

Abstract
Synaptic neurotransmission is modified at cortical connections throughout life. Varying the amplitude of the postsynaptic response is one mechanism that generates flexible signaling in neural circuits. The timing of the synaptic response may also play a role. Here, we investigated whether weakening and loss of an entire connection between excitatory cortical neurons was foreshadowed in the timing of the postsynaptic response. We made electrophysiological recordings in rat primary somatosensory cortex that was undergoing experience-dependent loss of complete local excitatory connections. The synaptic latency of pyramid–pyramid connections, which typically comprise multiple synapses, was longer and more variable. Connection strength and latency were not correlated. Instead, prolonged latency was more closely related to progression of connection loss. The action potential waveform and axonal conduction velocity were unaffected, suggesting that the altered timing of neurotransmission was attributable to a synaptic mechanism. Modeling studies indicated that increasing the latency and jitter at a subset of synapses reduced the number of action potentials fired by a postsynaptic neuron. We propose that prolonged synaptic latency and diminished temporal precision of neurotransmission are hallmarks of impending loss of a cortical connection.
Keywords: latency, neural code, plasticity, presynaptic, rewiring, synapse
Introduction
Sensory stimuli typically elicit sparse neural firing in the neocortex (Newsome et al., 1989; Vinje and Gallant, 2000; Brecht et al., 2003; Olshausen and Field, 2004; de Kock et al., 2007; Kerr et al., 2007; Barth and Poulet, 2012). The contribution of this sparse firing to sensory processing is debated (Wolfe et al., 2010). However, it is thought that both the number of action potentials and their timing carry information about the sensory input (Rieke et al., 1997; Ahissar et al., 2000; Pinto et al., 2000; Panzeri et al., 2001; Arabzadeh et al., 2006; von Heimendahl et al., 2007; Stüttgen and Schwarz, 2008; Jadhav et al., 2009; London et al., 2010).
In general, the cortical circuits that generate sparse firing comprise a distributed network of local pyramidal neurons and inhibitory interneurons. Each connection is formed by a single axon, which typically makes multiple synapses with the postsynaptic neuron (Markram et al., 1997; Somogyi et al., 1998; Cheetham et al., 2007). Despite being multisynaptic, both thalamocortical inputs and recurrent excitatory connections in the neocortex are weak (Bruno and Sakmann, 2006; Cheetham et al., 2007; Lefort et al., 2009). Therefore, coincident activity of multiple excitatory neurons is required to cause a cortical pyramidal neuron to fire an action potential (Crochet et al., 2011). Accordingly, the timing of excitatory synaptic activity is crucial to firing of pyramidal neurons in the neocortex.
The classic signature of cortical reorganization is altered neural firing, which may affect either the number or timing of sensory-evoked action potentials (Jenkins et al., 1990; Recanzone et al., 1992a; b; Benedetti et al., 2009). The plasticity mechanisms that underpin altered neural firing can be subdivided into altered neuronal excitability, changes in synaptic strength, and rewiring of connections (Zhang and Linden, 2003; Feldman, 2009; Barnes and Finnerty, 2010). Much work has focused on how changes in synaptic strength modify neural firing (Hebb, 1949; Bienenstock et al., 1982; Margolis et al., 2014). However, reorganization of touch-evoked neural firing in somatosensory cortex (Margolis et al., 2012) is also accompanied by extensive rewiring of local excitatory circuits (Albieri et al., 2014). Changes in synaptic strength and rewiring-induced increases or decreases in synapse number both affect the amount of excitatory input onto a neuron. Far less attention has been paid to the timing of those synaptic inputs. In particular, it remains unclear whether modifications to the timing of synaptic transmission play a role during cortical reorganization.
We investigated how the timing of synaptic transmission is affected when entire pyramid-to-pyramid (Pyr→Pyr) connections are lost in reorganizing somatosensory cortex. We show that the latency of the postsynaptic potential is longer and more temporally imprecise. Modeling studies indicate that prolonging the synaptic latency effectively reduces the contribution of a connection to spiking in a cortical microcircuit without physical loss of that connection. We propose that prolonged synaptic latency and diminished temporal precision of neurotransmission are hallmarks of impending loss of a cortical connection.
Materials and Methods
Whisker trimming.
All procedures were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986. Rats of either sex were studied. We trimmed the lower two (D and E rows, γ and δ outliers) or upper three (A–C rows and α, β, and γ outliers) rows of whiskers (Fig. 1Ai). Whisker trimming was performed daily from P30 for 2–8 d. Age-matched sham-trimmed littermates served as controls. For 26+ d trims, whisker trimming began at P19 (Cheetham et al., 2007).
Figure 1.
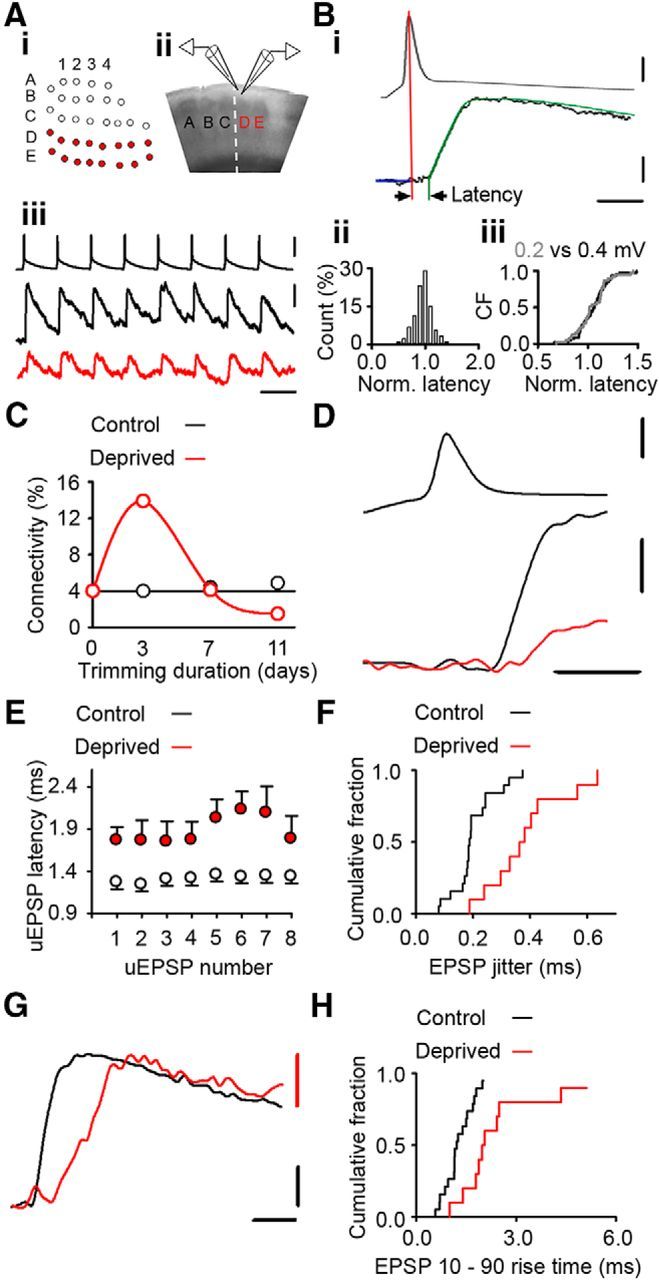
Altered timing of neurotransmission at excitatory connections in deprived cortex. Ai, Schematic of a D-E whisker row trim [white circles, untrimmed whiskers (spared); red circles, trimmed whiskers (deprived)]. Aii, Brain slice showing position of recording electrodes in L2/3 of deprived D barrel column. Aiii, Twenty Hertz train of presynaptic action potentials (upper trace) and evoked postsynaptic uEPSPs recorded from synaptically connected neurons in control (black, middle trace) and 4–8 d deprived (red, lower trace) cortex. Calibrations: 50 mV, 0.1 mV; 50 ms. Bi, Example latency measurement: peak of action potential, vertical red line; uEPSP fit, green; baseline, blue line. Arrowheads indicate the latency measurement. Calibrations: 0.1 mV, 30 mV; 2.5 ms. Bii, Histogram of normalized latency times measured from simulated uEPSPs with known latency. Biii, Empirical distribution functions of normalized latencies for 0.2 (gray) and 0.4 mV (black) simulated uEPSPs were not statistically different (p = 0.930, t test). C, Connectivity between L2/3 pyramidal neurons varies with duration of whisker deprivation [2–3 d, 13/93 (14.0%) tested connections; 4–8 d, 8/195 (4.1%) tested connections; p = 0.012, χ2 test]. D, Expanded trace depicting increased synaptic latency at weakened connections in deprived cortex. Top, Action potential. Bottom, Overlay of example uEPSPs in control (black) and deprived (red) cortex. Calibrations: 50 mV, 1 mV; 2 ms. E, uEPSP synaptic latency in deprived cortex (red) is increased after 4–8 d deprivation (uEPSP1: deprived 4–8 d = 1.7 ± 0.2 ms, n = 10; control 1.2 ± 0.1 ms, n = 19; p = 0.007, t test) and throughout a 20 Hz train stimulus train (uEPSP 2–8: deprived 1.7 [1.3–2.6] ms, n = 10; control 1.3 [1.0–1.5] ms, n = 19, p = 0.014, MWRST). F, Empirical distribution plot of the uEPSP1 jitter in control (black) and deprived (red) cortex. The jitter is greater in deprived cortex than in control cortex (deprived 0.38 ± 0.04 ms, n = 10; control 0.20 ± 0.02 ms, n = 19; p < 0.001, t test). G, EPSP trace from deprived cortex (red) scaled to amplitude of control trace (black; average of 50 responses) illustrates increased rise time in deprived cortex. Calibrations: 0.1 mV; 2 ms. H, Empirical distribution plots of uEPSP1 rise time in deprived (red) and control (black) connections. The uEPSP rise time is greater in deprived cortex (deprived 2.0 [1.7–3.0] ms, n = 10; control 1.2 [1.0–1.7] ms, n = 19; p = 0.001, MWRST).
Brain slice preparation and electrophysiological recording.
Brain slices from P32–P57 rats were cut across the whisker barrel rows (65° to the midline; Fig. 1Aii). We made whole-cell voltage recordings from synaptically connected pairs of L2/3 pyramidal neurons (Pyr→Pyr) or pyramidal to fast-spiking (FS) interneuron pairs (Pyr→FS) at 36–37°C as described previously (Cheetham et al., 2007; Albieri et al., 2014; Fig. 1Aii). Capacitance neutralization was optimized (3–6 pF) to minimize filtering of the voltage signal by the pipette. Electrophysiological recordings were amplified, low-pass filtered (synaptic responses 3 kHz, action potentials 10 kHz; Multiclamp 700A; Molecular Devices) and digitized (16 bit, 20 kS per channel; National Instruments PCI 6221 DAQ card) using custom-written software (LabView; National Instruments). Presynaptic neurons were stimulated with eight-pulse trains at 20 Hz or 40 Hz and the evoked response was recorded in the postsynaptic neuron (Fig. 1Aiii). For six connections, recordings were made sequentially in normal ACSF, which contains 2 mm CaCl2 and 1 mm MgCl2, followed by 1 mm CaCl2 and 2 mm MgCl2 or normal ACSF containing the selective NMDA receptor antagonist APV (50 μm). Spontaneous activity was recorded in the absence of drugs. Recordings of synaptically evoked action potentials were made by injecting somatic current to depolarize the neuron's membrane potential close to threshold. Recordings of mEPSPs were made in the presence of TTX (1 μm) and picrotoxin (100 μm) as described previously (Cheetham et al., 2007). Antidromic stimulation was evoked by placing a concentric bipolar stimulating electrode in the lower part of L2/3 above L4 and stimulating with the synaptic blockers APV (50 μm) and CNQX (20 μm) added to the ACSF.
Electrophysiological analysis.
Unitary EPSPs (uEPSPs) were analyzed using custom-written software (LabView; National Instruments) as described previously (Cheetham et al., 2007). Briefly, uEPSP parameters were measured by fitting two functions to the uEPSP. One function (a + bte−ct + dte−gt) was fitted to the uEPSP over a 3–15 ms time window that encompassed the uEPSP peak (Fig. 1Bi). An exponential curve was fitted to the baseline membrane potential over a 3–10 ms window before uEPSP onset and was extrapolated over the period of the uEPSP fit. uEPSP onset was taken as the time at which the baseline function intercepted the EPSP function. Synaptic latency was defined as the time between the peak of the action potential in the presynaptic neuron and the uEPSP onset in the postsynaptic neuron (Fig. 1Bi; Katz and Miledi, 1965; Sabatini and Regehr, 1996). Synaptic latency was measured for individual uEPSPs at each connection. uEPSP amplitude, 10–90% rise time, time to peak, and half-width of the uEPSP were extracted from the fits (EPSP function–baseline function). Failures were defined as traces that contained no evoked uEPSP. Release probability was assessed by measuring the probability of failure and the CV (SD divided by mean of individual uEPSPs) for the first stimulus in the train. Potency was defined as the mean uEPSP amplitude excluding failures. The minimum uEPSP amplitude for each connection was defined as the mean of the three smallest responses (mean ± SEM, 23 ± 3 trials).
The start of the action potential was defined to be the point when the rate of change of membrane potential (dVm/dt) exceeded 20 V · s−1. The action potential half-width was measured at the midpoint on the action potential waveform between action potential initiation and the peak of the action potential. Spontaneous activity was recorded in 5 s epochs. Action potentials (APs) evoked by spontaneous activity (PSP-evoked APs) were analyzed by two experimenters who identified the point of PSP inflection from the membrane potential baseline blind to experimental conditions. The spike latency was measured from the point of PSP onset to the peak of the action potential driven by the PSP (Fig. 8C). The firing frequency was calculated as the number of spikes divided by the total recording time.
Figure 8.
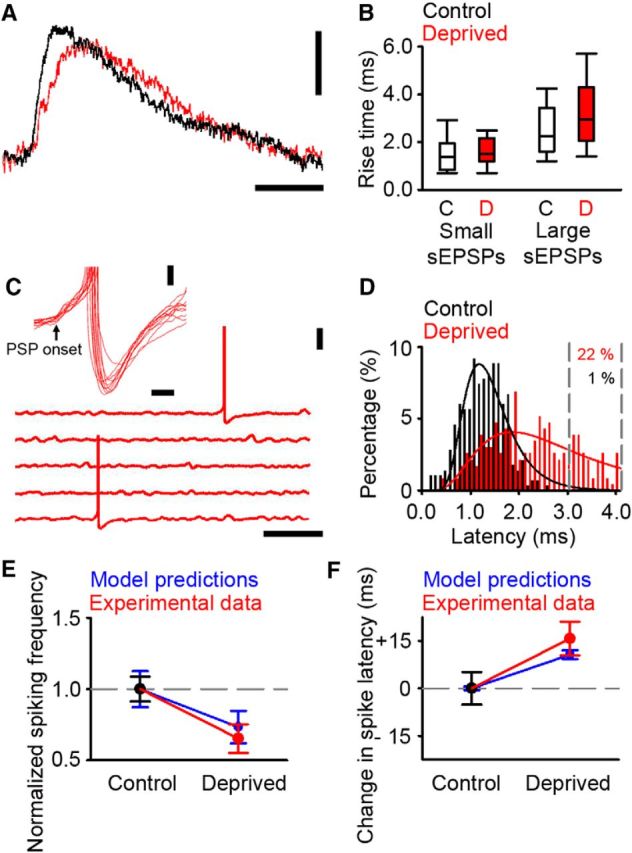
Altered firing in deprived cortex during ongoing activity in vitro. A, Example traces of sEPSP in control (black) and deprived (red) cortex. Calibrations: 0.5 mV, 10 ms. B, Rise time of small sEPSPs (presumed mEPSPs) in control (white fill) and deprived (red fill) cortex were similar (control 1.4 [0.8–2.0] ms, n = 40; deprived 1.5 [1.2–2.2] ms, n = 101; p = 0.206, MWRST). Large sEPSPs have longer rise times in deprived cortex (control 2.3 [1.6–3.5] ms, n = 81; deprived 3.0 [2.1–4.3] ms, n = 168; p < 0.001, MWRST). C, Example of PSP-evoked action potentials in 5 s traces of ongoing synaptic activity recorded from neurons in deprived cortex. Neurons were depolarized with constant somatic current injection to values that were close to threshold. Calibrations: 20 mV, 1 s. Inset, Examples of PSP-evoked action potentials in deprived cortex aligned to the onset of the PSP (black arrow). Calibrations: 2 mV, 20 ms. D, Latency histogram adapted from Figure 4F for putative single-synapse latencies in control (black) and deprived (red) cortex. Gray dashed lines denote the percentage of the deprived responses with latency values that are >99% of the control distribution. The percentage of deprived responses in this range is 22%. E, Comparison of change in firing frequency predicted by the model with the firing frequency found in vitro. Model and experimental data were normalized with respect to their respective control conditions. F, Comparison of the change in spike latency predicted by the model compared with the delay found in vitro. Time differences are expressed with respect to the control spike latency.
Latency measurement.
To assess the accuracy of our latency measurements, we simulated the unitary response evoked at a Pyr→Pyr connection in L2/3 of primary somatosensory cortex. The presynaptic trace comprised two action potentials separated by 50 ms that had been recorded from the presynaptic neuron of an L2/3 Pyr→Pyr connection. The postsynaptic trace comprised epochs of background “noise” containing membrane fluctuations and spontaneous synaptic activity recorded in whole-cell mode from an L2/3 pyramidal neuron onto which we superimposed two simulated EPSPs. The simulated EPSPs were derived from a fit to the average EPSP response evoked by 50 trials at one Pyr→Pyr connection in L2/3 of primary somatosensory cortex. The simulated EPSPs were added at known time intervals after the peak of the presynaptic action potentials (EPSP1, 1.55 ms; EPSP2, 1.95 ms) to mimic a unitary connection with variable EPSP latency. The amplitude of the simulated EPSP was either 0.2 mV or 0.4 mV. The simulated EPSPs were then measured with the experimenter blind to the synaptic latency and amplitude. We first asked how accurate were latency measurements of small, 0.2 mV, simulated EPSPs when superimposed on background noise. The measured latencies of 0.2 mV simulated EPSPs were similar to the known values (Fig. 1Bii; normalized EPSP latency, 1.05 ± 0.03). Similarly, the measured latencies of 0.4 mV simulated EPSPs were comparable to the known synaptic latency (normalized EPSP latency, 1.05 ± 0.03). Importantly, there was no difference in the latency of 0.2 mV simulated EPSPs and 0.4 mV simulated EPSPs (Fig. 1Biii). Therefore, our latency measurements do not become longer when measuring smaller uEPSPs.
Estimating putative single-synapse responses with low extracellular calcium concentration.
Single-synapse responses at excitatory Pyr→Pyr connections in deprived cortex were estimated by reducing the extracellular calcium concentration from 2 to 1 mm to reduce the probability of release so that failures of transmission occurred in ∼50% of trials. We compared the distributions of the evoked uEPSP for single Pyr→Pyr connections recorded sequentially in 2 and 1 mm calcium to determine whether single-synapse responses could be identified in recordings with 2 mm calcium in the bath. Histograms of evoked uEPSP amplitudes showed peaks in both the normal and low calcium conditions. A log normal function (comparable to that found in our simulations) was fitted to the first peak in each histogram (Fig. 4A). The range of uEPSP amplitudes encompassed by the normal calcium fit and low-calcium fit were similar in all of the eight connections that we tested. We concluded that putative single-synapse responses could be identified at weakened Pyr→Pyr connections. Therefore, we pooled the putative single-synapse responses. We focused on the putative single-synapse responses that were in the same amplitude range as our mEPSP recordings because mEPSPs are thought to be from the release of a single vesicle of neurotransmitter and, hence, are examples of single-synapse responses (Fig. 4D). The lower response bound was set as 85 μV. This is the lower bound that we used for mEPSP recordings previously (Cheetham et al., 2007) and is 2.5 times the root mean square noise on baseline traces. The upper response bound was set at the maximum mEPSP amplitude recorded. The upper and lower bounds encompassed 85% of all putative single-synapse responses (Fig. 4E).
Figure 4.

Increased latency of putative single-synapse EPSPs in deprived cortex. A, Top, Example traces recorded in 2 mm Ca2+ (gray) or 1 mm Ca2+ (blue). Bottom, Histogram of EPSP amplitudes from a control connection recorded in 2 mm Ca2+ and 1 mm Mg2+. Blue Gaussian fit is taken from putative single-synapse responses in B (R2 = 0.65, p < 0.001), and black Gaussian is the fit to the 2 mm calcium data over the same amplitude range as the blue Gaussian fit (R2 = 0.78, p < 0.001). Calibrations: 0.15 mV, 10 ms. B, Top, Example putative single-synapse response recorded in low calcium with the average response (black) superimposed. Bottom, Histogram of EPSP amplitudes recorded in 1 mm Ca2+ from the connection in A. Calibrations: 0.15 mV, 10 ms. C, Gaussian or log normal fits to postsynaptic responses recorded in low calcium for all the connections used to estimate putative single-synapse response amplitude (R2 values range from 0.65 to 0.91). Inset, Pooled low-calcium data from the six connections with log normal fit in black (R2 = 0.94, p < 0.001). D, Cumulative fraction plot of 1250 mEPSP amplitudes recorded from 25 L2/3 pyramidal neurons with 2 mm extracellular calcium. Dashed gray lines signify the lower boundary (LB; 85 μV) and upper boundary (UB; 0.28 mV) of the mEPSP range. E, Cumulative fraction plot of putative single-synapse uEPSP responses recorded in 1 mm extracellular calcium. The dashed gray lines, which signify the LB and UB taken from the mEPSP data in D, enclose >85% of the putative single-synapse uEPSP responses recorded in low calcium. F, Latency histogram for putative single-synapse latencies in control (black; log normal fit, R2 = 0.88, p < 0.001) and deprived cortex (red; log normal fit, R2 = 0.66, p < 0.001). G, Average latency for putative single-synapse responses in deprived cortex (1.86 [1.30–2.75] ms, n = 10 connections) is greater than that in control cortex (1.30 [1.11–1.53] ms, n = 19 connections, p = 0.017, MWRST). H, EPSP jitter for putative single-synapse responses in deprived cortex (0.51 [0.45–0.74] ms, n = 10 connections) is greater than that in control cortex (0.25 [0.23–0.38] ms, n = 19 connections, p < 0.001, MWRST). I, EPSP 10–90 rise times for putative single-synapse responses recorded were similar in deprived and control cortex (deprived: 1.21 ± 0.04 ms, n = 110 responses; control: 1.34 ± 0.07 ms, n = 110 responses; p < 0.096, t test).
Electron microscopy and 3D reconstruction of synapses onto dendritic spines.
Tissue from layer 2/3 of rat somatosensory cortex was prepared for electron microscopy as described previously (Knott et al., 2011). Briefly, brains were fixed via cardiac perfusion with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 m PBS, pH 7.4. Vibratome sections were cut at 80 μm thickness through the somatosensory cortex, heavy metal stained with reduced osmium and uranyl acetate, dehydrated, and embedded in Durcupan resin. Five hundred serial electron microscopy images were recorded using focused ion beam-scanning electron microscopy (FIBSEM; Zeiss NVision 40) at a resolution of 6 nm per pixel, with image sizes of 15 × 12 μm (Knott et al., 2011). The image spacing was 12 nm. The FIJI software, running TrakEM2 software (Cardona et al., 2012), was used to mark the position of vesicles in 25 synapses found on dendritic spines of various sizes. The vesicles were plotted with a ball object and the presynaptic membrane drawn with the AreaList function. These synaptic models were then imported into the Blender software (www.blender.org). The Synaptic Vesicle Density Measure add-on (downloadable at http://cvlab.epfl.ch/NeuroMorph) for Blender was then used to calculate the nearest distance from each vesicle to the presynaptic density.
Modeling the effect of altered timing of neurotransmission at L2/3 Pyr→Pyr connections on spiking output.
Modeling was conducted in AnimatLab (http://www.animatlab.com/) using synaptic parameters derived from our experiments. Inputs evoked synaptic responses in a leaky integrate and fire model cell with a resting membrane potential of −65 mV. Gaussian noise was injected into the model cell so that its membrane potential fluctuated below firing threshold. Inputs with high temporal precision were modeled with a mean latency of 1 ms and SD (jitter) of 0.1 ms, which recapitulated our findings in control cortex. The connections in deprived cortex, which showed impaired temporal fidelity, were modeled by increasing the latency of the response to either 1.5 ms or 2.0 ms or 2.5 ms and by increasing the jitter from 0.1 ms to 0.3 ms or 0.4 ms or 0.6 ms, which recapitulated our findings in deprived cortex. Distributions of latency times with a mean latency and jitter of interest were generated using random numbers in MATLAB (rand function). Both the increase in latency and the increase in jitter were similar to those measured after 4–8 d of whisker deprivation. The response to repetitive stimulation was modeled with eight trains of 10 spikes in the presynaptic neurons. The effect of suprathreshold total synaptic input was modeled by increasing the total synaptic input to the postsynaptic neuron to either 2.5 mV or 5 mV or 7.5 mV above threshold. The proportion of temporally impaired putative single synapses was estimated as the percentage of deprived putative single synapses that were greater than three SDs, i.e., 99%, of the control distribution (Fig. 8D).
Statistics.
Normally distributed data were described by their mean ± SEM, and were analyzed using t tests or ANOVA. Data that failed normality and/or equal variance tests were expressed as median [interquartile range]. Where possible, these data underwent a natural log transform to normalize their distributions and/or equalize their variances before performing t tests or ANOVA. The Holm–Sidak method was used for multiple comparisons in ANOVA. The Mann–Whitney rank sum test (MWRST) was used to compare medians. Measures of connectivity were compared using the χ2 test. All tests were two-tailed with α = 0.05.
The uEPSP latency was modeled statistically under the generalized linear model framework in R (R Project for Statistical Computing; http://www.r-project.org/) using the Gamma error family and the following formula:
where E[uEPSP latency] is the expected value of the uEPSP latency; days deprivation is the duration of whisker trimming; and α, β, and γ are parameters (coefficients) of the model.
Results
Whisker trimming induces a biphasic shift in local connectivity between L2/3 pyramidal neurons (Albieri et al., 2014). This biphasic response comprises an initial increase in local excitatory connectivity after 2–3 d of whisker trimming (14.0%, 13/93), which is followed by a marked reduction in local excitatory connectivity by 6–8 d (Fig. 1C). The strength of Pyr→Pyr connections in L2/3 of somatosensory cortex is markedly reduced over the period when local excitatory connections are lost (Albieri et al., 2014). Here, we investigate how the timing of synaptic transmission is altered at connections that are being weakened and are likely to be lost.
Delayed synaptic responses with impaired temporal precision at excitatory connections in deprived cortex
We explored whether the temporal features of synaptic transmission at Pyr→Pyr connections in L2/3 are modified in deprived primary somatosensory cortex. The timing of synaptic responses was changed in multiple ways after 4–8 d deprivation. First, the latency of the first synaptic response evoked by a train of presynaptic action potentials was increased (Fig. 1D,E). The prolongation of EPSP latency was not transient, but persisted throughout repetitive (20 Hz) firing of the presynaptic pyramidal neuron (Fig. 1E). Second, the SD of the synaptic latency, termed the jitter, was greater (Fig. 1F). Third, the kinetics of uEPSPs were altered with a longer uEPSP rise time (Fig. 1G,H). The passive membrane properties of L2/3 pyramidal neurons were not affected by whisker deprivation (Table 1), suggesting that changes in passive membrane properties did not underlie the observed changes in uEPSP kinetics. Action potential firing in the postsynaptic neuron is most likely to occur near the peak of the EPSP. Therefore, we measured the time from the peak of the presynaptic action potential to the peak of the evoked EPSP. We found that the time from the presynaptic action potential to the peak of the evoked uEPSP in Pyr→Pyr connections was increased by 60% in 4–8 d deprived cortex (deprived 5.5 [4.5–7.0] ms, n = 10; control 3.7 [3.0–4.4] ms, n = 19; p = 0.004, MWRST). Collectively, our data show that, after 4–8 d deprivation, evoked synaptic responses at Pyr→Pyr connections in deprived cortex are delayed and are less temporally precise.
Table 1.
Passive membrane properties of pairs of synaptically connected L2/3 pyramidal neurons
| Passive membrane property | Control (n = 22 neurons) | Deprived (n = 10 neurons) |
|---|---|---|
| Presynaptic resting membrane potential (mV) | −73 ± 2 | −73 ± 2 |
| Presynaptic input resistance (MΩ) | 64 ± 4 | 60 ± 6 |
| Presynaptic membrane time constant (ms) | 13.9 ± 1.0 | 13.8 ± 1.6 |
| Presynaptic membrane capacitance (pF) | 240 ± 20 | 230 ± 30 |
| Postsynaptic resting membrane potential (mV) | −74 ± 1 | −73 ± 2 |
| Postsynaptic input resistance (MΩ) | 62 ± 3 | 69 ± 10 |
| Postsynaptic membrane time constant (ms) | 14.4 ± 1.0 | 14.3 ± 1.3 |
| Postsynaptic membrane capacitance (pF) | 220 (200–290) | 200 (180–250) |
Deprivation did not affect (one-way ANOVA or ANOVA on ranks) presynaptic resting membrane potential (p = 0.908, F(1,30) = 0.014), presynaptic input resistance (p = 0.573, F(1,30) = 0.325), presynaptic membrane time constant (p = 0.907, F(1,30) = 0.014), presynaptic membrane capacitance (p = 0.878, F(1,30) = 0.024), postsynaptic resting membrane potential (p = 0.545, F(1,30) = 0.375), postsynaptic membrane time constant (p = 0.968, F(1,30) = 0.002), postsynaptic membrane capacitance (p = 0.329, H(1,30) = 0.952), or postsynaptic input resistance (p = 0.380, F(1,30) = 0.795).
Prolonged latency and impaired temporal precision occur in concert with weakening and loss of excitatory connections in deprived cortex
We characterized the functional properties of weakened L2/3 Pyr→Pyr connections in deprived cortex following 4–8 d whisker trimming. The probability of failure was greater (deprived 0.57 [0.57–0.82], n = 10; control 0.05 [0.02–0.15], n = 19; p < 0.001, MWRST; Albieri et al., 2014) and the CV of uEPSP1 amplitude was greater than in control cortex (Fig. 2Ai). The average uEPSP amplitude after EPSP failures were removed, termed the potency, was lower compared with control cortex (Fig. 2Aii). We next explored the distribution of uEPSP responses. To enable comparison, we normalized uEPSP amplitudes to the minimum uEPSP amplitude for each connection, and then pooled the normalized responses. Following 4–8 d whisker trimming, the distribution of normalized uEPSP amplitude was shifted toward one (Fig. 2B). Our data show that L2/3 Pyr→Pyr connections, which have been weakened by 4–8 d of deprivation, fail more frequently and, when they are active, generate smaller uEPSP responses.
Figure 2.

Altered timing of neurotransmission occurs in concert with loss of excitatory connections in deprived cortex. Ai, uEPSP1 coefficient of variation was greater at connections in 4–8 d deprived cortex (deprived 1.43 [1.22–2.41], n = 10; control 0.45 [0.29–0.59], n = 19; p < 0.001, MWRST). Aii, Mean uEPSP1 potency is reduced in deprived cortex (deprived 0.20 [0.15–0.27] mV, n = 10, control 0.41 [0.29–0.62] mV, n = 19; p = 0.002, MWRST). B, Histogram of normalized uEPSP1 amplitude is shifted toward values of 1 in 4–8 d deprived cortex (red) compared with 2–3 d deprived cortex (black; deprived 4–8 d = 1.4, [1.1–1.7], n = 100; deprived 2–3 d = 2.0 [1.5–2.8], n = 435; p < 0.001, MWRST). C, uEPSP1 latency in deprived cortex is longer after 4–8 d deprivation compared with 2–3 d deprivation, 26+ d deprivation, and controls (uEPSP latency: 4–8 d, 1.7 ± 0.2 ms, n = 10; 26+ d, 1.1 ± 0.1 ms, n = 6, p = 0.017; 2–3 d, 1.3 ± 0.1 ms, n = 15, p = 0.022; control, 1.2 ± 0.1 ms, n = 19, p = 0.015; one-way ANOVA). Error bars indicate SEM. D, uEPSP1 amplitude in deprived cortex is reduced following 4–8 d deprivation compared with 2–3 d deprivation, 26+ d deprivation, or controls (uEPSP1 amplitude: 4–8 d, 0.13 ± 0.04 mV, n = 10 connections; 2–3 d, 0.40 ± 0.12 mV, n = 15 connections, p = 0.010; 26+ d, 0.51 ± 0.10 mV, n = 6 connections, p = 0.003; control, 0.49 ± 0.11 mV, n = 19 connections, p < 0.001; one-way ANOVA on log transformed data). Error bars indicate SEM. E, spiking evoked by 500 ms depolarizing current pulses in the presynaptic pyramidal neuron (Ei) and postsynaptic FS interneuron (Eii) of a Pyr→FS connection. Calibrations: 50 ms, 50 mV n. Eiii, Action potentials in the presynaptic pyramidal neuron (triangle) elicit uEPSPs in the postsynaptic FS interneuron (circle). Calibrations: 50 ms, 50 mV, 1 mV. F, The latency of uEPSPs in FS interneurons is similar in control (0.4 ± 0.1 ms, n = 13) and 4–8 d deprived cortex (0.4 ± 0.1 ms n = 8, p = 0.878, t test). G, uEPSP latency of L2/3 Pyr→Pyr connections increases with the duration of deprivation. The expected uEPSP latency from the statistical model for 0–8 d deprivation is shown by the red curve, uEPSP latency = 1.1891 + 0.0107(days of deprivation)2 + 0.1041(uEPSP amp). Cyan, uEPSP latencies after 26 d of deprivation. H, uEPSP amplitude and latency do not covary in control (r = −0.09, p = 0.721) or deprived (r = 0.04, p = 0.853) cortex. I, Brain slice showing position of recording electrodes in L2/3 of spared C barrel column (orange) adjacent to the deprived D barrel column (red). J, uEPSP1 latency in spared cortex is similar to controls whereas latency of deprived connections is longer (control 1.22 ± 0.07 ms, n = 19; 26+ d spared 1.16 ± 0.10 ms, n = 14; 4–8 d deprived 1.66 ± 0.16 ms, n = 10; control vs spared p = 0.640, control vs deprived p = 0.011, one-way ANOVA). K, uEPSP1 jitter in spared cortex is similar to controls whereas jitter of deprived connections is greater (control 0.20 ± 0.02 ms, n = 19; 26+ d spared 0.23 ± 0.03 ms, n = 14; 4–8 d deprived 0.38 ± 0.04 ms, n = 10; control vs spared p = 0.450, control vs deprived p < 0.001, one-way ANOVA).
We have shown previously that weakening and loss of L2/3 Pyr→Pyr connections in deprived cortex starts when whisker deprivation lasts more than 3 d (Albieri et al., 2014). Therefore, we explored whether prolonged synaptic latency was restricted to the period when L2/3 excitatory connections were being weakened and lost rapidly. We pooled our data into groups based on changes in connectivity. Pyr→Pyr connectivity is increasing after 2–3 d deprivation, which contrasts with the rapid loss of connections after 4–8 d deprivation. We found that synaptic latency was prolonged after 4–8 d deprivation compared with either 2–3 d deprivation or controls (Fig. 2C). We next compared the 4–8 d connections with L2/3 Pyr→Pyr connections recorded after 26+ d of deprivation. Local excitatory connectivity is low after 26+ d of deprivation (Cheetham et al., 2007). However, the average uEPSP amplitude of the remaining deprived L2/3 Pyr→Pyr connections is similar to uEPSP amplitude in both control cortex and in 2–3 d deprived cortex and is greater than the uEPSP amplitude of L2/3 Pyr→Pyr connections after 4–8 d deprivation (Fig. 2D). We found that the uEPSP latency was longer for the weaker 4–8 d Pyr→Pyr connections than for the 26+ d deprived, 2–3 d deprived, and control Pyr→Pyr connections (Fig. 2C). We concluded that the prolonged synaptic latency is restricted to the time period during which excitatory connections are being weakened and lost.
We next investigated whether altered timing of neurotransmission was a general feature of all excitatory connections in deprived cortex after 4–8 d deprivation. L2/3 pyramidal neurons form reciprocal connections with local FS interneurons (Fig. 2E), which are not weakened by 4–8 d of deprivation (4–8 d deprived, 1.7 [0.94–2.0] mV, n = 8 connections; control, 1.2 [0.94–1.9] mV, n = 13 Pyr→FS connections; p = 0.587, MWRST; Albieri et al., 2014). Therefore, we examined whether L2/3 Pyr→FS connections also exhibited prolonged latencies. We found that the uEPSP latencies in FS interneurons were similar in deprived and control cortex (Fig. 2F). We concluded that the prolongation of synaptic latency was not a general feature of excitatory connections in deprived cortex after 4–8 d deprivation. Instead, our data indicate that increased synaptic latency is specific to excitatory connections that are being weakened and lost by altered sensory experience.
Prolonged latency at Pyr→Pyr connections increases with the duration of deprivation, but is not tightly coupled to connection strength
We explored whether the prolongation of uEPSP latency was related to the weakening of connection strength or whether it was more closely linked with connection loss. We reasoned that if the prolonged synaptic latency and the weakening of Pyr→Pyr connections were closely related mechanistically, then there would be tight coupling of uEPSP amplitude and latency at each connection. On the other hand, if the prolongation of synaptic latency and the weakening were mediated by distinct mechanisms, then the correlation would be weak.
Connection loss is most pronounced after 4–8 d of deprivation. Therefore, we incorporated the duration of deprivation into our analysis of uEPSP latency and uEPSP amplitude (see Materials and Methods). We found that the uEPSP latency increased progressively over the 0–8 d deprivation period (coefficient = 0.01 ms/d2, p = 0.008; Fig. 2G). In contrast, uEPSP latency did not vary with uEPSP amplitude (coefficient = 0.08 ms/mV, p = 0.461; Fig. 2H). We concluded that the uEPSP latency became more prolonged during the 0–8 d deprivation period. However, increases in uEPSP latency were not tightly coupled to connection weakening in deprived cortex.
Our data had shown no relationship between connection weakening and latency. Therefore, we explored whether the lack of relationship was more general and asked whether uEPSP latency was modified by strengthening of Pyr→Pyr connections. We have shown previously that our whisker-trimming protocol (Fig. 1Aii) causes L2/3 Pyr→Pyr connections in spared cortex to strengthen progressively over weeks (Cheetham et al., 2007). We found that the uEPSP latency and the jitter of L2/3 Pyr→Pyr connections in spared cortex after 26+ d deprivation were both similar to controls (Fig. 2I–K). We concluded that strengthening of local excitatory connections was not accompanied by shortening of the uEPSP latency. Collectively, our data suggest that uEPSP latency at L2/3 Pyr→Pyr connections does not covary with connection strength.
Prolonged synaptic latency and impaired temporal precision are not related to the probability of release or tonic activity of NMDA receptors
We explored whether the altered temporal characteristics of neurotransmission in deprived cortex could be reproduced by modifying synaptic transmission acutely. Our findings showed that the failure rate, the CV of the uEPSP amplitude, and the synaptic latency were all increased at connections in 4–8 d deprived cortex. Both the increase in failure rate and the higher CV of the uEPSP amplitude could be explained, at least in part, by a reduced probability of release. It has been proposed that synaptic latency is inversely related to the probability of release at some excitatory cortical connections (Boudkkazi et al., 2007). Therefore, we investigated whether the increase in synaptic latency at L2/3 Pyr→Pyr connections in 4–8 d deprived cortex could be recapitulated in control cortex by acutely decreasing the probability of release.
We tested the effect of reducing the probability of release by first making baseline recordings of uEPSPs at L2/3 Pyr→Pyr connections in control cortex. We then reduced the calcium concentration in the recording bath so that we had recordings of the same connection before and after reducing the probability of release. Decreasing the extracellular calcium concentration during recording significantly increased probability of failure (Fig. 3A) and reduced uEPSP1 efficacy (2 mm: 0.39 ± 0.09 mV; 1 mm: 0.07 ± 0.02 mV; n = 6; p < 0.001, paired t test). However, EPSP latency (Fig. 3B), EPSP jitter (2 mm: Ca2+ 0.27 ± 0.08 ms; 1 mm: Ca2+ 0.29 ± 0.07 ms; n = 6 connections, p = 0.313, paired t test), and EPSP rise time (Fig. 3C) did not change. We further investigated the relationship between synaptic latency and probability of release by increasing the presynaptic stimulation frequency from 20 to 40 Hz, which reduces the time available for replenishment of the readily releasable pool of synaptic vesicles and, thus, lowers the probability of release during the train (Zucker and Regehr, 2002). Raising the presynaptic firing rate to 40 Hz caused the second uEPSP in the train, EPSP 2, to exhibit a greater failure rate (Fig. 3D) and reduced synaptic efficacy (20 Hz = 0.29 ± 0.05 mV, 40 Hz = 0.25 ± 0.04 mV, n = 9; p = 0.003, paired t test). However, EPSP latency (Fig. 3E) and rise time (Fig. 3F) were not changed. We concluded that the altered timing of neurotransmission at weakened connections in deprived cortex is not related to the reduced probability of release of these connections.
Figure 3.
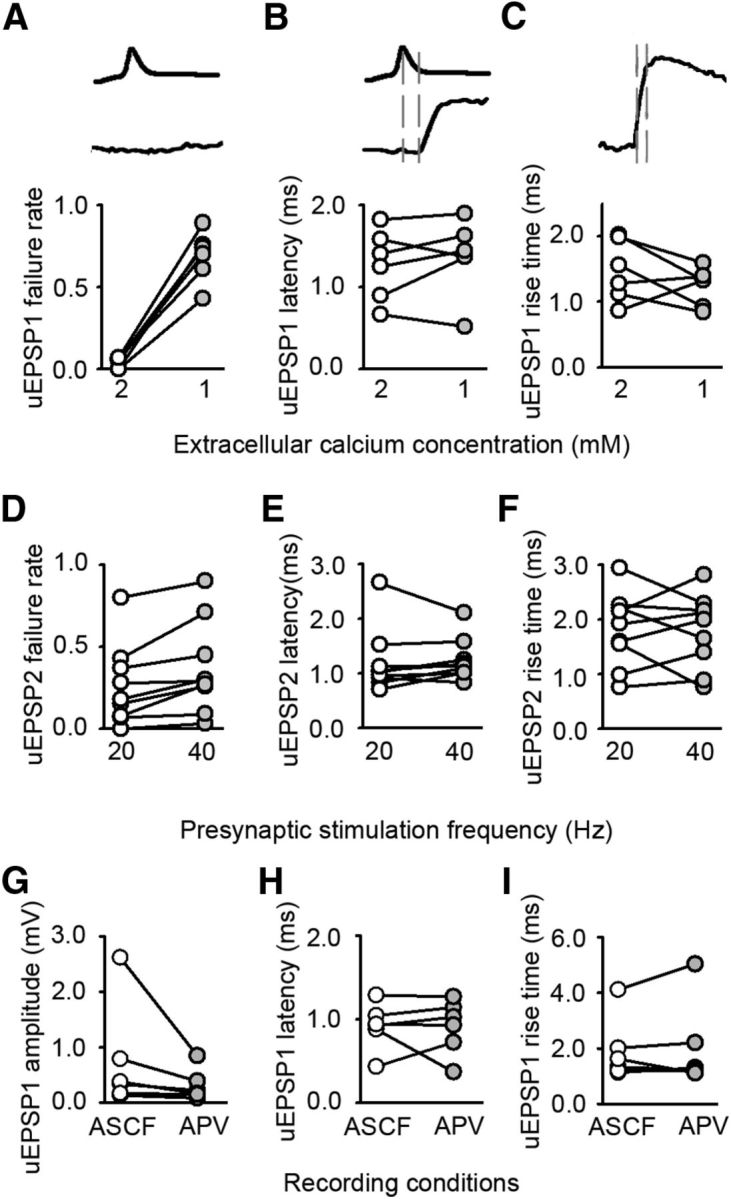
Altered timing of neurotransmission in deprived cortex is independent of release probability and NMDA receptor activation. A–C, Effect of decreasing the extracellular calcium concentration on uEPSP1. A, Failure rate (2 mm 0.04 ± 0.01; 1 mm 0.69 ± 0.06; n = 6, p < 0.001, paired t test). B, Latency (2 mm 1.3 ± 0.2 ms; 1 mm 1.4 ± 0.2 ms; n = 6, p = 0.380, paired t test), C, Rise time (2 mm 1.5 ± 0.2 ms; 1 mm 1.2 ± 0.1 ms; n = 6, p = 0.255, paired t test). D–F, Effect of increasing the frequency of presynaptic firing from 20 to 40 Hz on uEPSP2. D, Failure rate (20 Hz = 0.26 ± 0.08, 40 Hz = 0.37 ± 0.09, p = 0.007, paired t test, n = 9). E, Latency (20 Hz = 1.2 ± 0.2 ms, 40 Hz = 1.2 ± 0.1 ms, p = 0.651, paired t test, n = 9). F, Rise time (20 Hz = 1.8 ± 0.2 ms, 40 Hz = 1.8 ± 0.2 ms, p = 0.861, paired t test, n = 9). G–I, Effect of blocking NMDA receptors on uEPSP1. G, Amplitude (before APV 0.36 [0.16–1.3] mV, after APV 0.19 [0.13–0.51] mV, n = 6 connections; p = 0.031, Wilcoxon signed rank test). H, Latency (before APV 0.92 ± 0.11 ms, after APV 0.91 ± 0.13 ms, n = 6 connections; p = 0.949, paired t test). I, Rise time (before APV 1.90 ± 0.46 ms, after APV 2.02 ± 0.63 ms, n = 6 connections; p = 0.574, paired t test).
Presynaptic NMDA receptors are not usually present at mature cortical synapses, but their expression can be induced by short periods of deprivation in adulthood (Larsen et al., 2014). Therefore, we tested whether blocking NMDA receptors could recapitulate the altered timing of excitatory synaptic transmission. Antagonizing NMDA receptors with 50 μm APV reduced uEPSP1 amplitude at L2/3 Pyr→Pyr connections (Fig. 3G). However, there was no change in either latency (Fig. 3H) or rise time (Fig. 3I) of the synaptic response. Hence, although NMDA receptor blockade acutely decreased uEPSP amplitude, uEPSP latency and rise time were left unchanged.
Impaired temporal precision of putative single-synapse responses
Cortical connections between pairs of neurons typically consist of multiple synapses (Cheetham et al., 2007, 2008). Therefore, we investigated whether the impaired temporal precision that we observed at weakened Pyr→Pyr connections was evident at the level of putative single-synapse responses. Reducing the extracellular calcium concentration to reduce the probability of release resulted in a shift in the response amplitude histograms from multiple peaks in 2 mm calcium (Fig. 4A) to a single peak in low calcium (Fig. 4B). The range of amplitudes of the uEPSPs in low calcium (Fig. 4C) was very similar to the range of mEPSP amplitudes recorded from L2/3 pyramidal neurons (Fig. 4D). We used the lower and upper bounds of the mEPSP amplitudes to set limits for the low-calcium uEPSPs that were studied further to characterize the temporal fidelity of putative single-synapse uEPSP responses (see Materials and Methods; Fig. 4E). We found the latency of putative single-synapse uEPSPs in deprived cortex was longer than putative single-synapse uEPSPs in control cortex (Fig. 4F). Similarly, we found the jitter of the putative single-synapse latency was greater at L2/3 Pyr→Pyr connections in deprived cortex (Fig. 4G,H). However, the rise time of putative single synapses in deprived cortex was not prolonged (Fig. 4I). Collectively, our results suggest that impaired temporal fidelity is evident at a sizeable proportion of synapses that form Pyr→Pyr connections with prolonged latency. Notably, however, the rise time of putative single-synapse responses was preserved. We drew two conclusions from these findings. First, the preservation of putative single-synapse response rise time indicated that slowed kinetics of postsynaptic AMPA receptors did not contribute greatly to the increased rise time of uEPSPs in deprived cortex. Second, the increase in latency, jitter, and rise time of deprived uEPSPs was most likely attributable to the release of neurotransmitter being delayed and less synchronous across the multiple synapses that form each L2/3–L2/3 connection.
Action potential waveform and axonal conduction velocity are not affected by whisker deprivation
We measured synaptic latency as the time between the peak of the presynaptic action potential at the soma and the onset of the postsynaptic uEPSP. Traditionally, synaptic latency has been partitioned into time for action potential propagation along the presynaptic axon to the presynaptic bouton and the synaptic delay, which includes those processes occurring at the synapse leading to the postsynaptic response (Katz and Miledi, 1965; Sabatini and Regehr, 1996). Hence, prolongation of the synaptic latency could be attributable to slower axonal conduction. Additionally, it has been proposed that changes in the shape (half-width and amplitude) of the action potential waveform could affect synaptic latency (Boudkkazi et al., 2011).
To investigate these issues, we first measured the waveform of the presynaptic action potential. Neither the half-width (Fig. 5A) nor the amplitude (Fig. 5B) of action potentials at the soma was modified by whisker trimming. We next estimated the axonal conduction velocity. We measured the time it took for an antidromic action potential evoked by extracellular axonal stimulation to invade the soma with excitatory synaptic antagonists in the recording bath (Fig. 5C,D). The axonal conduction velocity was similar in control and deprived cortex (Fig. 5E). Our findings indicate that the altered timing of excitatory neurotransmission in deprived cortex is not attributable to changes in either action potential waveform or axonal conduction velocity. Instead, the prolonged synaptic latency and the temporal imprecision are more likely to be attributable to mechanisms at the synapse.
Figure 5.
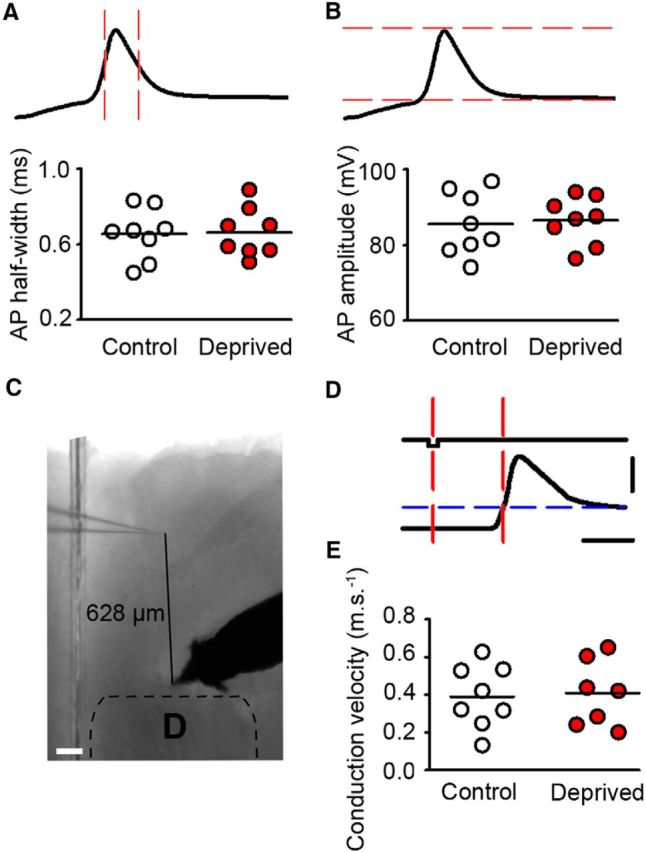
AP waveform and axonal conduction velocity of L2/3 pyramidal neurons are unchanged in deprived cortex. A, AP half-width is the time between the points midway between AP threshold and AP peak (intersection of upstroke and downstroke of the action potential with the red, vertical dashed lines). Cluster plot of average action potential half-widths for neurons in control (white) and deprived cortex (red; deprived 0.7 ± 0.1 ms, n = 8 neurons, control 0.7 ± 0.1 ms, p = 0.917, t test, n = 8 neurons). Horizontal lines denote grand mean. B, Image depicts measurement of AP amplitude (between red dashed lines). Cluster plot of average AP amplitude for neurons in control (white) and deprived cortex (red; deprived 86 ± 2 mV, n = 8, control 85 ± 3 mV, p = 0.786, t test, n = 8). Horizontal lines denote grand mean. C, Antidromic stimulation setup: 5× bright-field image of brain slice illustrating position of recording and stimulating electrode in L2/3 of D barrel column. Scale bar, 100 μm. D, Antidromic stimulation paradigm. Top trace, 0.2 ms negative current injection. Bottom trace, A whole-cell recording of the antidromic AP invading the soma. AP threshold is denoted by the blue dashed line. Axonal conduction velocity was measured between the current injection midpoint and the AP threshold (red dashed lines). E, Cluster plot of axonal conduction velocities in control (open circles) and deprived cortex (red circles; control 0.39 ± 0.05 ms, n = 8 neurons, deprived 0.40 ± 0.06 ms, p = 0.870, t test, n = 7 neurons).
Location of synaptic vesicles
Our data found no relationship between postsynaptic AMPA or NMDA receptors with the impaired temporal precision of neurotransmission. Therefore, we reasoned that the locus of the altered timing of neurotransmission may be presynaptic. As a result, we considered whether there was a structural analog of prolonged synaptic latency in the presynaptic bouton. We focused on small synapses because they are more likely to be lost (Holtmaat and Svoboda, 2009). Prolonged latency may occur at small synapses because the synaptic vesicles are further away from the presynaptic active zone, where vesicle exocytosis occurs. To explore this possibility, we examined the distance of synaptic vesicles from the active zone. We studied asymmetric, presumed excitatory synapses on dendritic spines with FIBSEM (Fig. 6A). The presynaptic boutons of 25 synapses containing a total of 2729 vesicles were reconstructed in three dimensions and the distance of each synaptic vesicle to the presynaptic active zone was measured (see Materials and Methods; Fig. 6B). The total number of vesicles at a synapse scaled with the area of the active zone (Fig. 6C). However, the median distance of vesicles from the active zone was not greater at smaller synapses (Fig. 6D).
Figure 6.

Spatial distribution of synaptic vesicles is not modified by synapse size. A, EM section through an asymmetric synapse onto a dendritic spine head. Calibration: 200 nm. B, Computer 3D reconstruction of the synapse shown in A. Synaptic vesicles are represented as spheres and the active zone is imaged as a mesh. The nearest distance of the center of each vesicle (sphere) to the active zone (mesh) is measured using an add-on written in Blender software. C, The number of synaptic vesicles is linearly correlated with the area of the active zone (R2 = 0.56, p < 0.001). D, The median distance of vesicles from the active zone does not change with size of the active zone (R2 = 0.01, p < 0.001). E, Schematic of criteria for docked vesicles. Left, Vesicle (average diameter, 35 nm) apposed to the active zone. Right, Vesicle is 10 nm from the active zone. F, The number of docked vesicles is linearly related to the area of the active zone (R2 = 0.63, p < 0.001). G, Median distance of docked vesicles from the active zone is not correlated with active zone size (R2 = 0.01, p < 0.001).
It is thought that the majority of neurotransmission is performed by docked vesicles that lie close to the active zone (Verhage and Sorensen, 2008). We reasoned that a structural analog of the increase in uEPSP latency and jitter may be present only in the docked vesicles. Here, we used the term docked vesicles to refer to vesicles that are within 10 nm of the presynaptic membrane (Fig. 6E; Verhage and Sorensen, 2008; Xiao et al., 2013). This approximates to the distance over which SNARE complexes are functional (Li et al., 2007). We found that the number of docked vesicles scaled with the area of the active zone (Fig. 6F). However, the median distance of docked synaptic vesicles from the active zone was not greater at small synapses. We concluded that the spatial distribution of synaptic vesicles was not affected by synapse size. Further investigation of the structural analogs of prolonged latency at deprived synapses requires that synapses with prolonged latency are labeled so that they can be distinguished from transient synapses or other small synapses with normal neurotransmission.
Increased synaptic latency and impaired temporal precision modify spiking output
Our data show prolonged synaptic latencies and impaired temporal precision at weakened Pyr→Pyr connections. We explored how changes in the temporal parameters of excitatory neurotransmission would affect the spiking output of an L2/3 pyramidal neuron with simple integrate-and-fire neurons combined with our experimental data (see Materials and Methods). The synaptic inputs to the integrate-and-fire neurons were arranged to generate a reduced and highly idealized model of the summation of EPSPs and action potential firing in a single L2/3 pyramidal neuron that occurs in vivo (Crochet et al., 2011). The model consisted of an input layer of neurons, which fired simultaneously, and synapsed onto a control cortex neuron and a deprived cortex neuron (Fig. 7A). Hence, the control cortex neuron and deprived cortex neuron both receive the same presynaptic action potential input and have the same number of synapses with the same synaptic weight projecting onto them. Gaussian noise was added to the resting membrane potential of both postsynaptic neurons to simulate background membrane potential fluctuations. Axonal conduction velocity was assumed to be constant, as we had found experimentally. We then varied the temporal properties of synaptic neurotransmission onto the deprived neuron using experimentally obtained measures to investigate how these temporal parameters affected postsynaptic firing independently of changes in synaptic weight.
Figure 7.

Low temporal fidelity of synaptic inputs reduces spiking output. A, Diagram of model showing the input layer projecting onto two postsynaptic neurons via high-fidelity connections (black) or low-fidelity connections (red). B, Example AP generated in the input layer and resultant EPSPs in the postsynaptic neurons connected by either high-fidelity (black) or low-fidelity (red) connections. Calibrations: 20 mV, 5 mV, 5 ms. C, Example traces from simulations showing AP firing and EPSPs. The frequency of action potentials is reduced in neurons receiving low-fidelity inputs (red). Calibrations: 20 mV, 300 ms. D, Summary of repeated simulations shows reduced spiking output at low-fidelity “deprived” connections (red) compared with “control” high-fidelity connections (low-fidelity deprived: 0.87 ± 0.06 Hz, n = 96 trials; high-fidelity control: 1.26 ± 0.06 Hz, n = 96 trials; p < 0.001, t test). E, Reduced temporal summation at low-fidelity inputs (red) compared with high-fidelity inputs (black). Dashed line indicates firing threshold for APs. Calibrations: 10 mV, 20 ms. F, EPSPs evoked by low-fidelity inputs recapitulate the greater rise time of deprived connections (deprived: 2.2 ± 0.10 ms, control 1.4 ± 0.07 ms, n = 20 connections; p < 0.001, t test). G, Increased EPSP jitter reduces firing rate at connections with an average latency of either 1.5 ms (red) or 2.0 ms (green) or 2.5 ms (blue; variation in firing rate: jitter, F(3,84) = 6.07, p < 0.001; latency, F(2,84) = 1.56, p = 0.216, n = 14 trials, two-way ANOVA). Error bars indicate SEM. H, Effect of increasing the fraction of low-fidelity inputs on spiking frequency of the postsynaptic neuron (quadratic fit: R2 = 0.99 p < 0.001). I, Effect of proportion of low-fidelity inputs on the latency of the output spike. J, Firing rates for low-fidelity neurons are decreased compared with high-fidelity neurons when the simulated total synaptic input takes the neuron to threshold or exceeds threshold by 2.5 mV (p = 0.002, n = 10 trials, t test) or by 5.0 mV (p = 0.001, n = 10 trials, t test), but are similar when the total synaptic input is 7.5 mV above threshold (p = 1.000, n = 10 trials, t test). K, Rise times of simulated suprathreshold EPSPs (total synaptic input, 5 mV above threshold) are greater in simulated deprived neurons than in simulated control neurons (rise time: control 1.84 ± 0.04 ms, deprived 2.40 ± 0.07 ms, p < 0.001, n = 149 EPSPs, t test). Inset, Percentage of EPSPs that induce firing is reduced in deprived cortex (suprathreshold responses: control, 141/300 (47%); deprived, 111/300 (37%); p = 0.016, z-test).
In the first set of simulations, all input layer neurons were connected to the control neuron by synapses with high temporal fidelity (1 ms latency, 0.1 ms jitter). In contrast, all connections from input neurons to the deprived neuron had low temporal fidelity modeled as longer synaptic latency and increased jitter to reproduce our experimental findings (see Materials and Methods; Fig. 7B). Prolonging the mean synaptic latency of all inputs simply delays the synaptic response. However, reducing the temporal precision of synaptic inputs by increasing their jitter reduced firing of the deprived neuron compared with the control neuron (Fig. 7C,D; low-fidelity deprived: 0.87 ± 0.06 Hz, n = 96 trials; high-fidelity control: 1.26 ± 0.06 Hz, n = 96 trials; p < 0.001, t test). This occurred because the increased jitter on the inputs led to reduced temporal summation of the responses evoked by individual inputs in the postsynaptic neuron. The outcome was that the EPSP in the deprived neuron had a longer rise time and lower amplitude (Fig. 7E,F). Furthermore, the decrease in spiking output became larger as the jitter of the low-fidelity inputs was increased (Fig. 7G; 0.1 ms jitter = 1.18 ± 0.13 Hz, 0.6 ms jitter = 0.52 ± 0.07 Hz, p < 0.001, n = 14 trials, t test). Our simulations suggested that increasing the jitter of synaptic responses diminished the temporal summation of those responses and resulted in reduced neural firing in the postsynaptic neuron.
The first simulations looked at an extreme situation where all synaptic latencies were prolonged. This enabled us to study the effects of increasing the jitter on firing output. In the second set of simulations, we varied the fraction of low-fidelity inputs to the deprived neuron. Hence, the deprived neuron received some inputs with a short latency and other inputs with prolonged latency. This mimics more closely the situation in vivo. Replacing the high-fidelity inputs onto a control neuron with low-fidelity inputs resulted in a rapid decrease in the number of action potentials fired by the postsynaptic neuron (Fig. 7H). This decrease in neural firing stabilizes as the proportion of low-fidelity inputs increases >50% (Fig. 7H). The effect on the time between firing in the presynaptic neurons and firing in the postsynaptic neuron was different. The delay in firing of the postsynaptic neuron increased most rapidly when a small proportion of high-fidelity inputs was replaced by low-fidelity inputs and then increased more slowly as the proportion of low-fidelity inputs was increased >20% (Fig. 7I). Hence, the predictions of the model vary with the proportion of low-fidelity inputs that the neuron is already receiving.
Our previous simulations have used parameters such that the summation of all inputs just brings the neuron to the firing threshold. We explored the effects of increasing the total synaptic drive above the firing threshold. The total synaptic drive is expressed as the sum of all EPSPs if they were to occur simultaneously. Effectively, the total synaptic drive is the maximum EPSP response that could occur during a simulation. EPSP amplitudes will be lower during trials because each trial incorporates temporal dispersion. We found that the decrease in firing caused by low-fidelity inputs persisted when the total synaptic input was a few millivolts (2.5 and 5 mV) above threshold, but it was lost when the summed synaptic input was 7.5 mV above threshold (Fig. 7J). We investigated the effect of increasing the temporal dispersion of the simulated EPSP by calculating its rise time. The 5 mV suprathreshold inputs generated simulated EPSPs with a slower rise time (Fig. 7K), which would account for the decrease in firing. We concluded that decreases in firing would only occur in neurons where spiking is driven by EPSPs that exceed threshold minimally. Notably, any reduction in firing caused by the low-fidelity inputs would not be apparent with strong, suprathreshold inputs. Recordings in awake behaving mice show that spiking of L2/3 pyramidal neurons is sparse (∼10% of the population), and when neurons do spike they commonly fire only one action potential near the peak of the EPSP (Crochet et al., 2011). Hence, local excitatory inputs to L2/3 neurons are probably rarely strong enough to overcome the reduced spiking that arises from low-fidelity inputs.
Validating model predictions
The model makes predictions about how spiking changes when the proportion of low-fidelity inputs are altered. Although the model was intended to reflect neural activity in vivo, it can be extended to brain slice preparations. L2/3 pyramidal neurons receive a high frequency of spontaneous EPSPs (sEPSPs) in vitro. The sEPSPs comprise mEPSPs and action potential-evoked EPSPs. A simple prediction is that low-fidelity synaptic inputs in deprived cortex will result in action potential-evoked sEPSPs having prolonged rise times. We divided sEPSPs into two groups based on their amplitudes: small (0.08–0.28 mV), which correspond to mEPSPs (Fig. 4D), and large (0.29–0.90 mV), which represent multisynaptic EPSPs evoked by action potentials. We found that large sEPSPs had longer rise times (Fig. 8A,B). In contrast, the rise time of small sEPSPs were similar in deprived and control cortex (Fig. 8B). These findings indicated that a sizeable proportion of action potential-evoked sEPSPs in deprived cortex comprised low-fidelity inputs.
The model predicted that neurons in deprived cortex with a sizeable proportion of low-fidelity input would exhibit reduced neural firing and increased spike latency. Therefore, we depolarized L2/3 pyramidal neurons to near threshold by injecting somatic current. Under these conditions, collisions between sEPSPs summate and cause the neurons to fire action potentials at ∼ 0.2 Hz in vitro (Fig. 8C; see Materials and Methods). We assessed the accuracy of the model's predictions by comparing the predicted changes in spike latency and firing rate from the model with the measured values in vitro. To generate predications from the model we first estimated the proportion of low-fidelity inputs received by L2/3 pyramidal neurons in deprived cortex. We estimated this proportion as 22% from the putative single-synapse responses recorded after 4–8 d of deprivation (Fig. 8D; see Materials and Methods). We then entered this estimate of the proportion of low-fidelity inputs into the model to calculate predicted effect sizes for changes in spike latency and firing frequency. The model predicted that the spike latency would increase by 7.1 ms and the mean firing frequency would decrease by 27%. Our in vitro data sat within the 95% confidence interval of the model's predictions for firing frequency and for spike latency (Fig. 8E,F). Hence, we concluded that the model's predictions were reasonable under our in vitro experimental conditions.
Discussion
We investigated how the timing of synaptic responses was altered in neocortex undergoing rapid loss of local excitatory connections. Our major finding is that the latency of the postsynaptic potential is prolonged and temporally imprecise at Pyr→Pyr connections that are being weakened and lost. Modeling studies revealed that increasing the latency and jitter of a subset of excitatory inputs to a postsynaptic neuron reduced the number of spikes fired by that neuron. Our results indicate that prolonging the synaptic latency minimizes the functional contribution of a connection to a neural circuit without requiring the complete destruction of the connection. We propose that the combination of prolonged synaptic latency and diminished temporal precision of neurotransmission constitute a temporal signature of impending loss of an excitatory cortical connection.
Prolonged latency and impaired temporal precision of neurotransmission accompanies connection disassembly
Altering tactile experience by trimming a subset of whiskers induced a biphasic change in local excitatory connectivity in deprived primary somatosensory cortex (Cheetham et al., 2007; Albieri et al., 2014). Pyr→Pyr connections initially tripled in number over a few days and then underwent a sustained loss. This process mirrors the reorganization of the whisker cortical map (Albieri et al., 2014). Prolonged latency and temporal imprecision of excitatory neurotransmission was only found during the period when the experience-dependent weakening and loss of local excitatory connections were progressing rapidly. The latency of putative single-synapse responses was prolonged and more variable over the same period. Hence, our results suggest that the majority of synapses forming a connection exhibit altered timing. This finding explains the increase in latency, jitter, and rise time of multisynaptic uEPSPs.
The initial response to deprivation involved a marked increase in local excitatory connectivity in deprived cortex after 2–3 d. Despite the prevalence of newly formed connections, the temporal properties of excitatory synaptic transmission were unchanged. This suggests that the synaptic responses at newly formed connections rapidly adopt the timing characteristics of established connections. Similarly, the latency and jitter of excitatory synaptic transmission did not differ from controls after prolonged deprivation. We concluded that the timing of neurotransmission at Pyr→Pyr connections is regulated, but the normal regulatory regime is interrupted in mature neocortex when entire connections are being weakened before their loss. Henceforth, we refer to this process of weakening before loss as connection disassembly.
Altered timing of neurotransmission at Pyr→Pyr connections undergoing connection disassembly is not part of a pervasive change in the timing of excitatory neurotransmission in the neocortex since the latency of Pyr→FS interneurons in L2/3 of deprived somatosensory cortex remained normal. We concluded that altered timing not only occurs over a short time window, but also affects specific local excitatory circuits.
Possible mechanisms for prolonged latency and impaired temporal precision of neurotransmission
The synaptic latency is traditionally subdivided into two components: the axonal conduction delay, which is the time taken for the action potential to travel from the action potential initiation site to the presynaptic bouton, and the synaptic delay defined as the time from depolarization of the presynaptic bouton to the onset of the postsynaptic response (Katz and Miledi, 1965). Abnormalities in either step could result in altered timing of synaptic transmission. We found no changes in the conduction velocity or action potential waveform at the soma. We cannot exclude the possibility that the action potential waveform is modified at the presynaptic bouton. Accordingly, this presynaptic mechanism could contribute to the altered timing of neurotransmission, but probably not by affecting calcium entry to the presynaptic terminal (see below).
The synaptic delay is thought to be mainly required for presynaptic processes mediating neurotransmitter release (Katz and Miledi, 1965; Sabatini and Regehr, 1999). Traditionally, the synaptic delay has been considered an invariant property of neurotransmission (Katz and Miledi, 1965; Barrett and Stevens, 1972). However, the latency of uEPSPs has been reported to vary at some central connections (Cope and Mendell, 1982; Waldeck et al., 2000; Boudkkazi et al., 2007) and direct measurements of the synaptic delay at large synapses show that it is mutable during development (Fedchyshyn and Wang, 2007). In mature neocortex, however, we found that the latency of uEPSPs did not change when the probability of release was decreased or with repetitive stimulation. The discrepancy between our findings and those where the uEPSP latency covaries with the probability of release may be attributable to the age of the tissue studied, particularly as neurotransmission at neocortical connections alters progressively during development (Feldmeyer and Radnikow, 2009); recording from a different cortical layer (L5 rather than L2/3); or different cortical area studied (Cheetham and Fox, 2010).
Our findings indicate that calcium entry to the presynaptic bouton does not play a key role in determining the latency of synaptic responses evoked by action potentials at L2/3 Pyr→Pyr connections in mature neocortex. It may be the case that the mechanism underpinning vesicle release is perturbed downstream of calcium entry (Eggermann et al., 2012). However, addressing this issue would require development of new high-resolution technologies to dissect the dynamics of presynaptic function at small synapses in mature neocortex.
Prolonged latency and increased temporal imprecision occur in concert with connection loss
Weakened excitatory connections in deprived cortex had a prolonged latency and increased jitter. Surprisingly, however, we found that uEPSP latency was not tightly coupled to connection strength. Instead, uEPSP latency increased over the 4–8 d deprivation period as did connection loss. These data suggest the altered timing of excitatory neurotransmission in deprived cortex was more tightly linked to connection loss than changes in connection strength. How might this work? We have shown previously that synapse size and uEPSP amplitude covary over days (Cheetham et al., 2014). Hence, a connection's synapses become smaller as the connection is weakened. One possibility is that once synapse size drops below a threshold then connection disassembly is triggered. In this scenario, connection disassembly is an extra process distinct from weakening of connection strength. We propose that the prolonged uEPSP latency signifies that connection disassembly has been activated and the connection is likely to be lost.
Impact of prolonged latency and impaired temporal precision of neurotransmission on spiking of pyramidal neurons
Whisker deprivation led to both weakening of L2/3 Pyr→Pyr connections and altered timing of neurotransmission. Our model enabled us to study how spiking output was affected by altered timing of synaptic transmission in isolation from weakening of connection strength. Increasing the proportion of low-fidelity synaptic inputs without changing synaptic strength reduced the temporal summation of postsynaptic potentials and was sufficient to decrease spiking of the postsynaptic neuron. In contrast, the timing of the remaining spikes changed markedly only when there were few low-fidelity inputs. Hence, both the weakening and increase in latency have the same effect: they reduce the contribution of an excitatory connection to the spiking of the postsynaptic neuron.
In vivo, excitatory synaptic activity is followed rapidly (within milliseconds) by inhibition, which narrows the time window during which a postsynaptic spike can occur (Pouille and Scanziani, 2001; Wehr and Zador, 2003). If a neuron receives a combination of short-latency and long-latency excitatory inputs, then fast GABAergic inputs will have a greater inhibitory effect on the longer latency inputs. Accordingly, weakened excitatory inputs in deprived cortex will be inhibited more because they have a longer latency. Hence, the combination of prolonged synaptic latency and inhibition will reduce the contribution of a weakened excitatory connection to postsynaptic spiking even further.
Prolonged latency and impaired temporal precision of neurotransmission and reorganization of cortical microcircuits
Loss of tactile inputs changes the number of sensory-evoked action potentials (Diamond et al., 1994) and the distribution of sensory-evoked neural firing in L2/3 of somatosensory cortex (Margolis et al., 2012). The altered neural firing is accompanied by extensive rewiring of local excitatory circuits (Albieri et al., 2014). We have proposed that the rewiring enables new pyramidal neurons to be recruited into a microcircuit and other neurons to be excluded (Albieri et al., 2014). However, rewiring potentially poses difficulties for sensory processing based on sparse firing. A key question is how does rewiring promote adaptation while simultaneously minimizing disruption to spiking output?
Our findings suggest a strategy for how excitatory connections are lost from a microcircuit. Weakening a Pyr→Pyr connection and prolonging its response latency minimizes the contribution of that connection to the spiking output of the postsynaptic neuron. Effectively, the connection is functionally excluded from the microcircuit, but without physical destruction of the connection. The connection disassembly process proceeds over days, offering a time window when connection disassembly can be reversed, if spiking output is affected detrimentally or if sensory input is restored. Hence, prolonging the response latency is not only an efficient strategy, it also incorporates fault tolerance.
Footnotes
This work is supported by the Medical Research Council (S.J.B., G.A., S.H.B.), the King's College Medical Research Trust (C.E..C.), and a Wellcome Trust Senior Clinical Fellowship (G.T.F.).
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Ahissar E, Sosnik R, Haidarliu S. Transformation from temporal to rate coding in a somatosensory thalamocortical pathway. Nature. 2000;406:302–306. doi: 10.1038/35018568. [DOI] [PubMed] [Google Scholar]
- Albieri G, Barnes SJ, de Celis Alonso B, Cheetham CE, Edwards CE, Lowe AS, Karunaratne H, Dear JP, Lee KC, Finnerty GT. Rapid bidirectional reorganization of cortical microcircuits. Cereb Cortex. 2014 doi: 10.1093/cercor/bhu098. doi: 10.1093/cercor/bhu098. Advance online publication. Retrieved May 16, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabzadeh E, Panzeri S, Diamond ME. Deciphering the spike train of a sensory neuron: counts and temporal patterns in the rat whisker pathway. J Neurosci. 2006;26:9216–9226. doi: 10.1523/JNEUROSCI.1491-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SJ, Finnerty GT. Sensory experience and cortical rewiring. Neuroscientist. 2010;16:186–198. doi: 10.1177/1073858409343961. [DOI] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Poulet JF. Experimental evidence for sparse firing in the neocortex. Trends Neurosci. 2012;35:345–355. doi: 10.1016/j.tins.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Benedetti BL, Glazewski S, Barth AL. Reliable and precise neuronal firing during sensory plasticity in superficial layers of primary somatosensory cortex. J Neurosci. 2009;29:11817–11827. doi: 10.1523/JNEUROSCI.3431-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudkkazi S, Carlier E, Ankri N, Caillard O, Giraud P, Fronzaroli-Molinieres L, Debanne D. Release-dependent variations in synaptic latency: a putative code for short- and long-term synaptic dynamics. Neuron. 2007;56:1048–1060. doi: 10.1016/j.neuron.2007.10.037. [DOI] [PubMed] [Google Scholar]
- Boudkkazi S, Fronzaroli-Molinieres L, Debanne D. Presynaptic action potential waveform determines cortical synaptic latency. J Physiol. 2011;589:1117–1131. doi: 10.1113/jphysiol.2010.199653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht M, Roth A, Sakmann B. Dynamic receptive fields of reconstructed pyramidal cells in layers 3 and 2 of rat somatosensory barrel cortex. J Physiol. 2003;553:243–265. doi: 10.1113/jphysiol.2003.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno RM, Sakmann B. Cortex is driven by weak but synchronously active thalamocortical synapses. Science. 2006;312:1622–1627. doi: 10.1126/science.1124593. [DOI] [PubMed] [Google Scholar]
- Cardona A, Saalfeld S, Schindelin J, Arganda-Carreras I, Preibisch S, Longair M, Tomancak P, Hartenstein V, Douglas RJ. TrakEM2 software for neural circuit reconstruction. PLoS One. 2012;7:e38011. doi: 10.1371/journal.pone.0038011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham CE, Fox K. Presynaptic development at L4 to l2/3 excitatory synapses follows different time courses in visual and somatosensory cortex. J Neurosci. 2010;30:12566–12571. doi: 10.1523/JNEUROSCI.2544-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham CE, Hammond MS, Edwards CE, Finnerty GT. Sensory experience alters cortical connectivity and synaptic function site specifically. J Neurosci. 2007;27:3456–3465. doi: 10.1523/JNEUROSCI.5143-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham CE, Hammond MS, McFarlane R, Finnerty GT. Altered sensory experience induces targeted rewiring of local excitatory connections in mature neocortex. J Neurosci. 2008;28:9249–9260. doi: 10.1523/JNEUROSCI.2974-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham CE, Barnes SJ, Albieri G, Knott GW, Finnerty GT. Pansynaptic enlargement at adult cortical connections strengthened by experience. Cereb Cortex. 2014;24:521–531. doi: 10.1093/cercor/bhs334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope TC, Mendell LM. Parallel fluctuations of EPSP amplitude and rise time with latency at single Ia-fiber-motoneuron connections in the cat. J Neurophysiol. 1982;47:455–468. doi: 10.1152/jn.1982.47.3.455. [DOI] [PubMed] [Google Scholar]
- Crochet S, Poulet JF, Kremer Y, Petersen CC. Synaptic mechanisms underlying sparse coding of active touch. Neuron. 2011;69:1160–1175. doi: 10.1016/j.neuron.2011.02.022. [DOI] [PubMed] [Google Scholar]
- de Kock CP, Bruno RM, Spors H, Sakmann B. Layer- and cell-type-specific suprathreshold stimulus representation in rat primary somatosensory cortex. J Physiol. 2007;581:139–154. doi: 10.1113/jphysiol.2006.124321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond ME, Huang W, Ebner FF. Laminar comparison of somatosensory cortical plasticity. Science. 1994;265:1885–1888. doi: 10.1126/science.8091215. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca(2)(+) channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Activity-dependent changes in temporal components of neurotransmission at the juvenile mouse calyx of Held synapse. J Physiol. 2007;581:581–602. doi: 10.1113/jphysiol.2007.129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Radnikow G. Developmental alterations in the functional properties of excitatory neocortical synapses. J Physiol. 2009;587:1889–1896. doi: 10.1113/jphysiol.2009.169458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO. Organization of behavior: a neuropsychological theory. New York: Wiley; 1949. [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Jadhav SP, Wolfe J, Feldman DE. Sparse temporal coding of elementary tactile features during active whisker sensation. Nat Neurosci. 2009;12:792–800. doi: 10.1038/nn.2328. [DOI] [PubMed] [Google Scholar]
- Jenkins WM, Merzenich MM, Ochs MT, Allard T, Guíc-Robles E. Functional reorganization of primary somatosensory cortex in adult owl monkeys after behaviorally controlled tactile stimulation. J Neurophysiol. 1990;63:82–104. doi: 10.1152/jn.1990.63.1.82. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The measurement of synaptic delay, and the time course of acetylcholine release at the neuromuscular junction. Proc R Soc Lond B Biol Sci. 1965;161:483–495. doi: 10.1098/rspb.1965.0016. [DOI] [PubMed] [Google Scholar]
- Kerr JN, de Kock CP, Greenberg DS, Bruno RM, Sakmann B, Helmchen F. Spatial organization of neuronal population responses in layer 2/3 of rat barrel cortex. J Neurosci. 2007;27:13316–13328. doi: 10.1523/JNEUROSCI.2210-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G, Rosset S, Cantoni M. Focussed ion beam milling and scanning electron microscopy of brain tissue. J Vis Exp. 2011;53:e2588. doi: 10.3791/2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen RS, Smith IT, Miriyala J, Han JE, Corlew RJ, Smith SL, Philpot BD. Synapse-specific control of experience-dependent plasticity by presynaptic NMDA receptors. Neuron. 2014;83:879–893. doi: 10.1016/j.neuron.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort S, Tomm C, Floyd Sarria JC, Petersen CC. The excitatory neuronal network of the C2 barrel column in mouse primary somatosensory cortex. Neuron. 2009;61:301–316. doi: 10.1016/j.neuron.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- London M, Roth A, Beeren L, Hãusser M, Latham PE. Sensitivity to perturbations in vivo implies high noise and suggests rate coding in cortex. Nature. 2010;466:123–127. doi: 10.1038/nature09086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Lütcke H, Schulz K, Haiss F, Weber B, Kügler S, Hasan MT, Helmchen F. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat Neurosci. 2012;15:1539–1546. doi: 10.1038/nn.3240. [DOI] [PubMed] [Google Scholar]
- Margolis DJ, Lütcke H, Helmchen F. Microcircuit dynamics of map plasticity in barrel cortex. Curr Opin Neurobiol. 2014;24:76–81. doi: 10.1016/j.conb.2013.08.019. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome WT, Britten KH, Movshon JA. Neuronal correlates of a perceptual decision. Nature. 1989;341:52–54. doi: 10.1038/341052a0. [DOI] [PubMed] [Google Scholar]
- Olshausen BA, Field DJ. Sparse coding of sensory inputs. Curr Opin Neurobiol. 2004;14:481–487. doi: 10.1016/j.conb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Panzeri S, Petersen RS, Schultz SR, Lebedev M, Diamond ME. The role of spike timing in the coding of stimulus location in rat somatosensory cortex. Neuron. 2001;29:769–777. doi: 10.1016/S0896-6273(01)00251-3. [DOI] [PubMed] [Google Scholar]
- Pinto DJ, Brumberg JC, Simons DJ. Circuit dynamics and coding strategies in rodent somatosensory cortex. J Neurophysiol. 2000;83:1158–1166. doi: 10.1152/jn.2000.83.3.1158. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Recanzone GH, Merzenich MM, Jenkins WM, Grajski KA, Dinse HR. Topographic reorganization of the hand representation in cortical area 3b owl monkeys trained in a frequency-discrimination task. J Neurophysiol. 1992a;67:1031–1056. doi: 10.1152/jn.1992.67.5.1031. [DOI] [PubMed] [Google Scholar]
- Recanzone GH, Merzenich MM, Schreiner CE. Changes in the distributed temporal response properties of SI cortical neurons reflect improvements in performance on a temporally based tactile discrimination task. J Neurophysiol. 1992b;67:1071–1091. doi: 10.1152/jn.1992.67.5.1071. [DOI] [PubMed] [Google Scholar]
- Rieke F, Warland D, de Ruyter van Steveninck R, Bialek W. Spikes: exploring the neural code. Ed 1. Cambridge, MA: The MIT; 1997. [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of synaptic transmission. Annu Rev Physiol. 1999;61:521–542. doi: 10.1146/annurev.physiol.61.1.521. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/S0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Stüttgen MC, Schwarz C. Psychophysical and neurometric detection performance under stimulus uncertainty. Nat Neurosci. 2008;11:1091–1099. doi: 10.1038/nn.2162. [DOI] [PubMed] [Google Scholar]
- Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Vinje WE, Gallant JL. Sparse coding and decorrelation in primary visual cortex during natural vision. Science. 2000;287:1273–1276. doi: 10.1126/science.287.5456.1273. [DOI] [PubMed] [Google Scholar]
- von Heimendahl M, Itskov PM, Arabzadeh E, Diamond ME. Neuronal activity in rat barrel cortex underlying texture discrimination. PLoS Biol. 2007;5:e305. doi: 10.1371/journal.pbio.0050305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Wolfe J, Houweling AR, Brecht M. Sparse and powerful cortical spikes. Curr Opin Neurobiol. 2010;20:306–312. doi: 10.1016/j.conb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Xiao L, Michalski N, Kronander E, Gjoni E, Genoud C, Knott G, Schneggenburger R. BMP signaling specifies the development of a large and fast CNS synapse. Nat Neurosci. 2013;16:856–864. doi: 10.1038/nn.3414. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]