as a robust framework for genome assembly [9]. SNP detection was revolutionized by the rapid development of high-throughput next-generation sequencing (NGS) technologies, which generate millions of high quality sequences rapidly and cost-effectively. Through large-scale genome sequencing, high-density haplotype maps containing millions of SNPs have been constructed for rice [10] and maize [11]. However, genome-wide SNP discovery in polyploidy crops is still challenging owing to their large, highly repetitive sequence and three or more subgenomes nature. The large genome size increases the whole genome sequencing cost, and the polyploidization makes the identification of SNPs more complicated, since a large number of homoeologous and paralogous sequences in genomes results in the discovery of many non-allelic SNPs, which are of no use in genetic studies. In order to discover SNPs at a genome-wide level and to reduce the complexity of plant genomes, several techniques, such as complexity reduction of polymorphic sequences (CRoPS) [12], restriction site associated DNA (RAD) [13], genotyping-by-sequencing (GBS) [14], and sequence-based genotyping (SBG) [15], have been used in SNP discovery. Additionally, SNPs within genes can be explored via transcriptome sequencing [5], which has been successfully applied to SNP detection in many species including maize [16], wheat [17], and rape [18]. Nevertheless, these techniques are targeted and based on small areas of genomes, such that the information gained on SNPs cannot cover sequence variation across the whole genome.
In most eukaryotes, the centromere is an essential chromosomal region and usually contains high-copy-number retrotransposons and satellite repeats, which are highly diverse and difficult to map, clone, and sequence. The precise position of the centromere determines the relative positions and linkage relationship between markers in or near the centromere region. Even though the identification of centromeres on genetic maps has been difficult due to highly repetitive sequence and low recombination rate in centromeric region [19, 20], centromeres have been localized to genetic maps in several plants, such as rice [21], maize [22], Arabidopsis [23], and G. raimondii [24].
Cotton is an important textile and oil crop throughout the world. The cotton species include two commercially important cultivated allotetraploid species, G. hirsutum L. and G. barbadense L., which have a genome size of 2.5 Gb and large numbers of repetitive sequences derived from an allopolyploidization event between G. herbaceum L. (A1) and G. raimondii (D5) Ulbrich approximately 1 to 2 million years ago (MYA) [25]. Due to its complexity, studies on the structure of the cotton genome have lagged behind those of other important crops such as rice and maize. Interspecific crosses between G. hirsutum and G. barbadense have been widely used to develop inter-specific maps because of the lack of polymorphisms between the intra-species [26–33]. However, these maps are not dense enough to support high-resolution mapping and genetic association studies. It is therefore important to build a well-saturated genetic map to allow deeper genetic research and breeding of this important fiber crop. SNP discovery in cotton was only initiated in recent years, and only a small number of SNP markers have been identified in the cotton genome, therefore few SNP markers were available in existing genetic maps constructed [34–36].
Here, we constructed an ultra-dense genetic map of allotetraploid cotton consisting of nearly 5 million SNPs (Additional file 1) with the largest amount of sequence variation identified in two cultivated allotetraploid cottons to date. This genetic map was used to improve the Upland cotton reference genome assembly, to detect genome rearrangements, and to identify centromeric regions of allotetraploid cotton. Ultimately, this high-precision and high-resolution genetic linkage map will serve as a valuable genomic resource for enhancing our understanding of polyploid genome structure and evolution, and for tagging genome-wide linkage disequilibrium and association studies that target genes to specific functions or traits.
Results and discussion
Sequencing and mining of potential SNPs in tetraploid cotton
A total of 59 interspecific F2 individuals (THF2) were developed from a cross between two cultivated tetraploid cotton species; G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124. For SNP discovery, two parents were sequenced at relatively high coverage using the Illumina sequencing approach. In total, 154.7 Gb of high quality short read sequences (61.9-fold genome coverage) were generated for TM-1, and 97.9 Gb (39.1-fold genome coverage) for Hai7124 (Table 1). First, an initial draft of the TM-1 genome was obtained from the TM-1 assembly project, which consisted of 17,188 scaffolds (>10 kb, accounting for 92.0 % of all sequence scaffolds). The size of the assembly was 2.2 Gb, with a scaffold N50 size of 210 kb. Further, all high quality reads from TM-1 and Hai7124 were aligned to the scaffolds of TM-1 to allow the detection of putative SNPs using BWA [37] and Samtools [38] softwares. The alignment results showed that 72.5 % and 38.7 % of the short reads (MQ >20) from TM-1 and Hai7124, respectively, were uniquely aligned to the reference genome (Additional file 2), and these locus-specific and high-quality reads were used to detect the interspecific SNPs between TM-1 and Hai7124. Finally, we identified 6,476,899 putative interspecific SNPs, including 5,054,468 simple SNPs, 1,421,857 hemi-SNPs, and 574 complex SNPs, classified as described by Trick [18]. These simple SNPs accounted for nearly 78 % of the total polymorphic SNPs and are potential markers for genotyping of the mapping population.
Table 1.
Summary of sequencing data from two parents and the mapping population in tetraploid cotton
| Sample | Insert size (bp) | Read length (bp) | Raw data (Gbp) | HQ data (Gbp) | ||
|---|---|---|---|---|---|---|
| Total data (Gbp) | Valid sequence depth (×) | Total data (Gbp) | Valid sequence depth (×) | |||
| TM-1 | 180 and 300 | 2 × 100 | 185.0 | 74.0 | 154.7 | 61.9 |
| Hai7124 | 300 | 2 × 100 | 111.8 | 44.7 | 97.9 | 39.1 |
| THF2 a | 300 | 2 × 100 | 938.0 | 375.2 | 784.0 | 313.6 |
aTHF2: 59 F2 populations from a cross between TM-1 and Hai7124. The estimated genome size is 2.5G. The cutoff value for the PHRED quality score for high-quality filtering is 20. The cutoff value for the percentage of read length that should be of given quality is 70
A total genome sequence of 784 Gb was generated for 59 F2 individuals; 13.3 Gb for each individual on average, equivalent to 5.3× coverage of the cotton genome (Table 1, Additional file 3). Reads of the 59 F2 individuals were mapped against the primary genome sequence and the uniquely mapped reads of high mapping quality (MQ >20) were retained for genotyping. Accurate genotyping of allopolyploid genomes based on short reads with low genome coverage was difficult due to two major obstacles: First, sequencing error and heterozygous regions in the genome led to inaccurate genotyping at given SNP sites, and second, excessive missing genotypes existed due to low genome coverage. To overcome these challenges, we performed a two-step genome-wide genotyping process. First, SNP positions were identified in the initial genotype of 59 F2 individuals using a minimum read depth of four and minimum read per allele of one as thresholds. A similarity score was calculated between SNP sites in each scaffold based on the initial genotype, and the SNP sites with high similarity scores were classified into a block in order to avoid mis-assembly in further analysis. Then, a sliding window approach was used to evaluate a group of consecutive SNPs in the same block for genotyping. The minimum read depth in a window containing adjacent SNP sites was 40 to avoid genotyping errors with a single allele at low genome coverage. The genotype of each sliding window was determined based on Bayesian theory (see Materials and Methods). First step screening showed that the missing genotype rate of SNP sites within the initial genotype was 43.95 % and the accuracy of genotype calling at SNP sites for the F2 population was 80.07 % (9.87 % A-H miss calls and 10.06 % B-H miss calls; A for homozygous TM-1 genotype, B for homozygous Hai7124 genotype, and H for heterozygous genotype), while in the second step screening, the missing genotype rate of SNP sites was reduced to 5.21 % when the sliding window approach was used and the accuracy of genotype calling at SNP sites was improved to 99.28 %. It is obvious that the accuracy and efficiency of the sequence-based genotyping of the mapping population were greatly improved through the use of the two-step method. As a result, 4,999,048 of 5,054,468 simple SNPs involved in 4,049 recombination bins were genotyped successfully, where a recombination bin represents a class of SNP loci without a recombination node and missing genotype detected. In our study, hemi and complex SNPs detected between two parents were not used for map construction because of their intricate nature that likely represents a mixture of two homoeologous or paralogous sequences due to the sequencing depth of the F2 individuals. Modern breeding has enhanced gene flow and post-domestication introgressions through deliberate hybridization between these two species. For example, the reciprocal introgressions between G. hirsutum and G. barbadense cultivars have developed Acala and Pima cultivar families. Therefore, many of these interspecific SNPs can be used in such introgressions in future cotton breeding.
Construction of sequence-based ultra-dense genetic and physical maps
To effectively order the linkage groups, 441 framework SSR markers that were able to amplify 519 discrete polymorphic loci and were distributed evenly on 26 chromosomes were selected, with 10 cM intervals between the markers based on our microsatellite-based, gene-rich linkage map [33]. The ultra-dense genetic map was constructed based on data from 59 F2 individuals and oriented by integrating the reference framework SSRs using Joinmap 3.0 [39]. The map consists of 4,049 recombination bins that include 519 framework SSR loci and 4,999,048 SNP loci, and covers 4,042 cM with an average inter-bin genetic distance of 1.0 cM (Table 2, Additional file 4). Analysis of the SNP calling accuracy and mapping quality using CheckMatrix [10] indicated that all markers in the composite map were assigned to correct linkages in the correct order (Fig. 1). Overall, this SNP array and high-density linkage map is the sole genetic map with the maximum number of SNPs in allotetraploid cotton constructed so far. It will be useful for genomic studies and molecular breeding in cotton.
Table 2.
Characteristics of the 26 linkage groups in allotetraploid cotton
| Linkage group | No. bins | No. SSRs | cMa | Inter-bin average distance (cM) | No. scaffolds | Size (Mb) | No. SNPs |
|---|---|---|---|---|---|---|---|
| A01 | 157 | 13 | 179 | 1.14 | 240 | 115.9 | 169,307 |
| A02 | 125 | 13 | 122 | 0.98 | 368 | 110.8 | 245,157 |
| A03 | 150 | 19 | 146 | 0.98 | 326 | 116.7 | 138,849 |
| A04 | 119 | 19 | 115 | 0.97 | 333 | 93.0 | 194,383 |
| A05 | 213 | 32 | 213 | 1.00 | 313 | 113.2 | 136,212 |
| A06 | 144 | 24 | 154 | 1.07 | 448 | 132.3 | 295,265 |
| A07 | 163 | 13 | 151 | 0.93 | 376 | 101.1 | 315,158 |
| A08 | 162 | 18 | 168 | 1.04 | 444 | 130.9 | 294,517 |
| A09 | 153 | 28 | 151 | 0.99 | 233 | 87.0 | 117,361 |
| A10 | 175 | 15 | 163 | 0.93 | 362 | 121.8 | 255,072 |
| A11 | 202 | 28 | 205 | 1.01 | 407 | 126.3 | 260,274 |
| A12 | 165 | 16 | 160 | 0.97 | 431 | 112.6 | 371,191 |
| A13 | 168 | 17 | 157 | 0.94 | 354 | 115.8 | 350,161 |
| A subgenome | 2,096 | 255 | 2,087 | 1.00 | 4,635 | 1477.4 | 3,142,907 |
| D01 | 136 | 27 | 125 | 0.92 | 107 | 64.2 | 138,376 |
| D02 | 143 | 19 | 128 | 0.90 | 124 | 71.1 | 245,387 |
| D03 | 121 | 18 | 128 | 1.06 | 101 | 55.4 | 182,200 |
| D04 | 111 | 12 | 118 | 1.06 | 75 | 57.1 | 77,329 |
| D05 | 213 | 30 | 214 | 1.01 | 145 | 66.0 | 133,959 |
| D06 | 141 | 24 | 145 | 1.03 | 117 | 67.4 | 208,772 |
| D07 | 145 | 16 | 145 | 1.00 | 116 | 58.4 | 128,679 |
| D08 | 144 | 18 | 136 | 0.95 | 115 | 69.4 | 144,574 |
| D09 | 146 | 25 | 157 | 1.08 | 122 | 53.8 | 116,224 |
| D10 | 167 | 18 | 156 | 0.94 | 142 | 68.2 | 149,762 |
| D11 | 174 | 21 | 176 | 1.01 | 130 | 72.6 | 152,575 |
| D12 | 169 | 17 | 174 | 1.03 | 121 | 63.0 | 94,876 |
| D13 | 143 | 19 | 153 | 1.07 | 96 | 64.5 | 83,428 |
| D subgenome | 1,953 | 264 | 1,956 | 1.00 | 1,511 | 831.1 | 1,856,141 |
| Total | 4,049 | 519 | 4,043 | 1.00 | 6,146 | 2308.5 | 4,999,048 |
aGenetic distance was calculated by JoinMap 3.0 [39] using the maximum likelihood mapping method
Fig. 1.
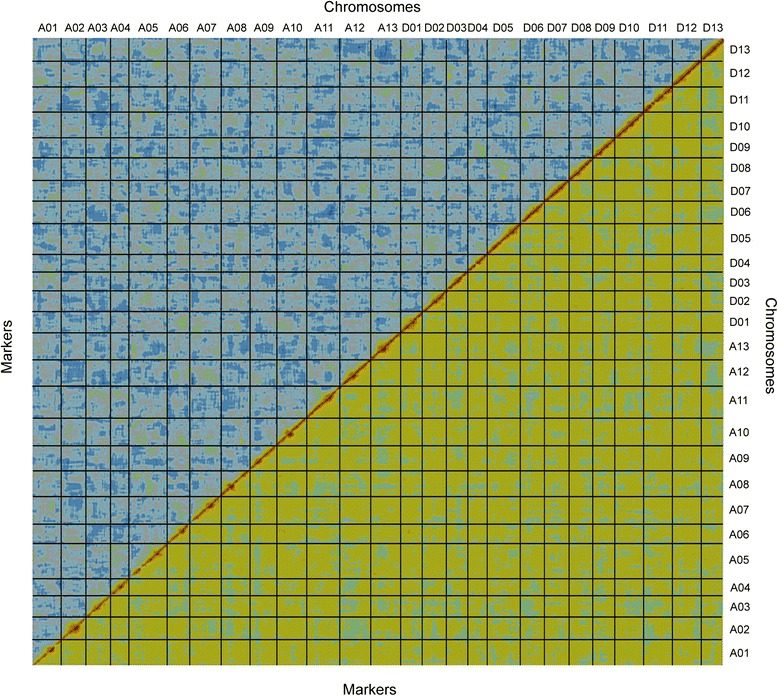
Graphical representation of the high-quality linkage map of tetraploid cotton. Alignment of 4,049 bins along the 26 chromosomes reveals a pairwise BIT score (top-left diagonal) and REC score (low-right diagonal) in all pairs of marker bins, plotted here using CheckMatrix [82]. Red represents tight linkage; yellow represents weak linkage; and blue represents no linkage. The red along the diagonal and the lack of red off the diagonal indicate the high quality and strong statistical support of the map
The inter-specific map was further compared to a high density intra-specific map which was developed to tag QTLs in Upland cotton using RAD sequencing technology [40]. We identified 21,109 SNPs between Upland cotton cultivars, Prema and 86–1, and used these for the genotyping of 161 recombinant inbred lines. Prema is an Acala cultivar which has introgressed segments from G. thurberi and G. barbadense. Finally, the highest density intra-specific linkage map comprising 4,153 loci over 3500-cM has been developed to date in Upland cotton [40]. The SNP alignments revealed their high consistency in marker order between these two maps (Additional file 5), suggesting the high degree of collinearity between these two cultivated tetraploid cottons.
Assessing and validating the genomes of tetraploid cottons
Although the emergence of NGS technologies provides a rapid and inexpensive method for genome-wide assembly, the assembly of complex and polyploid genomes such as cotton is still challenging. It is difficult to distinguish homoeologous genomic segments with a high degree of sequence similarity, large gene families, and extensive segmental duplications such as multiple ‘paralogs’. In the present research, the ultra-dense SNP map was further used to validate the initial assembly of the TM-1 genome (scaffold N50: 210 kb) based on the Illumina short reads. We detected 90 instances of mis-assemblies of the scaffolds, which covered 36.0 Mb and accounted for 1.6 % of the entire assembled genome (Additional file 6).
The population size is relatively small for use in resolving smaller scaffolds. Therefore, in addition to the paired-end (PE) reads of 2, 5, and 10 kb libraries used to generate the assembly, the longer scaffolds were enhanced by assembling 174,454 pairs of Sanger-sequenced bacterial artificial chromosome (BAC)-end sequences comprising 116.5 megabases (Mb) [41]. After the BAC-end sequences were merged into the validated assembly, we generated an updated version, V1.0, in which the length of the scaffolds was increased (N50: 1.9 Mb). In the V1.0 assembly, 128 misassembled scaffolds (406.2 Mb, accounting for 16.2 % of the assembly) were detected (Additional file 6). Through comparing the mis-assembled types in the different assembly versions, we found that about half of the scaffold mis-assemblies resulted from homoeologous chromosomes (46.9 % to 52.1 %). More interchromosomal mis-assemblies were detected in the A subgenome chromosomes (21.9 % to 29.1 %) than in the D subgenome, most likely due to the greater number of repetitive sequences in the A subgenome.
After these false mis-assemblies were broken, the final assembly of the TM-1 genome was produced (V1.1, PRJNA248163) [41]. According to the ultra-dense SNP map, which contains 4,999,048 SNP loci, the scaffolds that were >500 kb in length were anchored, and the percentage of anchored scaffolds gradually decreased as the length of the scaffold decreased. Finally, 6,146 scaffolds (covering 2.3 Gb) were anchored and concatenated into 26 pseudo-chromosomes, accounting for 94.9 % (2.4 Gb) of the assembled sequence; 1,477 Mb (4,635 scaffolds) in the A subgenome and 831 Mb (1,511 scaffolds) in the D subgenome. The size of the 26 pseudo-chromosomes varied from 55.4 Mb (D03) to 132.3 Mb (A06) [41].
Allopolyploid plants often undergo major changes in genome structure and function induced by the combination of divergent genomes from two or more related parental species [42]. Many studies have revealed that inter-genomic chromosomal rearrangements, differential gene loss (loss of some duplicates but not others), gene conversion, divergence, and functional diversification of duplicated genes often arise with the onset of polyploidization [43]. Therefore, several assistant strategies such as genetic mapping [3], physical mapping, fluorescent in situ hybridization (FISH) [44], and flow-sorting [45] technology, have been employed to aid in whole genome assembly. For example, a chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome has been produced by sequencing isolated chromosome arms through flow-sorting technology [45]. However, for most polyploidy species with small chromosomes that are difficult to classify and isolate using flow cytometry and sorting, this method is impracticable. In our method, an ultra-dense genetic linkage map is tremendously beneficial in the assembly of large and complex polyploidy genomes; as emphasized by Lewin, ‘every genome sequence needs a good map’ [46]. In fact, the production of high-quality genome assemblies in many species has benefited appreciably from good maps.
Structural variations in allotetraploid cotton genomes
Generally, ancestors of the A genome species, G. herbaceum or G. arboreum, and the D genome species, G. raimondii, contributed the constituent A and D subgenomes of tetraploid cotton, respectively [25]. Using two sequenced diploid genomes, G. arboreum (A2) [47] and G. raimondii (D5) [48], two extant progenitor relatives of tetraploid cotton, the integration of the molecular genetic map with the physical map can be realized. However, this collinearity was not obvious within the A-progenitor genome [47], partly due to numerous examples of mis-assemblies, and partly because G. arboreum is an important cultivated diploid species and may have undergone some of its own chromosomal rearrangements during its evolution and improvement. When the ultra-dense tetraploid genetic map was aligned with the G. raimondii genome [48], we found a high degree of collinearity, especially in the D subgenome (Fig. 2). Therefore, only the G. raimondii genome was used for comparative genomic analysis.
Fig. 2.
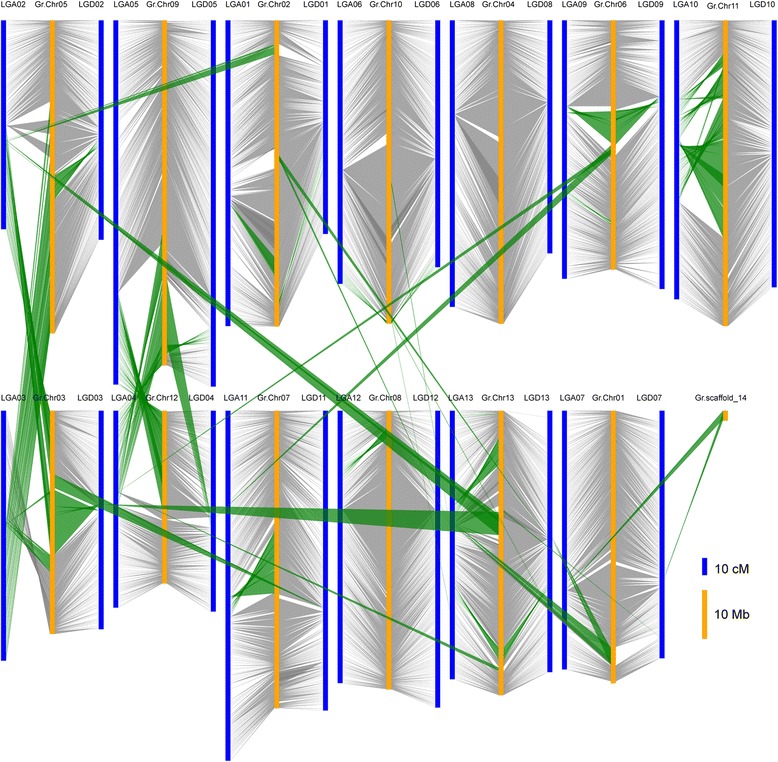
Comparison of the genetic map of tetraploid cotton and the corresponding physical locations on the pseudo molecules of the D genome sequence of G. raimondii. TM-1 scaffolds in the 26 linkage groups (blue) were aligned to the D genome sequence of G. raimondii (yellow). Genome variations (green blocks) were identified
Although the comparative genomic results showed a high degree of collinearity between the genetic map and the physical map, some instances of chromosomal rearrangement were detected between the A, D subgenome and the D genome. Two postpolyploidization reciprocal translocations were displayed between A02 and A03, and between A04 and A05, in the At subgenome (Fig. 2),which broke the chromosomes as follows; A02/A03 (61.90-122.31 cM), corresponding to pseudo molecule Chr03 in G. raimondii (GrChr03) at 33736–28298512 bp (28.26 Mb, accounting for 61.75 % of GrChr03), and A03/A02 (66.75-146.42 cM) corresponding to GrChr05 at 17656841–64067579 bp (46.41 Mb, accounting for 72.36 % of GrChr05); A04/A05 (0–54.66 cM) corresponding to GrChr09 at 43817852–70655127 bp (26.84 Mb, accounting for 37.96 % of GrChr09), and A05/A04 (160.43-213.26 cM) corresponding to GrChr12 at 591184–15299554 bp (14.71 Mb, accounting for 41.51 % of GrChr12) (Additional file 7). Furthermore, we identified 15 simple translocations between the A or D subgenome and the D genome, for example, D11 had a simple translocation of 2.75 Mb from GrChr03 (accounting for 6.01 % of GrChr03 at 13111322–15861215 bp). The A and D subgenomes displayed 19 possible inversions that were slightly different from those found by Wang et al. [32] and Rong et al. [27]. These 19 inversions, with sizes in the range of 0.05 Mb to 37.57 Mb, were found in 16 chromosomes, accounting for 0.09 % to 59.93 % of the corresponding chromosomes. Detailed information on these chromosomal rearrangement events with their locations on both the genetic map and the physical map of G. raimondii are presented in Additional file 7. From our comparison of the genetic and physical maps of the D genome sequence, we detected some structural variations not only in the A subgenome, but also in the D subgenome. However, we were unable to determine whether these structural changes occurred in the tetraploid after polyploidization and domestication or in the diploids in the last 1 to 2 million years, since the exact donor species that led to the formation of the tetraploid cotton species 1 to 2 MYA no longer exist.
Two reciprocal translocations revealed in the present study are in agreement with the results from prior studies [27, 32]. Three of 19 possible inversions were almost in agreement with the results of Wang et al. [32] and 15 simple translocations are first reported in the study, although we cannot exclude the possibility that some of the newly observed chromosomal structural rearrangements resulted from misassemblies in the G. raimondii reference genome sequence. Furthermore, the complex chromosomal rearrangements (inversions and simple translocations) observed here need to be accounted for in the ‘translation’ of information from the D genome in G. raimondii to allotetraploid cotton genomes. Genome variations such as inversions and translocations are considered important factors in species evolution. In the process of evolution from diploid to tetraploid cotton, these chromosomal structural rearrangements may have caused a series of changes in phenotype. These structural variations in the genome may help to elucidate the relationships between the phenotype and genotype of important agronomic traits in tetraploid cotton.
Comparison of the genetic and physical maps of allotetraploid cotton
A total of 4,999,048 mapped SNP loci (1 SNP/0.5 kb) were unevenly distributed on 26 chromosomes, with more loci in the A subgenome (3,142,907) than the D subgenome (1,856,141) (Fig. 3a). On average, each chromosome had 192,271 SNP loci, ranging from as high as 371,191 SNPs on A12, to as low as 77,329 on D04. To explore the patterns of SNP distribution in TM-1 and Hai7124, SNP frequency was plotted at 50 kb intervals along each pseudomolecule (Fig. 3a). Nine SNP-poor regions (at least 20 contiguous intervals with low SNP frequency) comprising a total of 54.0 Mb and accounting for 2.3 % of the whole genome were detected on chromosomes A01, A05, A06, A08, D04, and D11 (Additional file 8). The SNP-poor regions may have been due to historical introgression between G. hirsutum and G. barbadense. In chromosome A01, for example, some QTLs related to fiber quality from interspecific hybrid populations have been mapped to the SNP-poor region [49–51]. Several investigators have also postulated that an introgression event was responsible for the creation of the Sea Island germplasm [52, 53] in which the introgressed segments play a role in conferring beneficial traits such as high fiber quality or photoperiod neutrality [54]. Furthermore, modern breeding has enhanced gene flow and post-domestication introgressions through deliberate hybridization of these two species. For example, the reciprocal introgressions between G. hirsutum and G. barbadense cultivars have developed the Acala and Pima cultivar families [55]. Moreover, the vast majority of the mapped SNPs (95.2 %) were located in intergenic regions, and smaller portions were found in intron, followed by exon, regions (Fig. 3b). This suggests that SNP discovery via transcriptome sequencing is limited.
Fig. 3.
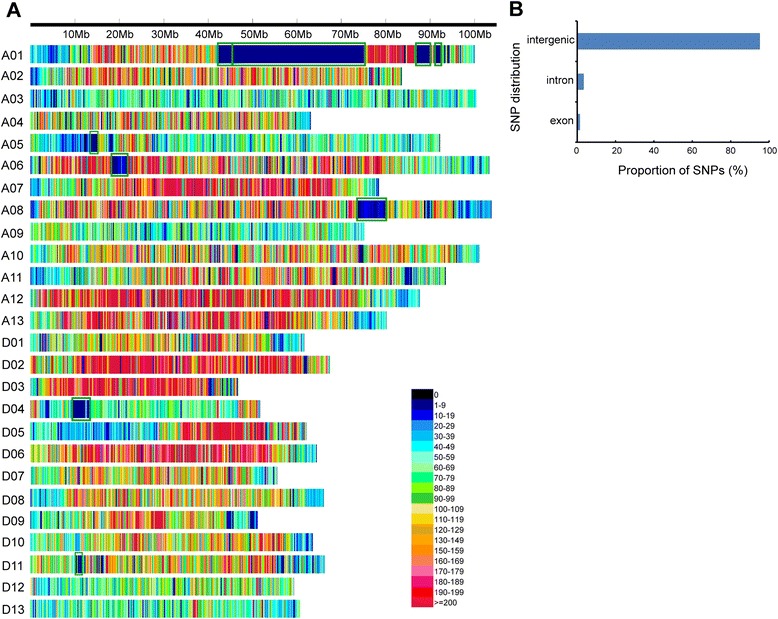
SNP distribution in the cotton genome. a Distribution of SNPs in the 26 chromosomes. The x-axis represents the physical distance along each chromosome, split into 50 kb windows. The green rectangles indicate SNP-poor regions. b Proportion of SNPs found in intergenic regions, introns, and exons
To facilitate genetic analysis, we converted the SNP linkage map into a skeleton bin map (Fig. 4). The two subgenomes had similar numbers of recombination bins in the skeleton bin map: 2,096 in the A subgenome and 1,953 in the D subgenome, making a total of 4,049. The average physical length of the recombination bins was 570 kb. The genetic and physical distances were not directly proportional, as shown in Fig. 5, where the plots of recombination bin placements on the 26 chromosomes are compared.
Fig. 4.
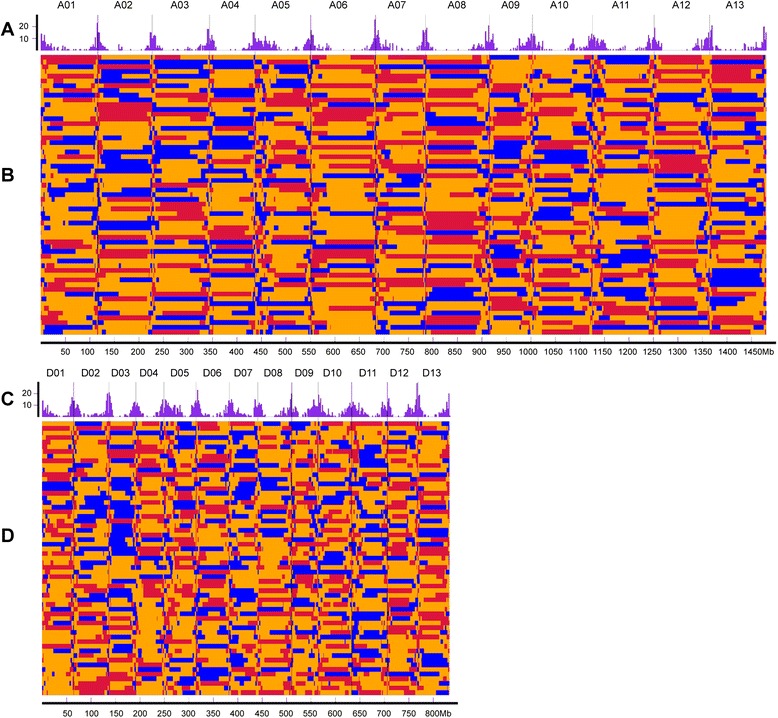
Recombination and bin map for 59 scored F2 individuals. a, c Frequency distribution of recombination nodes (RNs) per 2 Mb. The vertical scale indicates the number of recombination nodes (RNs); b, d Bin maps for the 59 scored F2 individual lines. Colored tracks represent the 59 individual lines of the THF2 population that were used for linkage map construction: red, alleles inherited from maternal parent (TM-1); blue, alleles inherited from paternal parent (Hai7124); yellow, alleles inherited from heterozygous genotype (TM-1 × Hai7124)F1. The horizontal scale indicates physical distance
Fig. 5.
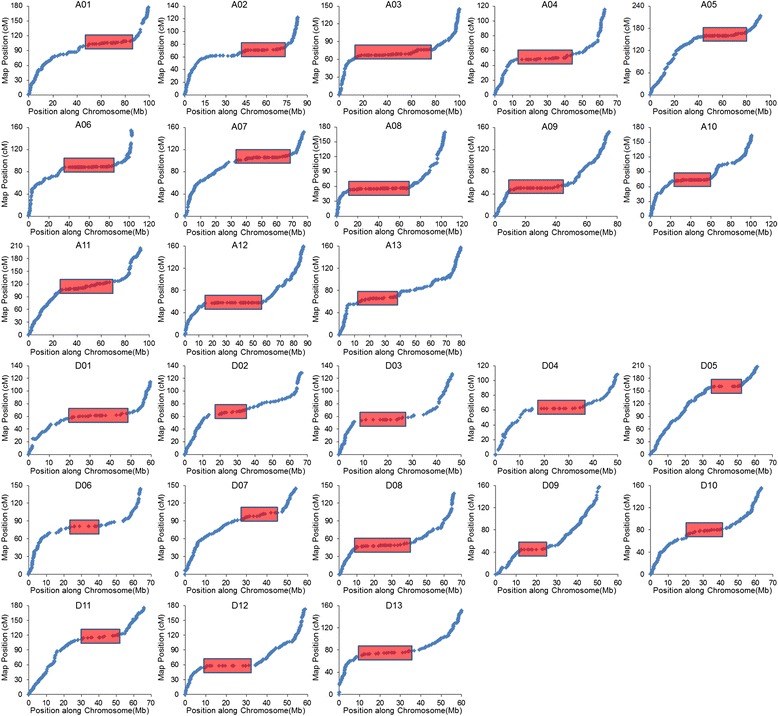
The marker placements for the genetic map on the G. hirsutum acc. TM-1 chromosomes. The marker order on the y-axis is derived from the genetic maps and the marker order on the x-axis is derived from the physical maps. Both the relative genetic and physical distances of the chromosomes on the plots are represented by the cells of different sizes according to the ratio of the chromosome lengths. Cumulative genetic distance in cM and physical distance in bp are indicated on x-axis and y-axis, respectively. The red rectangles represent the recombination suppression regions (nearly flat shaded) which are predicted to be pericentromeric regions
Recombination can generate genetic variation and induce genetic diversity. By comparing genetic and physical distances between adjacent markers, we further examined the relative changes in recombination rate along each chromosome. Consistently high recombination at chromosome ends and recombination suppression were clearly observed in different linkage groups. We found 26 recombination suppression regions in the 26 linkage groups (Fig. 5), which might be related to the heterochromatic regions [56]. These covered the repeat-rich centromeric or pericentromeric regions on each chromosome. We found that the recombination rates in hotspot regions were in the range of 1.4 to 5.7 cM/Mb, while in the suppression regions, recombination rates were in the range of 0 to 0.5 cM/Mb. Previous reports have revealed even more extreme variation in wheat [57, 58], where 1 Mb corresponds to a range of 0.05 to 8.47 cM, and in chromosome 4 of Arabidopsis [59], where 1 Mb corresponds to a range of 0 cM near the centromere to 20 cM on the short arm of the chromosome. Each chromosome was approximately subdivided into recombination hotspots (recombination-rich) and recombination suppression (recombination-poor) regions. The average ratio of genetic-to-physical distance was 1.75 cM/Mb for the whole genome of G. hirsutum. Genome-wide patterns of recombination have been described previously for some species, including Arabidopsis [59, 60] and rice [61]. This ratio is similar to those for watermelon (2.3 cM/Mb) [6], sorghum (1.43 cM/Mb) [62], and cucumber (3.2 cM/Mb) [63], but is lower than that for maize (5.5 cM/Mb) [64].
Genetic mapping of centromeric regions in allotetraploid cotton chromosomes
Little is known of the dynamics of centromeric DNA in polyploidy plants. Centromeres usually contain high-copy-number retrotransposons and satellite repeats, which are difficult to map, clone, and sequence. Few studies have focused on centromeres in cotton [24, 65]. In our previous study, we found that BAC 97G20 hybridized to the centromeric regions of all 52 chromosomes in domesticated Upland cotton [66]. Sequence analysis of BAC 97G20 showed that there were four long terminal repeat (LTR) retrotransposons, named GhCR1-4, with a strong similarity to the centromeric retrotransposons reported in other plants [24]. Of them, GhCR1 and GhCR3 had 93.6 % and 94.4 % similarity to centromere retroelements in Gossypium (CRG), CRG1 and CRG2, respectively. CRG1 and CRG2 were recently identified by co-localization immunostaining with antiserum to the centromere-specific histone CENH3 in cotton [65], indicating that GhCR1 and GhCR3 may belong to the same class as the CRGs.
Whole-genome sequence screening for four GhCR LTR homologies in G. hirsutum acc. TM-1 [41] identified 3,515 LTR homologies (about 270 for each chromosome on average) in the D subgenome; a much higher number than the 430 (about 33 for each chromosome on average) in the A subgenome (T-test: P value = 6.81e-07) (Additional file 9). This observation is consistent with the results of our FISH hybridization, which revealed that four of these retrotransposons were hybridized specifically to the centromere region in all of the chromosomes of G. hirsutum; however, much weaker signals were observed in the A subgenome than in the D subgenome. Of these retrotransposons, GhCR1 and GhCR3 LTRs had 1,857 and 1,375 homologous sequences, respectively; much higher than the 146 and 137 found in GhCR2 and GhCR4, respectively, in the TM-1 genome (Additional file 9). The putative centromeric regions in allotetraploid Upland cotton chromosomes are illustrated in Fig. 6. In the D subgenome, the centromeric regions were highly overlapped by GhCR1-5’LTR and GhCR3-5’LTR. With the exception of the predicted centromeric regions, which covered more than 4 Mb in D04 and D13, there were less than 2 Mb of centromeric regions in all other chromosomes (Fig. 6 and Additional file 10). In the A subgenome, however, no putative centromeric region was detected in A02, A07, A08, A11, or A13, with GhCRs absent in these chromosomes. In the other eight chromosomes, there was an extremely low level of homology with the predicted centromeric region sequences and a relatively large physical distance, ranging from 4.8 to 23.7 Mb, was detected; implying that there are large centromeric regions in the A subgenome (Fig. 6 and Additional file 10). In other plant species, the size of centromeric regions ranges from 9 Mb, in chromosome 1 of Arabidopsis, to 124 kb, in chromosome 4 of rice [67–70]. Although the sequences of centromeric regions show baffling diversification and species-specificity, the present report implies that the cotton centromeric regions span several hundred kilobases to several megabases. The centromeric regions identified in the study will serve as useful landmarks for fine-tuning the structure of the cotton genome.
Fig. 6.
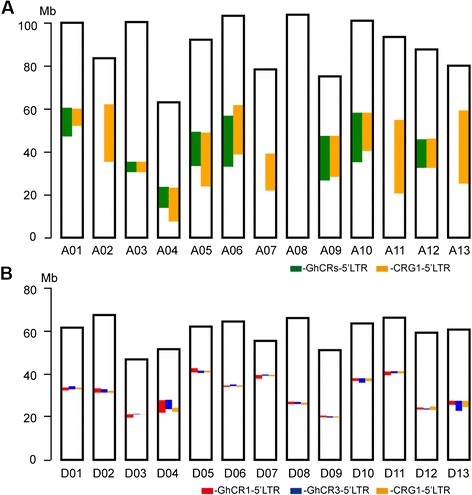
Schematic diagrams of centromeric regions of G. hirsutum acc. TM-1. a Centromeric regions of the A subgenome were identified by GhCRs-5’LTR (green) and CRG1-5’LTR (orange). b Centromeric regions of the D subgenome were identified by GhCR1-5’LTR (red), GhCR3-5’LTR (blue), and CRG1-5’LTR (orange)
In recent years, the study of centromeres has progressed rapidly in cotton [24, 65]. FISH analysis revealed that CRGs [65] and GhCRs [24] are both found in all 52 chromosomes in allotetraploid cottons and in D genome diploid cotton species, but not in A genome diploid species, such as G. arboreum and G. herbaceum. Whole-genome screening of the G. arboreum genome sequence [47] found only 17 homologies of CRGs (sequence identity >80 %) [65] and 119 GhCR LTRs in the A genome. Furthermore, the number of CRG homologies [65] and GhCR LTRs are much higher in the D subgenome than in the A subgenome in allotetraploid cotton. No reverse transcriptase (RT) domain from GhCRs LTR retrotransposon was found in the diploid A genome. However, one RT domain of GhCRs LRT was found in the A subgenome of TM-1. Phylogenetic analysis was performed using the Neighbor-joining (NJ) method in the MEGA 5.10 program [71] and showed that this RT domain could be derived from the D genome/D subgenome (Fig. 7). Together, these results revealed that these centromeric retrotransposons may have been derived from the D subgenome progenitor, invaded the A subgenome centromeres in allotetraploid cotton during allopolyploid formation, and were amplified during its evolutionary history. Extensive retrotransposon amplification must have occurred through the genome and, in particular, in the pericentromeric regions. Transposon activation following hybridization or polyploidy formation has been detected in some plants [65, 72, 73]. To limit genomic damage, plant hosts have evolved RNA interference-meditated mechanisms to tame their endogenous retroelements [74].
Fig. 7.
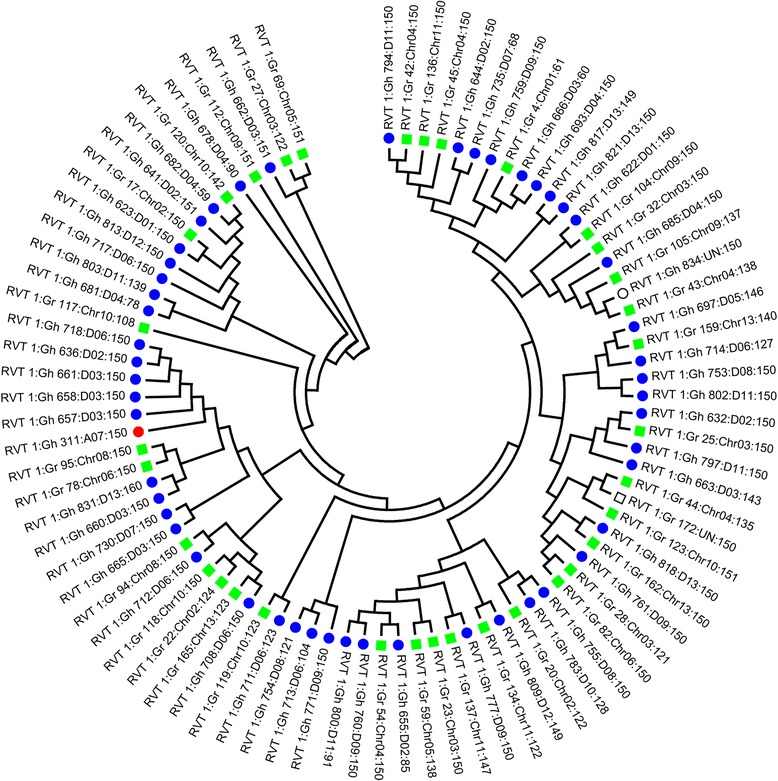
Neighbor-joining (NJ) phylogenetic tree based on RT amino acid sequences of GhCRs LTR from G. raimondii and G. hirsutum. Statistical support was evaluated by bootstrapping (1,000 replications); Green squares represent RTs from G. raimondii. Red and blue circles represent RTs from the A subgenome and D subgenome of TM-1, respectively. Hollow squares and circles represent RTs from scaffolds that were unanchored to chromosomes of G. raimondii and G. hirsutum, respectively
Conclusions
In this study, we constructed the first ultra-dense genetic map composed of approximately 5 million NGS sequence-derived SNP markers in allotetraploid cotton. This map has played an important role in genome assembly, the detection of genome rearrangements, and the identification of centromeric regions of polyploid cotton. The methodology used in this study can be applied to whole genome sequencing and the assembly of many other polyploid crops with large and complex genomes. Further, this complete and high resolution composite map is of practical importance for novel QTL discovery, gene mapping, map-based cloning, and marker assisted selection in cotton.
Materials and methods
Plant materials, DNA extraction, PCR amplification, and electrophoresis
The plant materials used consisted of G. hirsutum acc TM-1 and G. barbadense cv. Hai7124, two highly homozygous parents, as well as 59 F2 individuals derived from a cross between TM-1 as the recipient and Hai7124 as the donor parent. TM-1 is a genetic standard line of G. hirsutum developed through single plant selection.barbadense cv. Hai7124, grown extensively in China, was also a single plant selection offspring before it was used as a parent in the construction of the linkage map [75]. Genomic DNA were extracted from young leaf tissues following the methods described by Paterson et al. [76], with increased RNase A and proteinase K treatment to prevent RNA and protein contamination, before the extracts were subjected to Illumina sequencing technology and SSR-PCR amplification.
We selected 441 framework SSR markers from a complete linkage map [33], including 3,147 markers with 10-cM intervals between markers on 26 chromosomes, to anchor our framework map. Four hundred and forty-one SSR markers were employed to survey the 59 individuals in the F2 mapping population. SSR-PCR amplification and electrophoresis followed the methods described by Zhang et al. [75].
Library construction and sequencing
Genomic libraries were prepared following the manufacturer’s standard instructions for high-throughput DNA sequencing for subsequent cluster generation, and were sequenced on the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA). To construct paired-end libraries, DNA was fragmented by sonication, and DNA ends were blunted before adding an A base to each 3′ end. DNA adaptors with a single T-base 3′ end overhang were ligated to the above products. Ligation products were purified on 2 % agarose gels that each targeted a specific range of insert sizes. Quantification and quality assessment were carried out by running 1 μL of library on an Agilent DNA 1000 LabChip analyzer (Agilent Technology 2100 Bioanalyzer). We constructed G. hirsutum genome sequencing libraries with insert sizes of 180 bp and 300 bp, G. barbadense with 300 bp inserts, and each F2 individual with 300 bp inserts.
Data Set extraction
Base-calling files were obtained from raw fluorescent images from the Illumina HiSeq 2000 platform using CASAVA 1.8 software in FASTQ format [77]. Illumina reads were filtered using NGSQCTookit v2.3 [78] (parameters of -cutOffReadLen4HQ 70, −cutOffQualScore 20).
Sequence alignment to the temporary reference genome, SNP discovery, and genotyping
Alignment of the sequences to the TM-1 reference genome [41] was performed with BWA [37] using only sequences aligning to the reference genome with less than two mismatches based on a high quality sequence. Only sequences with a mapping score of at least 20 were used for SNP discovery.
For each parental sample, putative SNPs with respect to the TM-1 reference sequences were first called using samtools [38] and an in-house Perl script under the following conditions: minimum read depth at a given position was 10, minimum supporting reads for an allele was 3, and the allele represented at least 20 % of all the alleles observed. The interspecific SNPs (allelic polymorphisms), which can be broadly classified as simple SNPs, hemi-SNPs, and complex SNPs, were obtained from the two parental ‘putative SNPs’ lists using an in-house Perl script. Simple SNP types represent instances of either the reference sequence being genome-specific or sequence divergence of one subgenome so as to prevent alignment of one homoeolog to the reference sequence. Hemi-SNP types represent instances of reads from both subgenomes aligned to reference sequences with an allelic variation being present in one. Complex SNP types represent instances of an allelic variation having occurred at the site of an inter-homoeolog variation between the subgenomes. The plain-text format ‘pileup’ files encapsulating the alignment maps generated by samtools were inspected sequentially for each population sample over each of the SNP positions present in the SNP superset.
Using the mapping population, the initial genotypes of individuals were detected and classified into blocks based on similarity scores. The similarity score was calculated at SNP sites independently according to the following formula:
where Gij and Giz is the genotype of individuals i at the j-th SNP and at the z-th SNP, respectively, and s(GizGij) / n is the score of individuals i between the genotype at the z-th SNP and the genotype at the j-th SNP. n represents the number of individuals without the missing genotype at the j and z-th SNP. The similarity score is the sum of the scores of n individuals at the z-th SNP except for those with missing genotypes. The score of individual i at the j and z-th SNPs are: s = 1 if the genotypes of Giz and GijGij are identical (A vs. A, B vs. B, H vs. H); s = 0.5 if the genotypes of Giz and Gij are different and one is ‘H’ (A vs. H, B vs. H); and s = 0 if the genotypes of Giz and Gij are different and neither is ‘H’ (A vs. B). The minimum similarity score used for classifying similar SNP sites into a block was 0.7. There were two reasons that a minimum similarity score of 0.7 was used: (1) Markers with a high proportion of matches (>27/37, 72.97 %) were classified into a block in a similar study [5]; and (2) the accuracy of initial genotype calling at SNP sites for the F2 population was 80.07 %.
For SNP sites in the same scaffold and block, an approach combining the sliding window approach [79] and Bayesian inference [80] based on reads in the window was used to determine the genotypes of individuals that could reduce the error rate and the number of missing genotypes. For a genotype at a given w window, assume there are n reads at the SNP sites in the window. The error rate of parent 1 and parent 2 are E1 (0.058) and E2 (0.02), respectively. Under the condition of the allele from parent 1 (P1P1) or parent 2 (P2P2) or two parents (P1P2), the probability of observing n1 reads of parent 1 at the SNP sites in the window would follow a binominal distribution:
Let P(P1P1 | n1), P(P2P2 | n1), P(P1P2 | n1), be the posterior probabilities that the allele comes from parent 1, parent 2, or two parents given the observed n1 reads of parent 1 at the SNP sites in the window. These probabilities were calculated as:
The genotype of the allele was determined based on the highest posterior probability at the SNP sites in the window:
The prior probabilities for genotypes P1P1, P2P2 and P1P2 used for the Bayesian probability calculation were estimated based on the theoretical probabilities for an F2 population, that is, P1P1 : P2P2 : P1P2 = 0.25 : 0.25 : 0.5.
To evaluate the accuracy of the genotypes of the 59 individuals, the genotypes of 26 random SSR markers that can be uniquely mapped to scaffolds by the size of their PCR product were identified in each individual. We assumed that the SNP genotypes of the 59 individuals in 10 kb flank intervals of 26 SSR markers’ physical locations were consistent with the genotypes of the corresponding SSR markers.
Genetic mapping and validation
After the imputed genotype was visually scored by a sliding window, unambiguous SNP sites were used for the construction of a genetic linkage map. Using SNP sites, an ultra-dense genetic linkage map based on the imputed genotype of allotetraploid cotton was constructed by the following procedure. The co-segregating SNP sites were clustered as a recombination bin, defined as a class of SNP sites that had identical genotypes across the 59 individuals with no recombination existing between each pair of SNP sites. The resulting 4,049 recombination bins combining SSR genotypes were used for the construction of the genetic linkage map using JoinMap Version 3.0 [39] with a recombination frequency <0.4 and minimum logarithm of odds (LOD) scores of 6. Recombination frequency was converted to linkage distances (centimorgan, cM) using the Kosambi mapping function. The resulting linkage groups constituted the genetic linkage map and were assigned the standard nomenclature for cotton (A01 to A13, D01 to D13) on the basis of the incorporation of a framework of SSR markers from the existing linkage map [33] and previously published chromosome naming systems [81]. To validate the linkage map, an independent program, CheckMatrix using PyMap BIT and REC scores, as described by Kozik [82], was utilized. The recombination bin map was used to anchor scaffolds and calculate recombination rates.
Correcting the genome mis-assembly
Each scaffold within the SNP linkage map was divided into 201 bp segments based on the SNP positions. SNP segments were mapped to the assembled genome using BWA software [37]. The stable SNP segments were used to detect mis-assembly scaffolds and anchor scaffolds, which were identified if the following conditions were met: (1) the minimum match base was 201 bp for the TM-1 genome; (2) there was a unique alignment on the genome sequence; and (3) there were at least 10 consecutive SNP segments with the same genetic position.
Integration of the newly generated genetic linkage map and the physical map of the G. raimondii genome
We aligned the scaffolds from the ultra-dense genetic linkage map based on the SNPs of allotetraploid cotton to the whole G. raimondii genome using MUMmer software, version 3.23 [83], and the best hit was chosen in the case of multiple matches.
Estimation of recombination rates
The estimated recombination rate (cM/Mb) was calculated using in-house Perl scripts, which divided the genetic length of the segment in cM by the physical length of the segment in Mb. We defined recombination suppression regions as those where the recombination rate was less than 1.0 cM/Mb, and recombination hot regions as those where the recombination rate was greater than 1.0 cM/Mb [6].
Linkage mapping of cotton centromeric regions
Four LTR retrotransposons were identified from the centromere-specific BAC clone, 97G20 [84], which is closely associated with the centromere of cotton. The retrotransposons were designated as GhCR1, GhCR2, GhCR3, and GhCR4, and more generally as GhCRs. By high-resolution mapping, the GhCRs were located in the centromeric regions of all 52 chromosomes in the tetraploid cotton, G. hirsutum acc TM-1. To search the centromeric regions of each chromosome in TM-1, a sequence alignment method was applied. According to the structure of the retrotransposons, relatively conserved 5’LTR regions, designated as GhCR1-5’LTR, GhCR2-5’LTR, GhCR3-5’LTR, and GhCR4-5’LTR, were selected as query sequences to align with the TM-1 genome database. All the alignments were obtained by BLASTn analysis with its parameters set to the default values. Subsequently, GhCRs-5’LTR were screened for sequence similarity and e-value, with ≥80 % and ≤1e-20 set as the threshold values, respectively. To identify the centromeric regions of the A subgenome, the screened alignments of 13 chromosomes from GhCRs-5’LTR were mingled. They were then used to calculate the median size of the 95 % confidence interval for the median, which was defined as the centromeric region for each chromosome. To identify the centromeric regions of the D subgenome, the screened alignments of 13 chromosomes from GhCR1-5’LTR and GhCR3-5’LTR were used to analyze the centromeric regions. For further analyses, we used a multiple alignment consensus sequence that contained the GhCRs LTR retrotransposon RT domain. Phylogenetic analysis was performed using the NJ method in the MEGA 5.10 program [71]. Statistical support for the NJ tree was evaluated by bootstrapping, where the number of replications was 1,000.
Data access
The sequences have been deposited in DDBJ/EMBL/GenBank under the accessions (PRJNA274882). The SNP data between G. hirsutum acc TM-1 and G. barbadense cv. Hai7124 are available at http://mascotton.njau.edu.cn/. The F3 descended from F2 population will be available for mapping the QTL/genes related to important traits for predictive breeding in the near future.
Acknowledgements
This work was financially supported in part by grants from the Major State Basic Research Development Program of China (973 Program) (2011CB109300), National Key Technology Support Program of China (2015BAD02B00), The Fundamental Research Funds by the Central Universities, the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 project (B08025), and JCIC-MCP project.
Abbreviations
- BAC
Bacterial artificial chromosomes
- bp
Base pair
- BWA
Burrows-wheeler aligner
- Chr
Chromosome
- cM
centimorgan
- CR
Centromeric retrotransposon
- FISH
Fluorescent in situ hybridization
- Gb
Gigabase
- GBS
Genotyping-by-sequencing
- kb
kilobase
- LOD
Logarithms of odds
- LTR
Long terminal repeat
- Mb
Megabase
- MQ
Mapping quality
- NGS
Next-generation sequencing
- PCR
Polymerase chain reaction
- QTL
Quantitative trait loci
- RAD
Restriction site-associated genomic DNA
- SBG
Sequence-based genotyping
- SNP
Single nucleotide polymorphism
- SSR
Simple sequence repeats
- WGS
Whole genome shotgun
Additional files
Processing workflow for assigning, ordering and orienting scaffolds and validating misassembled scaffolds. First, sequencing the two parents (TM-1 and Hai7124) and an interspecific F2 population (THF2) derived from a cross between the two parents; second, tentatively assembling the WGS sequences of TM-1 using SOAPdenovo package [85]; third, SNP calling between two parents, genotyping, and constructing a high resolution SNP linkage map; fourth, validating the structural correctness of primarily assembled scaffolds of the TM-1 genome by the linkage map; fifth, further assembling the sequence scaffolds by integrating paired BAC-end sequences (BES) with default and stringent parameters, respectively; sixth, validating, anchoring and orienting of assembled scaffolds; finally, coupling construction of the ultra-dense genetic map for assembling and validating.
Information on reads mapped to the TM-1 reference sequence.
Distribution of HQ data in 59 individuals of (TM-1 × Hai7124)F 2 . To construct an ultra-dense linkage map in tetraploid cotton, population sequencing by low-depth (approximately 5× coverage of the genome) whole genome sequencing (WGS) of 59 F2 individuals was performed. The x axis indicates the 59 individuals of (TM-1 × Hai7124)F2. The y axis indicates the number of the high quality sequence reads (Gb).
Marker details for the ultra-dense SNP genetic linkage map and the corresponding framework SSRs linkage map. Table listing the position of SSR markers in corresponding chromosomes, SSR marker name, and the genotypes of 59 F2 individuals for the framework SSRs linkage map, the position of the marker or scaffold in the corresponding chromosome, the ID of recombination bin, the region ID of the scaffold, the scaffold name, the length of the scaffold, the number of SNPs, the genotypes of the 59 F2 individuals, and the number of SNPs per recombination bin for the ultra-dense genetic linkage map. Alleles: A/A (red) = homozygous TM-1 genotype (maternal parent); B/B (blue) = homozygous Hai7124 genotype (paternal parent); H/H (yellow) = heterozygous genotype (TM-1 × Hai7124)F1; C = not genotype A (dominant B-allele); D = not genotype B (dominant A-allele); Missing data (genotype unknown) are noted as “—”.
Comparison of two genetic maps of inter-specific SNPs and intra-specific SNPs. Intra-specific SNP tags in 104 linkage groups (purple) were aligned to TM-1 scaffolds in the 26 linkage groups based on inter-specific SNPs (blue).
Coordinates for splitting of the TM-1 genome assembly scaffolds. Tables S1 and S2, listing the misassembled scaffold name, the position of the misassembled scaffold mapped in different chromosomes, the breakout region of the misassembled scaffold, and the types of mis-assembly for the TM-1 genome initial assembly and version 1.0, respectively.
Genome variations identified via comparative mapping against D genome pseudomolecules.
SNP-poor regions between TM-1 and Hai7124.
Aligned G. hirsutum TM-1 genome with four sequences of GhCRs-5’LTR and CRG1-5’LTR.
Centromere mapping in the physical map of the TM-1 genome. Tables S1 and S2 listing centromere mapping in the physical map of the TM-1 A and D subgenomes, respectively.
Footnotes
Sen Wang and Jiedan Chen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TZZ conceptualized the research program and coordinated the project. SW, JDC, WPZ, YH, LJC, LF, QW, FNL, HTW, ZFS, SQC, CPC, XFZ, and BLZ conducted laboratory experiments and were involved in data analysis. SW and JDC performed bioinformatics analyses. TZZ, WZG, SW, JDC, CPC, XFZ, and BLZ supervised data generation and analysis. SW and JDC drafted the manuscript. TZZ and WZG revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Sen Wang, Email: 2009201059@njau.edu.cn.
Jiedan Chen, Email: chenjiedan2010@gmail.com.
Wenpan Zhang, Email: flying0802@163.com.
Yan Hu, Email: njauhuyan@njau.edu.cn.
Lijing Chang, Email: 2013201068@njau.edu.cn.
Lei Fang, Email: njaufanglei@njau.edu.cn.
Qiong Wang, Email: wangq0903@gmail.com.
Fenni Lv, Email: 2009201036@njau.edu.cn.
Huaitong Wu, Email: 2011201065@njau.edu.cn.
Zhanfeng Si, Email: 2012201068@njau.edu.cn.
Shuqi Chen, Email: chenshuqi@qq.com.
Caiping Cai, Email: cpcai@njau.edu.cn.
Xiefei Zhu, Email: fxx615@njau.edu.cn.
Baoliang Zhou, Email: baoliangzhou@njau.edu.cn.
Wangzhen Guo, Email: moelab@njau.edu.cn.
Tianzhen Zhang, Email: cotton@njau.edu.cn.
References
- 1.Gupta PK, Roy JK, Prasad M. Single nucleotide polymorphisms: A new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants. Curr Sci. 2001;80:524–35. [Google Scholar]
- 2.Ganal MW, Altmann T, Roder MS. SNP identification in crop plants. Curr Opin Plant Biol. 2009;12:211–7. doi: 10.1016/j.pbi.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW, Shoemaker RC, et al. High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics. 2010;11:38. doi: 10.1186/1471-2164-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huo N, Garvin DF, You FM, McMahon S, Luo MC, Gu YQ, et al. Comparison of a high-density genetic linkage map to genome features in the model grass Brachypodium distachyon. Theor Appl Genet. 2011;123:455–64. doi: 10.1007/s00122-011-1598-4. [DOI] [PubMed] [Google Scholar]
- 5.Bancroft I, Morgan C, Fraser F, Higgins J, Wells R, Clissold L, et al. Dissecting the genome of the polyploid crop oilseed rape by transcriptome sequencing. Nat Biotechnol. 2011;29:762–6. doi: 10.1038/nbt.1926. [DOI] [PubMed] [Google Scholar]
- 6.Ren Y, Zhao H, Kou Q, Jiang J, Guo S, Zhang H, et al. A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS One. 2012;7:e29453. doi: 10.1371/journal.pone.0029453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteras C, Formisano G, Roig C, Diaz A, Blanca J, Garcia-Mas J, et al. SNP genotyping in melons: genetic variation, population structure, and linkage disequilibrium. Theor Appl Genet. 2013;126:1285–303. doi: 10.1007/s00122-013-2053-5. [DOI] [PubMed] [Google Scholar]
- 8.Atwell S, Huang YS, Vilhjalmsson BJ, Willems G, Horton M, Li Y, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–31. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deokar AA, Ramsay L, Sharpe AG, Diapari M, Sindhu A, Bett K, et al. Genome wide SNP identification in chickpea for use in development of a high density genetic map and improvement of chickpea reference genome assembly. BMC Genomics. 2014;15:708. doi: 10.1186/1471-2164-15-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice Database Project. http://www.ncgr.ac.cn/RiceHapMap.
- 11.Lai JS, Li RQ, Xu X, Jin WW, Xu ML, Zhao HN, et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet. 2010;42:1027–U1158. doi: 10.1038/ng.684. [DOI] [PubMed] [Google Scholar]
- 12.Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, et al. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–6. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- 13.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Poecke RM, Maccaferri M, Tang J, Truong HT, Janssen A, van Orsouw NJ, et al. Sequence-based SNP genotyping in durum wheat. Plant Biotechnol J. 2013;11:809–17. doi: 10.1111/pbi.12072. [DOI] [PubMed] [Google Scholar]
- 16.Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS. SNP discovery via 454 transcriptome sequencing. Plant J. 2007;51:910–8. doi: 10.1111/j.1365-313X.2007.03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krasileva KV, Buffalo V, Bailey P, Pearce S, Ayling S, Tabbita F, et al. Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 2013;14:R66. doi: 10.1186/gb-2013-14-6-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trick M, Long Y, Meng J, Bancroft I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol J. 2009;7:334–46. doi: 10.1111/j.1467-7652.2008.00396.x. [DOI] [PubMed] [Google Scholar]
- 19.Round EK, Flowers SK, Richards EJ. Arabidopsis thaliana centromere regions: genetic map positions and repetitive DNA structure. Genome Res. 1997;7:1045–53. doi: 10.1101/gr.7.11.1045. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Wang J, Jiang J, Zhang Q. Mapping of centromeric regions on the molecular linkage map of rice (Oryza sativa L.) using centromere-associated sequences. Mol Gen Genet. 2000;263:165–72. doi: 10.1007/s004380050044. [DOI] [PubMed] [Google Scholar]
- 21.Singh K, Ishii T, Parco A, Huang N, Brar DS, Khush GS. Centromere mapping and orientation of the molecular linkage map of rice (Oryza sativa L.) Proc Natl Acad Sci U S A. 1996;93:6163–8. doi: 10.1073/pnas.93.12.6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolfgruber TK, Sharma A, Schneider KL, Albert PS, Koo DH, Shi J, et al. Maize centromere structure and evolution: sequence analysis of centromeres 2 and 5 reveals dynamic Loci shaped primarily by retrotransposons. PLoS Genet. 2009;5:e1000743. doi: 10.1371/journal.pgen.1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Copenhaver GP, Nickel K, Kuromori T, Benito MI, Kaul S, Lin X, et al. Genetic definition and sequence analysis of Arabidopsis centromeres. Science. 1999;286:2468–74. doi: 10.1126/science.286.5449.2468. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Cao Y, Wang K, Zhao T, Chen J, Pan M, et al. Identification of centromeric regions on the linkage map of cotton using centromere-related repeats. Genomics. 2014;104:587–93. doi: 10.1016/j.ygeno.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Brubaker C, Paterson A, Wendel J. Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome. 1999;42:184–203. doi: 10.1139/g98-118. [DOI] [Google Scholar]
- 26.Lacape JM, Nguyen TB, Thibivilliers S, Bojinov B, Courtois B, Cantrell RG, et al. A combined RFLP-SSR-AFLP map of tetraploid cotton based on a Gossypium hirsutum × Gossypium barbadense backcross population. Genome. 2003;46:612–26. doi: 10.1139/g03-050. [DOI] [PubMed] [Google Scholar]
- 27.Rong J, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, et al. A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium) Genetics. 2004;166:389–417. doi: 10.1534/genetics.166.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu J, Kohel RJ, Smith CW. The construction of a tetraploid cotton genome wide comprehensive reference map. Genomics. 2010;95:230–40. doi: 10.1016/j.ygeno.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Yu Y, Yuan D, Liang S, Li X, Wang X, Lin Z, et al. Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genomics. 2011;12:15. doi: 10.1186/1471-2164-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu JZ, Kohel RJ, Fang DD, Cho J, Van Deynze A, Ulloa M, et al. A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3 (Bethesda) 2012;2:43–58. doi: 10.1534/g3.111.001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blenda A, Fang DD, Rami JF, Garsmeur O, Luo F, Lacape JM. A high density consensus genetic map of tetraploid cotton that integrates multiple component maps through molecular marker redundancy check. PLoS One. 2012;7:e45739. doi: 10.1371/journal.pone.0045739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Zhang D, Wang X, Tan X, Guo H, Paterson AH. A whole-genome DNA marker map for cotton based on the D-genome sequence of Gossypium raimondii L. G3 (Bethesda) 2013;3:1759–67. doi: 10.1534/g3.113.006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao L, Yuanda L, Caiping C, Xiangchao T, Xiangdong C, Wei Z, et al. Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genomics. 2012;13:539. doi: 10.1186/1471-2164-13-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byers RL, Harker DB, Yourstone SM, Maughan PJ, Udall JA. Development and mapping of SNP assays in allotetraploid cotton. Theor Appl Genet. 2012;124:1201–14. doi: 10.1007/s00122-011-1780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hulse-Kemp AM, Ashrafi H, Zheng X, Wang F, Hoegenauer KA, Maeda AB, et al. Development and bin mapping of gene-associated interspecific SNPs for cotton (Gossypium hirsutum L.) introgression breeding efforts. BMC Genomics. 2014;15:945. doi: 10.1186/1471-2164-15-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Gao W, Guo H, Zhang X, Fang DD, Lin Z. Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping. BMC Genomics. 2014;15:1046. doi: 10.1186/1471-2164-15-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Ooijen JW, Voorrips R. JoinMap: version 3.0: software for the calculation of genetic linkage maps. Wageningen, the Netherlands: Plant Research International; 2001. [Google Scholar]
- 40.Wang YK, Ning ZY, Hu Y, Chen JD, Zhao R, Chen H, et al. Molecular mapping of restriction-site associated DNA markers in allotetraploid Upland cotton. PLoS One. 2015;10:e0124781. doi: 10.1371/journal.pone.0124781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang TZ, Hu Y, Jiang WK, Fang L, Guan XY, Chen JD, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fibre improvement. Nat Biotechnol. 2015;33:531–7. doi: 10.1038/nbt.3207. [DOI] [PubMed] [Google Scholar]
- 42.Chen ZJ. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol. 2007;58:377–406. doi: 10.1146/annurev.arplant.58.032806.103835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams KL, Wendel JF. Polyploidy and genome evolution in plants. Curr Opin Plant Biol. 2005;8:135–41. doi: 10.1016/j.pbi.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Chester M, Leitch AR, Soltis PS, Soltis DE. Review of the application of modern cytogenetic methods (FISH/GISH) to the study of reticulation (polyploidy/hybridisation) Genes. 2010;1:166–92. doi: 10.3390/genes1020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.International Wheat Genome Sequencing C A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 46.Lewin HA, Larkin DM, Pontius J, O’Brien SJ. Every genome sequence needs a good map. Genome Res. 2009;19:1925–8. doi: 10.1101/gr.094557.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 2014;46:567–72. doi: 10.1038/ng.2987. [DOI] [PubMed] [Google Scholar]
- 48.Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492:423–7. doi: 10.1038/nature11798. [DOI] [PubMed] [Google Scholar]
- 49.Park YH, Alabady MS, Ulloa M, Sickler B, Wilkins TA, Yu J, et al. Genetic mapping of new cotton fiber loci using EST-derived microsatellites in an interspecific recombinant inbred line cotton population. Mol Genet Genomics. 2005;274:428–41. doi: 10.1007/s00438-005-0037-0. [DOI] [PubMed] [Google Scholar]
- 50.Lacape JM, Llewellyn D, Jacobs J, Arioli T, Becker D, Calhoun S, et al. Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biology. 2010;10:132. doi: 10.1186/1471-2229-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen X, Guo W, Liu B, Zhang Y, Song X, Cheng Y, et al. Molecular mechanisms of fiber differential development between G. barbadense and G. hirsutum revealed by genetical genomics. PLoS One. 2012;7:e30056. doi: 10.1371/journal.pone.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephens SG. The origin of Sea Island cotton. Agric Hist. 1975;50:391–9. [Google Scholar]
- 53.Stephens SG. Some observations on photoperiodism and the development of annual forms of domesticated cottons. Econ Bot. 1976;30:409–18. doi: 10.1007/BF02904663. [DOI] [Google Scholar]
- 54.Wang GL, Dong JM, Paterson AH. The distribution of Gossypium hirsutum chromatin in G. barbadense germ plasm: molecular analysis of introgressive plant breeding. Theor Appl Genet. 1995;91:1153–61. doi: 10.1007/BF00220953. [DOI] [PubMed] [Google Scholar]
- 55.Lubbers EL, Chee PW. The worldwide gene pool of G. hirsutum and its improvement. In: Paterson AH, editor. Genetics and Genomics of Cotton. New York: Springer; 2009. pp. 23–52. [Google Scholar]
- 56.Cheng Z, Presting GG, Buell CR, Wing RA, Jiang J. High-resolution pachytene chromosome mapping of bacterial artificial chromosomes anchored by genetic markers reveals the centromere location and the distribution of genetic recombination along chromosome 10 of rice. Genetics. 2001;157:1749–57. doi: 10.1093/genetics/157.4.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gill KS, Gill BS, Endo TR, Taylor T. Identification and high-density mapping of gene-rich regions in chromosome group 1 of wheat. Genetics. 1996;144:1883–91. doi: 10.1093/genetics/144.4.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gill KS, Gill BS, Endo TR, Boyko EV. Identification and high-density mapping of gene-rich regions in chromosome group 5 of wheat. Genetics. 1996;143:1001–12. doi: 10.1093/genetics/143.2.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drouaud J, Camilleri C, Bourguignon PY, Canaguier A, Berard A, Vezon D, et al. Variation in crossing-over rates across chromosome 4 of Arabidopsis thaliana reveals the presence of meiotic recombination “hot spots”. Genome Res. 2006;16:106–14. doi: 10.1101/gr.4319006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright SI, Agrawal N, Bureau TE. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 2003;13:1897–903. doi: 10.1101/gr.1281503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu J, Mizuno H, Hayashi-Tsugane M, Ito Y, Chiden Y, Fujisawa M, et al. Physical maps and recombination frequency of six rice chromosomes. Plant J. 2003;36:720–30. doi: 10.1046/j.1365-313X.2003.01903.x. [DOI] [PubMed] [Google Scholar]
- 62.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–6. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 63.Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, et al. The genome of the cucumber, Cucumis sativus L. Nat Genet. 2009;41:1275–81. doi: 10.1038/ng.475. [DOI] [PubMed] [Google Scholar]
- 64.Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, et al. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet. 2007;3:e123. doi: 10.1371/journal.pgen.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo S, Mach J, Abramson B, Ramirez R, Schurr R, Barone P, et al. The cotton centromere contains a Ty3-gypsy-like LTR retroelement. PLoS One. 2012;7:e35261. doi: 10.1371/journal.pone.0035261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang K, Guo W, Yang Z, Hu Y, Zhang W, Zhou B, et al. Structure and size variations between 12A and 12D homoeologous chromosomes based on high-resolution cytogenetic map in allotetraploid cotton. Chromosoma. 2010;119:255–66. doi: 10.1007/s00412-009-0254-0. [DOI] [PubMed] [Google Scholar]
- 67.Hosouchi T, Kumekawa N, Tsuruoka H, Kotani H. Physical map-based sizes of the centromeric regions of Arabidopsis thaliana chromosomes 1, 2, and 3. DNA Res. 2002;9:117–21. doi: 10.1093/dnares/9.4.117. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Huang Y, Zhang L, Li Y, Lu T, Lu Y, et al. Structural features of the rice chromosome 4 centromere. Nucl Acids Res. 2004;32:2023–30. doi: 10.1093/nar/gkh521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagaki K, Cheng Z, Ouyang S, Talbert PB, Kim M, Jones KM, et al. Sequencing of a rice centromere uncovers active genes. Nat Genet. 2004;36:138–45. doi: 10.1038/ng1289. [DOI] [PubMed] [Google Scholar]
- 70.Gong Z, Wu Y, Koblizkova A, Torres GA, Wang K, Iovene M, et al. Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell. 2012;24:3559–74. doi: 10.1105/tpc.112.100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evo. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petit M, Guidat C, Daniel J, Denis E, Montoriol E, Bui QT, et al. Mobilization of retrotransposons in synthetic allotetraploid tobacco. New Phytol. 2010;186:135–47. doi: 10.1111/j.1469-8137.2009.03140.x. [DOI] [PubMed] [Google Scholar]
- 73.Liu B, Wendel JF. Retrotransposon activation followed by rapid repression in introgressed rice plants. Genome. 2000;43:874–80. doi: 10.1139/g00-058. [DOI] [PubMed] [Google Scholar]
- 74.Dooner HK, Weil CF. Give-and-take: interactions between DNA transposons and their host plant genomes. Curr Opin Genet Dev. 2007;17:486–92. doi: 10.1016/j.gde.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 75.Zhang J, Guo W, Zhang T. Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. × Gossypium barbadense L.) with a haploid population. Theor Appl Genet. 2002;105:1166–74. doi: 10.1007/s00122-002-1100-4. [DOI] [PubMed] [Google Scholar]
- 76.Paterson AH, Brubaker CL, Wendel JF. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep. 1993;11:122–7. doi: 10.1007/BF02670470. [DOI] [Google Scholar]
- 77.Pomraning KR, Smith KM, Bredeweg EL, Connolly LR, Phatale PA, Freitag M. Library preparation and data analysis packages for rapid genome sequencing. Methods Mol Biol. 2012;944:1–22. doi: 10.1007/978-1-62703-122-6_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–76. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang K, Song X, Han Z, Guo W, Yu JZ, Sun J, et al. Complete assignment of the chromosomes of Gossypium hirsutum L. by translocation and fluorescence in situ hybridization mapping. Theor Appl Genet. 2006;113:73–80. doi: 10.1007/s00122-006-0273-7. [DOI] [PubMed] [Google Scholar]
- 82.CheckMatrix. http://www.atgc.org/XLinkage/
- 83.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang K, Zhang W, Cao Y, Zhang Z, Zheng D, Zhou B, et al. Localization of high level of sequence conservation and divergence regions in cotton. Theor Appl Genet. 2012;124:1173–82. doi: 10.1007/s00122-011-1777-3. [DOI] [PubMed] [Google Scholar]
- 85.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]