

Abstract
To investigate the role of secreted protein acidic and rich in cysteine (SPARC) in age-related cardiac inflammation, we studied six groups of mice: young (3–5 mo old), middle-aged (10–12 mo old), and old (18–29 mo old) C57BL/6 wild-type (WT) and SPARC-null (Null) mice (n = 7–10/group). Cardiac function and structure were determined by echocardiography. The left ventricle was used for cytokine gene array and macrophage quantification by immunohistochemistry. Macrophage infiltration increased with age in WT (n = 5–6/group, P < 0.05 for young vs. old), but not in Null. Proinflammatory markers (Ccl5, Cx3cl1, Ccr2, and Cxcr3) increased in middle-aged and old WT, whereas they were increased only in old Null compared with respective young (n = 5–6/group, P < 0.05 for all). These results suggest that SPARC deletion delayed age-related cardiac inflammation. To further assess how SPARC affects inflammation, we stimulated peritoneal macrophages with SPARC (n = 4). SPARC treatment increased expression of proinflammatory macrophage M1 markers and decreased anti-inflammatory M2 markers. Echocardiography (n = 7–10/group) revealed an age-related increase in wall thickness of the left ventricle in WT (0.76 ± 0.02 mm in young vs. 0.91 ± 0.03 mm in old; P < 0.05) but not in Null (0.78 ± 0.01 mm in young vs. 0.84 ± 0.02 mm in old). In conclusion, SPARC deletion delayed age-related increases in macrophage infiltration and proinflammatory cytokine expression in vivo and in vitro. SPARC acts as an important mediator of age-related cardiac inflammation by increasing the expression of macrophage M1 markers and decreasing M2 markers.
Keywords: secreted protein acidic and rich in cysteine, aging, heart, inflammation, macrophage
cardiovascular disease is a leading cause of death in the elderly (10). While advanced age increases the chance of exposure to cardiovascular risk factors, accumulating studies show that aging itself is an independent risk factor for cardiovascular disease (18, 29). With advancing age, the left ventricle (LV) develops concentric remodeling and diastolic dysfunction, which is associated with myocyte loss and an increase in extracellular matrix (ECM) proteins, such as fibrillar collagen (18, 30, 38). In addition to ECM accumulation, inflammation is a key component of cardiac aging (13, 48). Higher levels of circulating proinflammatory cytokines are observed in elderly people, even in the absence of chronic disease (3, 8, 16, 50). In old mice, the cardiac expression of inflammatory genes increases compared with that in young mice (56). Cardiac inflammation triggers the release of fibrogenic cytokines and growth factors, leading to excessive accumulation of fibrillar collagens and adverse remodeling that, in turn, may cause cardiac dysfunction (51, 54). Understanding the mechanisms of age-related inflammation in the heart is an important step to prevent or delay cardiac aging and improve quality of life in the elderly.
Procollagen, biosynthesized by the fibroblast, is secreted into the extracellular space and processed to become a mature insoluble collagen fibril (41). Secreted protein acidic and rich in cysteine (SPARC), a collagen-binding matricellular protein, is involved in procollagen processing and promotes the formation of cross-linked collagen fibrils (4, 45). The expression of SPARC is observed primarily in cardiac fibroblasts, but is also expressed at lower levels by endothelial cells, cardiomyocytes, and macrophages. Expression of SPARC is particularly induced during morphogenesis and tissue remodeling (7). SPARC-null mice show less age-related increases in cardiac stiffness and collagen content than wild-type (WT) mice (5). SPARC also participates in various biological processes, such as cellular adhesion, proliferation, and migration by regulating cell-matrix interactions, growth factor signaling, and ECM assembly (6, 7).
Previous reports have demonstrated a role for SPARC in inflammation. Compared with WT mice, young SPARC-null mice exhibit less renal inflammation and fibrosis induced by angiotensin II infusion, as well as less inflammation in response to lipopolysaccharide (LPS)-induced inflammation in a footpad model (44, 49). Similarly, SPARC deletion decreases colonic inflammatory symptoms in dextran sodium sulfate-induced murine colitis (39). The function of SPARC in regulating inflammatory processes likely depends on cellular origin of SPARC expression. Sangaletti et al. (47) reported distinct effects on lung inflammation dependent on the expression of SPARC by bone marrow versus stromal cells. Plasma levels of SPARC positively correlate with hypersensitive C-reactive protein and white blood cell numbers in gestational diabetes mellitus (55). These reports support the idea that SPARC may be a key factor to promote inflammation. Since animal models exhibit increased SPARC levels in aged hearts (5, 21), it is possible that SPARC plays an important role in age-related cardiac inflammation. However, up to date, there are no studies investigating the role of SPARC in cardiac age-related inflammation. Accordingly, the aim of this study was to investigate if SPARC deletion may delay age-related cardiac inflammation.
MATERIALS AND METHODS
Animals.
Mice colonies were maintained at the Medical University of South Carolina (MUSC) animal care facility. All procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, The National Academies Press, Washington, DC, 2011) and were approved by the Institutional Animal Care and Use Committee at MUSC (approval ID: ACORP 511). We used C57BL/6 WT and SPARC-null (Null) mice to study age and genotype differences in the LV. Null mice were maintained on a C57BL/6 strain (>12 back-crossed generations). WT and Null mice were bred as homozygous breeding pairs and housed under identical conditions since birth in the same room and fed identical diets at MUSC. Environmental factors that might potentially influence phenotype with age were minimized. Three age groups were studied: young (3–5 mo old), middle-aged (10–12 mo old), and old (18–29 mo old), and both male and female mice were included in each group (n = 7–10/age/genotype). The generation and phenotype of Null mice have been previously described by Norose and colleagues (40).
Echocardiography and necropsy.
Echocardiographic measurements were performed to examine LV structure and function, using a 40-MHz mechanical scanning transducer (707B) and a Vevo 770 echocardiograph (VisualSonics, Toronto, Canada), as described previously (58). LV volumes, ejection fraction, and wall thickness were measured and calculated using the American Society of Echocardiography recommendations for chamber quantification (31). Animals were anesthetized with isoflurane and hearts were excised. The LV was separated from the right ventricle and divided into two equal pieces. One piece was snap frozen and used for RNA extraction, and the rest of the LV was fixed in zinc formalin and used for histology.
RNA extraction and quantitative real-time RT-PCR.
Total RNA was isolated using the TRIzol reagent (Invitrogen), and cDNA was synthesized using the RT2 First Strand Kit (Qiagen, 330401). To assess mRNA expression of cytokine molecules, quantitative real-time RT-PCR was performed using RT2 SYBR green Rox qPCR Master Mix (Qiagen, 330523) and 96-well cytokine Array plates (Qiagen, PAMM-011E). For cyclin-dependent kinase inhibitor 2A (Cdkn2a), chemokine (C-X3-C motif) ligand 1 (Cx3cl1), chemokine (C-X-C motif) ligand 5 (Cxcl5), interleukin-6 (IL-6), tumor necrosis factor-α (Tnfα), and Fizz-1 expression, Taqman gene expression assays were performed using specific primers (Mm00494449_m1, Mm00436454_m1, Mm00436451_g1, Mm00446190_m1, Mm00443260_g1, and Mm00445109_m1; Applied Biosytems). The relative gene expression of individual target molecules was calculated by normalization of the threshold cycle (Ct) values of the target genes to the Ct values of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase 1 (Hprt1), which did not change in expression in our samples. Hprt1 has been identified as the most stable housekeeping gene during the aging process (11).
Histology.
Paraffin-embedded LV sections were deparaffinized in Citric-Solv (Fisherbrand), rehydrated, and then boiled for 15 min in Antigen Retrieval Solution (Dako). The sections were stained with hematoxylin and eosin or incubated in 3% hydrogen peroxide to quench endogenous peroxide activity for immunohistochemistry analysis. A rat anti-mouse Mac-3 antibody (Cedarlane CL8943AP at 1:100 dilution) and a goat anti-mouse SPARC antibody (R&D AF942 at 1:100 dilution) were used to detect macrophages and SPARC, respectively, by the avidin-biotin complex method using the 3,3′-diaminobenzidine chromogen with eosin counterstain. Five images were acquired randomly from each sample, and the dimensions of five myocytes were measured using Image Pro Analyzer 7.0 (Media Cybernetics). Macrophage numbers and SPARC expression were determined by Mac-3- and SPARC-positive area per total stained (Image-Pro Analyzer 7.0).
Peritoneal macrophage isolation and SPARC treatment.
Peritoneal macrophages were isolated from young male WT, as described previously (57). After overnight incubation at 37°C, the cells were treated with or without recombinant mouse SPARC (20 μg/ml, R&D, lot: DJW0314031) in serum-free medium for 24 h. The medium was removed, and cells were washed with PBS and lysed directly in the plate for RNA extraction with TRIzol. All cell culture experiments were performed under a sterile tissue culture hood to avoid contamination. The cDNA was synthesized using a high-capacity RNA to cDNA kit (ABI, 4387406). Taqman gene expression assays (Applied Biosystems) were used to perform quantitative real-time RT-PCR for chemokine (C-C motif) ligand 5 (Ccl5; Mm01302427_m1), chemokine (C-C motif) ligand 3 (Ccl3; Mm00441259_g1), Tnfα (Mm00443260_g1), IL-12 (Mm00434174_m1), IL-6 (Mm00446190_m1), arginase 1 (Arg1; Mm00475988_m1), and mannose receptor C type 1 (Mrc1; Mm00485148_m1). Gene levels were normalized to Hprt1 (Mm01545399_m1). SPARC treatment had no effect on the expression of IL-1β (P = 0.73), which confirmed that changes in cytokine synthesis driven by SPARC were not due to LPS contamination.
Statistics.
Data are expressed as means ± SE. Genotype comparisons of WT and Null across ages were performed using two-way ANOVA followed by the Bonferroni multiple test. One-way ANOVA followed by the Student Newman-Keuls post hoc test was used for comparisons among young, middle-aged, and old mice. Two group comparisons of peritoneal macrophages were performed using paired t-test. A P < 0.05 was considered significant.
RESULTS
SPARC expression increased with age in LV.
Our laboratory previously reported that old mice have greater levels of myocardial SPARC expression than young mice by immunoblot analysis (5). To confirm age-dependent increases in SPARC in this study, we performed immunohistochemical analysis of the LV. In accordance with our previous data, SPARC protein expression significantly increased in the old WT LV compared with both young and middle-aged WT LV (Fig. 1). These results support the concept that aging is associated with increased SPARC expression in the LV.
Fig. 1.

Secreted protein acidic and rich in cysteine (SPARC) protein expression in the left ventricle (LV) increased with age. A: representative images of SPARC-positive cells in the LVs of young, middle-aged, and old wild-type (WT) mice. Old SPARC-null mice served as the negative control. B: percentage of SPARC-positive area per total area showing age-dependent increase in SPARC in WT mice. Values are means ± SE (n = 5–6/group).
SPARC deletion suppressed the age-dependent increase in wall thickness.
Previously, our laboratory reported that senescent WT mice (31.8 ± 0.4 mo old) exhibit LV dilation and decreased ejection fraction (32). In the present study, echocardiographic analysis showed no significant differences in end-diastolic volume or end-systolic volume among the groups. Our data support the concept that LV dilation generally occurs after 29 mo of age for healthy C57BL/6 mice. There were no significant differences in ejection fraction among the groups, indicating that myocyte contractility and global LV function are still preserved at the ages evaluated. Mean and relative wall thicknesses were increased in an age-related manner in WT, but not in Null (Table 1, n = 7–10/group), indicating that the WT mice had age-associated LV hypertrophy.
Table 1.
Body weight and echocardiography results
| WT |
Null |
|||||
|---|---|---|---|---|---|---|
| Young | Middle-aged | Old | Young | Middle-aged | Old | |
| BW, g | 26 ± 1 | 35 ± 2* | 44 ± 2*‡ | 26 ± 1 | 30 ± 2 | 32 ± 2*† |
| LVEDV, μl | 60 ± 3 | 65 ± 4 | 61 ± 4 | 57 ± 3 | 59 ± 3 | 66 ± 4 |
| LVESV, μl | 25 ± 2 | 30 ± 4 | 24 ± 3 | 24 ± 1 | 26 ± 1 | 29 ± 3 |
| LVEF, % | 59 ± 2 | 55 ± 4 | 61 ± 3 | 58 ± 2 | 56 ± 2 | 56 ± 4 |
| LVSV, μl | 35 ± 2 | 35 ± 2 | 37 ± 3 | 33 ± 2 | 33 ± 2 | 37 ± 3 |
| Mean wall thickness, mm | 0.76 ± 0.02 | 0.81 ± 0.03 | 0.91 ± 0.03*‡ | 0.78 ± 0.01 | 0.77 ± 0.04 | 0.84 ± 0.02 |
| Relative wall thickness | 0.40 ± 0.01 | 0.42 ± 0.02 | 0.48 ± 0.03*‡ | 0.42 ± 0.01 | 0.40 ± 0.01 | 0.42 ± 0.01 |
Values are means ± SE (n = 7–10/group).
WT, wild-type; BW, body weight; LV, left ventricle; EDV, end-diastolic volume; ESV, end-systolic volume; EF, ejection fraction; SV, stroke volume; mean wall thickness, the average of the interventricular septal wall thickness and LV posterior wall thickness; relative wall thickness, the mean LV wall thickness divided by the LV internal dimension at end diastole.
P < 0.05 vs. respective young.
P < 0.05 vs. respective middle-aged.
P < 0.05 vs. age-matched WT.
SPARC deletion diminished age-related myocyte hypertrophy.
Myocyte hypertrophy develops with advancing age (32, 56). We performed hematoxylin-eosin staining and measured individual myocyte sizes for each group. Myocyte size was increased in the middle-aged and old WT mice compared with young WT (Fig. 2). This age-dependent increase in myocyte size was blunted in Null mice. Our results indicated that SPARC promoted age-related myocyte hypertrophy.
Fig. 2.

SPARC deletion decreased age-related myocyte hypertrophy. A: representative images of hematoxylin-eosin-stained sections from the LVs of young, middle-aged, and old WT and SPARC-null mice. Scales indicate 50 μm. B: myocyte cross-sectional areas were quantified by measuring the area of five random cells per section. Arbitrary units show age-dependent increase in myocyte area in WT, but not in SPARC-null mice. Values are means ± SE (n = 5–6/group). *P < 0.05 vs. age-matched WT.
Cdkn2a, a biomarker of cellular senescence, was increased in old mice.
Cdkn2a has been used widely as a marker of mammalian aging (26). Cdkn2a mRNA was significantly increased in old mice compared with young, for both WT and Null (Fig. 3). These results indicate that Cdkn2a is a useful marker of cardiac aging.
Fig. 3.
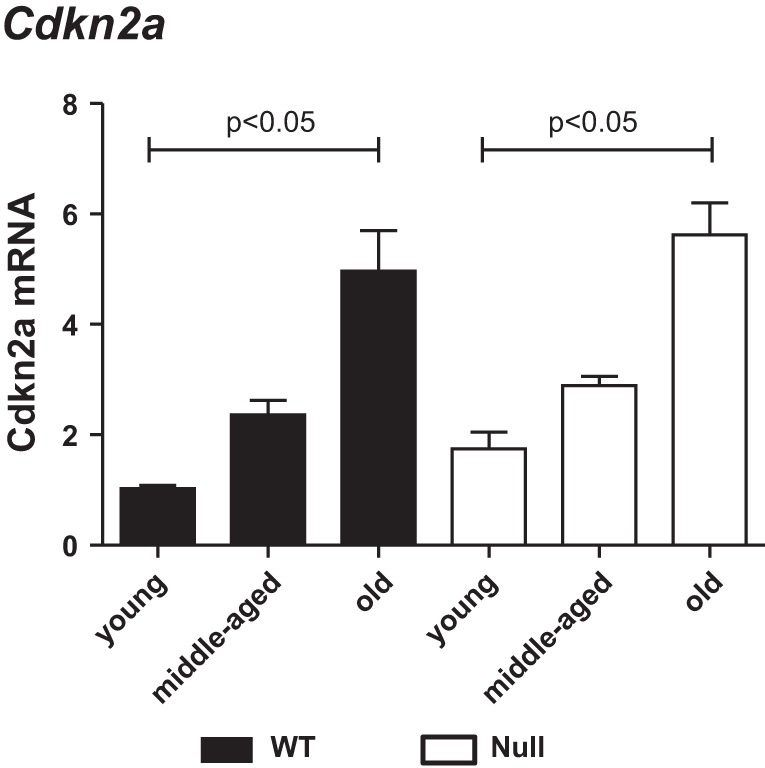
The cellular senescent marker, cyclin-dependent kinase inhibitor 2A (Cdkn2a), increased in the LV with age. Cdkn2a expression was similarly increased with age in the LVs of both WT (solid bars) and SPARC-null (open bars) mice. Expression was normalized to hypoxanthine-guanine phosphoribosyltransferase (Hprt1) levels and is shown as 2−ΔCt × 100 units, where ΔCt is change in threshold cycle. Values are means ± SE (n = 5–6/group).
SPARC deletion affected the age-dependent changes in the expression of proinflammatory cytokines.
Inflammatory cytokines are important contributors to cardiac aging (12, 56). To assess whether SPARC mediates cardiac inflammation, we measured the LV expression of inflammatory cytokines and receptors by gene array (Fig. 4). Of the 84 genes measured, 9 showed differences between age or genotype. The levels of Ccl5, Cx3cl1, C-C chemokine receptor type 2 (Ccr2) and chemokine (C-X-C motif) receptor 3 (Cxcr3) were significantly increased in middle-aged and old WT compared with young WT (P < 0.05 for all). Ccl5, Cx3cl1, Ccr2, and Cxcr3 demonstrated increases in middle-age over young myocardium, while SPARC expression was found to increase in old myocardium only. This result suggested that 1) proinflammatory cytokines might increase SPARC expression, and 2) age-dependent changes in cytokine profiles might be induced by both SPARC-dependent and -independent factors. In the absence of SPARC, only old mice showed a greater expression of these molecules compared with young mice. We validated the gene array results using a specific TaqMan gene expression assay for Cx3cl1 and confirmed the results from gene array (Fig. 5, left). These results suggest that SPARC deletion delayed age-dependent increases in the expression of these proinflammatory cytokines.
Fig. 4.

Age-related changes in the expression of proinflammatory cytokines differed in WT vs. SPARC-null myocardium. Age-dependent increases in cardiac expression of chemokine (C-C motif) ligand 5 (Ccl5), chemokine (C-X-C motif) ligand 1 (Cxcl1), C-C chemokine receptor type 2 (Ccr2), and chemokine (C-X-C motif) receptor 3 (Cxcr3) were delayed in SPARC-null (open bars) mice compared with WT (solid bars). Migration inhibitory factor (Mif) was increased with age in both WT and SPARC-null mice. C-C chemokine receptor type 1 (Ccr1) was decreased with age in WT mice. chemokine (C-X-C motif) ligand 5 (Cxcl5), chemokine (C-C motif) ligand 4 (Ccl4), and tumor necrosis factor receptor superfamily member 1b (Tnfrsf1b) were increased with age in SPARC-null mice. Each gene expression was normalized to Hprt1 and shown as 2−ΔCt × 100 units. Values are means ± SE (n = 5–6/group). *P < 0.05 vs. age-matched WT.
Fig. 5.
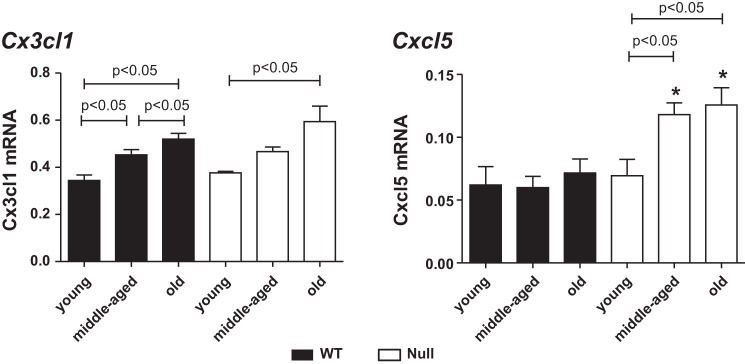
SPARC deletion shifted the inflammatory phenotype in the aging heart. Age-related increase in Cx3cl1 was delayed in SPARC-null (open bars) mice compared with WT (solid bars). Cxcl5 was increased with age in SPARC-null mice. Each gene expression was normalized to Hprt1 and shown as 2−ΔCt × 100 units. Values are means ± SE (n = 5–6/group). *P < 0.05 vs. age-matched WT.
In contrast, other inflammatory cytokines exhibited altered patterns of age and genotype-dependent expression. Expression of macrophage migration inhibitory factor (Mif) was increased in middle-aged and old mice compared with young mice in both WT and Null hearts. The expression of C-C chemokine receptor type 1 (Ccr1), one of the receptors of Ccl5, in old WT was lower than young and middle-aged WT, while there was no age-related change in Null mice. The increase in Ccr1 may be due to a negative feedback subsequent to the increase in Ccl5 (17, 22). No significant differences were found in the expression of Cxcl5, chemokine (C-C motif) ligand 4 (Ccl4), and tumor necrosis factor receptor superfamily member 1b (Tnfrsf1b) among all three groups in WT, although, in old Null conditions, higher levels of Cxcl5, Ccl4, and Tnfrsf1b were detected compared to young and middle-aged Null mice. Using a TaqMan gene-specific assay, we validated the gene array data for the Cxcl5 gene levels. There were no significant differences in Cxcl5 among the three age groups in WT, but there was an age-dependent increase in the Cxcl5 gene levels in the Null mice (Fig. 5, right). Hence, an increase in Cxcl5, Ccl4, and Tnfrsf1b might compensate for a delay in other chemokine/receptors, including Ccl5, Cx3cl1, Ccr2, and Cxcr3.
Macrophage infiltration was suppressed in Null mice.
Proinflammatory cytokines recruit macrophages, and macrophages release a variety of proinflammatory cytokines, which lead to a repetitive cycle of inflammation. To test if SPARC deletion could suppress macrophage infiltration, we quantified the expression of the macrophage marker, Mac-3. Levels of Mac-3 were enhanced with age in WT; however, SPARC deletion abolished age-dependent increase in macrophage numbers (Fig. 6). Our data suggest that SPARC facilitates age-related cardiac inflammation by promoting macrophage infiltration.
Fig. 6.

SPARC deletion inhibited age-related increase in macrophage infiltration. A: representative images of Mac-3-positive cells in the LV of young, middle-aged, and old WT and SPARC-null mice. B: percentage of Mac-3-positive area per total area showing age-dependent increase in macrophage infiltration in WT, but not in SPARC-null mice. Values are means ± SE (n = 5–6/group).
SPARC-stimulated peritoneal macrophages have increased M1 and decreased M2 macrophage polarization.
To further investigate the relationship between SPARC and inflammation, we isolated peritoneal macrophages from young mice and treated cells with SPARC (Fig. 7) to determine whether SPARC was sufficient to generate a proinflammatory status. Macrophage M1 markers, Ccl5, Ccl3, Tnfα, and IL-12, were enhanced, and M2 markers, Arg1 and Mrc1, were reduced by SPARC treatment. SPARC treatment had no effect on expression of IL-6 in vitro, another M1 macrophage marker, indicating that the major source of IL-6 in aged myocardium is not likely macrophages. Our results showed that SPARC treatment increased M1 and decreased M2 macrophage polarization in peritoneal macrophages, which could contribute to the proinflammatory effects of SPARC in advancing age. Thus SPARC may influence cardiac aging by suppressing M2 polarization, resulting in a proinflammatory phenotype.
Fig. 7.

SPARC treatment increased macrophage M1 polarization and decreased M2 polarization in peritoneal macrophages. Peritoneal macrophages from young (3–6 mo old) WT mice were stimulated with SPARC (20 μg/ml) for 24 h. SPARC increased the gene expression of macrophage M1 markers [Ccl5, chemokine (C-C motif) ligand 3 (Ccl3), tumor necrosis factor-α (Tnfα), and interleukin-12 (IL-12)] and decreased macrophage M2 markers [arginase 1 (Arg1) and mannose receptor C type 1 (Mrc1)]. SPARC treatment had no effect on the expression of IL-6, another M1 macrophage marker. Expression of each gene was normalized to Hprt1 levels and shown as 2−ΔCt units. Values are means ± SE (n = 4/group).
SPARC increased M1 macrophage polarization in vivo.
To confirm our in vitro results that SPARC promotes M1 macrophage polarization, we investigated the expression of IL-6 and Tnfα, M1 macrophage markers, in vivo. Levels of mRNA encoding IL-6 and Tnfα were higher in old WT vs. young and middle-aged WT. No age-dependent changes were found in Null mice, suggesting that SPARC, which increases in old age, enhanced M1 macrophage polarization in vivo (Fig. 8). The expression of Fizz-1, an M2 macrophage marker, was lower in middle-aged and old WT LV compared with young WT LV, whereas no age-related changes in Fizz-1 expression were detected in Null mice. This suggests that SPARC decreased M2 macrophage polarization in vivo (Fig. 8).
Fig. 8.

IL-6 and Tnfα, markers of M1 macrophage polarization, increased, and Fizz-1, a marker of M2 macrophage polarization, decreased with age only in the WT mice. Age-related increases in IL-6 and Tnfα were blunted in SPARC-null (open bars) mice compared with WT (solid bars). Gene expression was normalized to Hprt1 and shown as 2−ΔCt × 100 units. Values are means ± SE (n = 4–6/group).
DISCUSSION
Fibrosis and inflammation are major components of cardiac aging. SPARC plays a role in cardiac aging by facilitating the deposition of fibrotic ECM proteins (5). In the present study, we examined the roles of SPARC in aging heart with respect to inflammation. A recent clinical study reports the positive correlation between plasma SPARC and hypersensitive C-reactive protein and white blood cell numbers in gestational diabetes mellitus (55). Another recent report by Ng et al. (39) demonstrated that SPARC-null mice showed less inflammatory symptoms in a mouse model of colitis. These studies suggest that SPARC might enhance inflammation in certain settings. The significant findings of our study were as follows: 1) SPARC deletion suppressed age-related LV hypertrophy; 2) SPARC deletion shifted the inflammatory phenotype by affecting age-dependent changes in gene expression of some inflammatory cytokines; 3) macrophage infiltration was delayed in Null mice; and 4) SPARC treatment increased the expression of proinflammatory M1 markers and reduced the expression of anti-inflammatory M2 markers in isolated macrophages from young mice. This study shows for the first time that SPARC facilitates inflammation in cardiac aging. Although further studies are needed, suppression of SPARC may be a potential therapeutic target for cardiac aging.
Various changes in LV structure and function occur in the aging heart (30). These changes affect mechanical load and wall stress, leading to collagen accumulation. We found that wall thickness and myocyte size increased with age in the WT, but not in the Null, which provides evidence that lack of SPARC during cardiac aging decreases cardiac hypertrophy. Our laboratory previously reported that SPARC deletion decreases age-dependent cardiac collagen accumulation, which supports the idea that SPARC participates in cardiac remodeling through, at least in part, the regulation of procollagen processing (5). Cardiac ECM not only provides structural support for the myocardium, but also serves as a reservoir for the sequestration of cytokines and growth factors (23). Further investigation into age-dependent changes in the structure and composition of ECM is needed to clarify the role of ECM in age-dependent inflammatory cell recruitment and cardiac remodeling.
Low levels of chronic inflammation are associated with age-related diseases, including cardiovascular disease (13, 15). Inflammatory cytokines, therefore, are predicted to play a role in cardiac aging. Once inflammatory homeostasis is disturbed, inflammatory cytokines are released from diverse cells, such as myocytes, macrophages, endothelial cells, and fibroblasts (52). These cytokines will in turn recruit leukocytes. Ccl5, Cx3cl1, Ccr2, Cxcr3, and Mif were all elevated in the old WT mice compared with young. Ccl5, also known as regulated upon activation, normal T-cell expressed and secreted (RANTES), plays an important role in recruiting inflammatory cell subsets to inflammatory sites though the binding to Ccr1, Ccr3, or Ccr5 (17, 22). Cx3cl1, also called fractalkine, can function both as a membrane-bound form and a cleaved circulating form. The membrane-bound form mainly acts as an adhesion molecule and enables leukocyte adhesion (1). Ccr2 is a seven-transmembrane G-protein-coupled receptor for monocyte chemotactic protein-1, which induces migration of monocytes to inflammatory sites (14). Cxcr3, a receptor for Cxcl9, Cxcl10, and Cxcl11, is known to recruit proinflammatory Th1 cells to sites of inflammation (20, 33). Mif regulates the release of proinflammatory mediators (9). In the heart, Mif is rapidly released in response to a cellular stressor, such as a brief hypoxia (25, 42). Plasma Mif levels are increased in old healthy volunteers compared with young controls, and dietary nitrate supplementation caused a reduction of plasma Mif levels and improvement in vascular function (43). The age-dependent elevated expression of these cytokines and receptors could, therefore, cause the age-dependent increase in macrophage infiltration observed in WT. Our data suggest that these cytokines play important roles in cardiac aging. In support, plasma monocyte chemotactic protein-1 and Ccl5 have been reported to be increased with age in healthy subjects (36).
Interestingly, in WT hearts, expression of Ccl5, Cx3cl1, Ccr2, and Cxcr3 were increased in middle-aged mice, whereas increases in macrophage numbers and expression of the senescence-associated Cdkn2a were detected only in old hearts. Hence, an increase in the expression of these cytokines precedes recruitment of significant numbers of macrophages. One interpretation is that the process of cardiac aging begins in middle age and slowly progresses with age. We also found that, with the exception of Mif, the age-dependent increase in these proinflammatory cytokines/receptors was delayed in Null hearts. Accordingly, Null mice do not exhibit increased collagen in aged myocardium and do not exhibit age-dependent increases in stiffness (5). As cardiac expression of SPARC is shown to increase with age (5, 21), we propose the novel concept that, with age, SPARC acts to enhance macrophage infiltration, which eventually leads to increased cardiac remodeling and dysfunction.
We further evaluated the effects of SPARC on macrophage phenotypes to determine whether SPARC might have a direct effect on macrophage activation in vitro. Macrophages are classified into two major subtypes: M1 (proinflammatory or classically activated) and M2 (anti-inflammatory or alternatively activated) macrophages (34, 37). We hypothesized that SPARC would increase M1 and/or decrease M2 polarization. Supporting our hypothesis, SPARC treatment increased macrophage polarization toward an M1 phenotype. Remarkably, SPARC-treated macrophages displayed decreased M2 polarization. In agreement with these results, Arnold et al. (2) reported an increase in the number of M2, alternatively activated, macrophages in tumors grown in Null mice. Although the precise mechanisms are unclear, our data suggest that increased levels of SPARC in aged hearts leads to suppression of M2 macrophage polarization and augmentation of M1 macrophage polarization. Supporting our in vitro results, SPARC deletion blunted in vivo age-dependent increases in Tnfα, an M1 macrophage marker, and age-dependent decreases in Fizz-1, an M2 macrophage marker. SPARC treatment had no effect on IL-6 levels in cultured peritoneal macrophages, whereas levels of IL-6 mRNA were increased with age in WT LV, but not in Null LV. These results suggest that the major source of IL-6 in aged myocardium is likely not macrophages; other candidate cell types that express IL-6 include cardiomyocytes, fibroblasts, and endothelial cells.
The effect of SPARC was less than that of the positive control, the well-characterized M1 driver LPS + interferon-γ. For example, macrophage treatment with LPS (1 μg/ml) and interferon-γ (20 ng/ml) increased Tnfα and Ccl3 expression 100-fold, whereas SPARC induced 2-fold increases in these two genes (35). Of note, in addition to SPARC increasing Tnfα and Ccl3, SPARC also decreased Mrc1 expression by 62%, whereas IL-4, an M2 driver, induces a 3.5-fold increase (35). Therefore, the proinflammatory effects of SPARC might be related more to the decrease in M2 macrophage polarization rather than an increase in M1 polarization.
In the present study, we did not determine the precise mechanisms by which SPARC affected inflammation in the aging heart. Goldblum et al. (19) reported that SPARC reduces cultured endothelial cell adhesion to form a rounded conformation with intercellular gaps that could allow the passage of macromolecules. Treatment of human umbilical endothelial cells with SPARC similarly enables leukocytes to more easily transmigrate through the monolayer (24). SPARC itself interacts with several receptors, including β1-integrin, vascular cell adhesion molecule-1, and stabilin-1 (42, 53). The interaction of SPARC and vascular cell adhesion molecule-1 triggers leukocyte transmigration (24). Stabilin-1, a scavenger receptor, is expressed by M2 macrophages and transiently by microvascular endothelial cells during inflammation (27, 46). Stabilin-1 is required for receptor-mediated endocytosis of SPARC in macrophages (28). Although further studies are needed, one possibility is that binding of SPARC to stabilin-1 might influence macrophage polarization.
While we found evidence of colocalization of SPARC and macrophages in the myocardium, we also observed macrophages that did not stain positive for SPARC, suggesting age-dependent increases in macrophage infiltration might be caused by both SPARC-dependent and -independent mechanisms. SPARC was upregulated following alterations in the cytokine profiles that differed in old WT compared with the young and middle-aged WT, suggesting the increases in SPARC expression might result from changes in cytokine expression. Our data showed that Null mice exhibited a different cytokine profile, suggestive of both direct and indirect effects of SPARC on the inflammatory phenotype in the aging heart. One possibility is that changes in the ECM assembled in the absence of SPARC might influence macrophage recruitment. Effects of SPARC, mediated through ligand interactions, for example, might represent another stage at which SPARC influences inflammatory cell phenotype. Our present results, coupled with previous observations that collagen content increases with aging and is attenuated in the absence of SPARC (5), suggest that a logical future examination would be to examine extracellular mechanisms that are likely SPARC dependent.
There were some limitations in the present study. Our study did not show the mechanism(s) by which SPARC affected cytokine synthesis or macrophage polarization that are upstream of the current focus. In addition, although we demonstrated the direct effects of SPARC on macrophages, the in vivo results likely also reflect the effects of SPARC on other cell types, including myocytes, fibroblasts, and vascular cells. Further studies into the mechanisms by which SPARC deletion suppresses age-related cardiac hypertrophy are important to demonstrate whether modulating SPARC expression might be clinically beneficial.
In conclusion, we showed that SPARC deletion delayed age-dependent increase in cardiac inflammation in vivo, and SPARC induced the expression of M1 markers and suppressed the expression of M2 macrophage markers in vitro. SPARC is a key player in age-associated cardiac inflammation (Fig. 9).
Fig. 9.
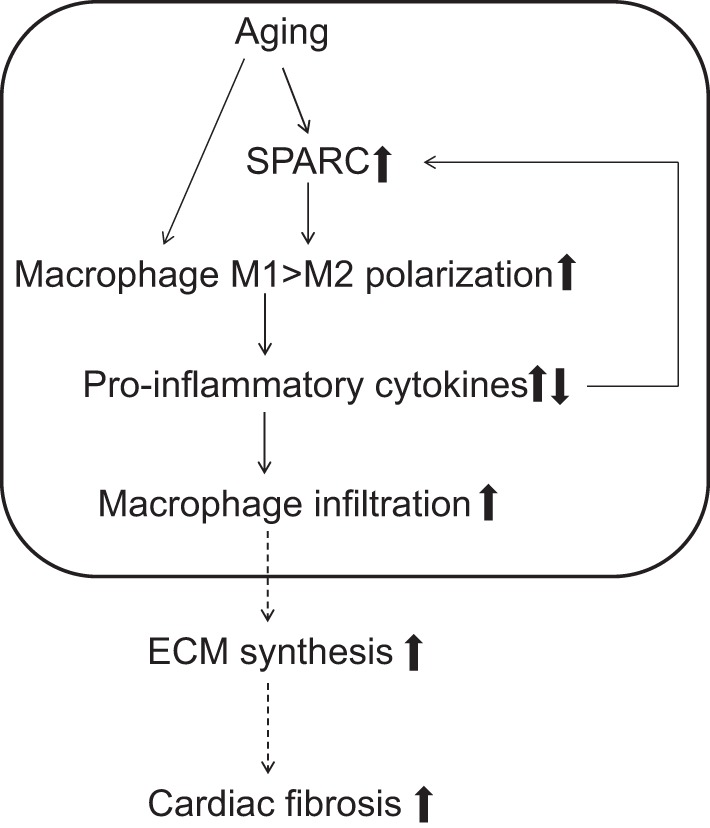
The role of SPARC in age-related cardiac inflammation. The diagram summarizes the present working hypothesis of the influence of SPARC in age-related cardiac inflammation. SPARC levels increase with advancing age, which induces proinflammatory macrophage M1 polarization. M1 macrophages produce proinflammatory chemokines, promoting macrophage infiltration. Increases in macrophage infiltration lead to a cycle wherein release of proinflammatory chemokines enhances further cardiac inflammation (boxed area highlights results from the present study). Cardiac inflammation induces the production of fibrogenic cytokines and growth factors, leading to excessive accumulation of extracellular matrix (ECM) and adverse remodeling.
GRANTS
We acknowledge support from the American Heart Association for 14SDG1886005, from the Biomedical Laboratory Research, Development Service of the Veterans Affairs Office of Research and Development Awards 1I01BX001385 and 5I01BX000505, and from the National Institutes of Health for HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center, HL-075360, HL-051971, and GM-104357.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
H.T., L.E.C.B., M.R.Z., M.L.L., and A.D.B. conception and design of research; H.T. and C.F.B. performed experiments; H.T., L.E.C.B., C.F.B., M.R.Z., M.L.L., and A.D.B. analyzed data; H.T., L.E.C.B., M.R.Z., and M.L.L. interpreted results of experiments; H.T. prepared figures; H.T. and M.L.L. drafted manuscript; H.T., L.E.C.B., C.F.B., M.R.Z., M.L.L., and A.D.B. edited and revised manuscript; H.T., L.E.C.B., C.F.B., M.R.Z., M.L.L., and A.D.B. approved final version of manuscript.
REFERENCES
- 1.Altin SE, Schulze PC. Fractalkine: a novel cardiac chemokine? Cardiovasc Res 92: 361–362, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, Castrillon DH, Sage EH, Puolakkainen P, Bradshaw AD, Brekken RA. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech 3: 57–72, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballou SP, Lozanski FB, Hodder S, Rzewnicki DL, Mion LC, Sipe JD, Ford AB, Kushner I. Quantitative and qualitative alterations of acute-phase proteins in healthy elderly persons. Age Ageing 25: 224–230, 1996. [DOI] [PubMed] [Google Scholar]
- 4.Bradshaw AD. The role of SPARC in extracellular matrix assembly. J Cell Commun Signal 3: 239–246, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Bonnema DD, Zile MR. Age-dependent alterations in fibrillar collagen content and myocardial diastolic function: role of SPARC in postsynthetic procollagen processing. Am J Physiol Heart Circ Physiol 298: H614–H622, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest 107: 1049–1054, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biol 19: 569–580, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Bruunsgaard H, Pedersen BK. Age-related inflammatory cytokines and disease. Immunol Allergy Clin North Am 23: 15–39, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3: 791–800, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheitlin MD. Cardiovascular physiology-changes with aging. Am J Geriatr Cardiol 12: 9–13, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Rider DA, Ruan R. Identification of valid housekeeping genes and antioxidant enzyme gene expression change in the aging rat liver. J Gerontol A Biol Sci Med Sci 61: 20–27, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, Jin YF, Lindsey ML. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res 96: 444–455, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev 8: 18–30, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson J, Miltz W, Mir AK, Wiessner C. Targeting monocyte chemoattractant protein-1 signaling in disease. Expert Opin Ther Targets 7: 35–48, 2003. [DOI] [PubMed] [Google Scholar]
- 15.De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des 15: 3003–3026, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Ershler WB, Sun WH, Binkley N, Gravenstein S, Volk MJ, Kamoske G, Klopp RG, Roecker EB, Daynes RA, Weindruch R. Interleukin-6 and aging: blood levels and mononuclear cell production increase with advancing age and in vitro production is modifiable by dietary restriction. Lymphokine Cytokine Res 12: 225–230, 1993. [PubMed] [Google Scholar]
- 17.Ferrandi C, Ardissone V, Ferro P, Ruckle T, Zaratin P, Ammannati E, Hauben E, Rommel C, Cirillo R. Phosphoinositide 3-kinase gamma inhibition plays a crucial role in early steps of inflammation by blocking neutrophil recruitment. J Pharmacol Exp Ther 322: 923–930, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Ferrari AU, Radaelli A, Centola M. Invited review: aging and the cardiovascular system. J Appl Physiol (1985) 95: 2591–2597, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Goldblum SE, Ding X, Funk SE, Sage EH. SPARC (secreted protein acidic and rich in cysteine) regulates endothelial cell shape and barrier function. Proc Natl Acad Sci U S A 91: 3448–3452, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res 317: 620–631, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horn MA, Graham HK, Richards MA, Clarke JD, Greensmith DJ, Briston SJ, Hall MC, Dibb KM, Trafford AW. Age-related divergent remodeling of the cardiac extracellular matrix in heart failure: collagen accumulation in the young and loss in the aged. J Mol Cell Cardiol 53: 82–90, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Houard X, Touat Z, Ollivier V, Louedec L, Philippe M, Sebbag U, Meilhac O, Rossignol P, Michel JB. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc Res 82: 532–541, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 57: 195–202, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Kelly KA, Allport JR, Yu AM, Sinh S, Sage EH, Gerszten RE, Weissleder R. SPARC is a VCAM-1 counter-ligand that mediates leukocyte transmigration. J Leukoc Biol 81: 748–756, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Koga K, Kenessey A, Powell SR, Sison CP, Miller EJ, Ojamaa K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid Redox Signal 14: 1191–1202, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114: 1299–1307, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kzhyshkowska J, Gratchev A, Goerdt S. Stabilin-1, a homeostatic scavenger receptor with multiple functions. J Cell Mol Med 10: 635–649, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kzhyshkowska J, Workman G, Cardo-Vila M, Arap W, Pasqualini R, Gratchev A, Krusell L, Goerdt S, Sage EH. Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. J Immunol 176: 5825–5832, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part I. Aging arteries: a “set up” for vascular disease. Circulation 107: 139–146, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part II: The aging heart in health: links to heart disease. Circulation 107: 346–354, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Lin J, Lopez EF, Jin Y, Van Remmen H, Bauch T, Han HC, Lindsey ML. Age-related cardiac muscle sarcopenia: Combining experimental and mathematical modeling to identify mechanisms. Exp Gerontol 43: 296–306, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med 184: 963–969, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res 112: 675–688, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mansfield AS, Nevala WK, Dronca RS, Leontovich AA, Shuster L, Markovic SN. Normal ageing is associated with an increase in Th2 cells, MCP-1 (CCL1) and RANTES (CCL5), with differences in sCD40L and PDGF-AA between sexes. Clin Exp Immunol 170: 186–193, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25: 677–686, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Martos R, Baugh J, Ledwidge M, O'Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115: 888–895, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Ng YL, Klopcic B, Lloyd F, Forrest C, Greene W, Lawrance IC. Secreted protein acidic and rich in cysteine (SPARC) exacerbates colonic inflammatory symptoms in dextran sodium sulphate-induced murine colitis. PLos One 8: e77575, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norose K, Clark JI, Syed NA, Basu A, Heber-Katz E, Sage EH, Howe CC. SPARC deficiency leads to early-onset cataractogenesis. Invest Ophthalmol Vis Sci 39: 2674–2680, 1998. [PubMed] [Google Scholar]
- 41.Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 64: 403–434, 1995. [DOI] [PubMed] [Google Scholar]
- 42.Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest 119: 3807–3816, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rammos C, Hendgen-Cotta UB, Pohl J, Totzeck M, Luedike P, Schulze VT, Kelm M, Rassaf T. Modulation of circulating macrophage migration inhibitory factor in the elderly. Biomed Res Int 2014: 582586, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rempel SA, Hawley RC, Gutierrez JA, Mouzon E, Bobbitt KR, Lemke N, Schultz CR, Schultz LR, Golembieski W, Koblinski J, VanOsdol S, Miller CG. Splenic and immune alterations of the Sparc-null mouse accompany a lack of immune response. Genes Immun 8: 262–274, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem 282: 22062–22071, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Salmi M, Koskinen K, Henttinen T, Elima K, Jalkanen S. CLEVER-1 mediates lymphocyte transmigration through vascular and lymphatic endothelium. Blood 104: 3849–3857, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Sangaletti S, Tripodo C, Cappetti B, Casalini P, Chiodoni C, Piconese S, Santangelo A, Parenza M, Arioli I, Miotti S, Colombo MP. SPARC oppositely regulates inflammation and fibrosis in bleomycin-induced lung damage. Am J Pathol 179: 3000–3010, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sikora E, Arendt T, Bennett M, Narita M. Impact of cellular senescence signature on ageing research. Ageing Res Rev 10: 146–152, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Socha MJ, Manhiani M, Said N, Imig JD, Motamed K. Secreted protein acidic and rich in cysteine deficiency ameliorates renal inflammation and fibrosis in angiotensin hypertension. Am J Pathol 171: 1104–1112, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei J, Xu H, Davies JL, Hemmings GP. Increase of plasma IL-6 concentration with age in healthy subjects. Life Sci 51: 1953–1956, 1992. [DOI] [PubMed] [Google Scholar]
- 51.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4: 44–52, 2011. [DOI] [PubMed] [Google Scholar]
- 52.Wilke A, Schonian U, Herzum M, Hengstenberg C, Hufnagel G, Brilla CG, Maisch B. [The extracellular matrix and cytoskeleton of the myocardium in cardiac inflammatory reaction]. Herz 20: 95–108, 1995. [PubMed] [Google Scholar]
- 53.Workman G, Sage EH. Identification of a sequence in the matricellular protein SPARC that interacts with the scavenger receptor stabilin-1. J Cell Biochem 112: 1003–1008, 2011. [DOI] [PubMed] [Google Scholar]
- 54.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199–210, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu L, Ping F, Yin J, Xiao X, Xiang H, Ballantyne CM, Wu H, Li M. Elevated plasma SPARC levels are associated with insulin resistance, dyslipidemia, and inflammation in gestational diabetes mellitus. PLos One 8: e81615, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yabluchanskiy A, Ma Y, Chiao YA, Lopez EF, Voorhees AP, Toba H, Hall ME, Han HC, Lindsey ML, Jin YF. Cardiac aging is initiated by matrix metalloproteinase-9-mediated endothelial dysfunction. Am J Physiol Heart Circ Physiol 306: H1398–H1407, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zamilpa R, Ibarra J, de Castro Bras LE, Ramirez TA, Nguyen N, Halade GV, Zhang J, Dai Q, Dayah T, Chiao YA, Lowell W, Ahuja SS, D'Armiento J, Jin YF, Lindsey ML. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function postmyocardial infarction. J Mol Cell Cardiol 53: 599–608, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zile MR, Baicu CF, Stroud RE, Van Laer AO, Jones JA, Patel R, Mukherjee R, Spinale FG. Mechanistic relationship between membrane type-1 matrix metalloproteinase and the myocardial response to pressure overload. Circ Heart Fail 7: 340–350, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]