

Abstract
We recently demonstrated that female mice are resistant to the development of obesity-induced hypertension through a sex hormone-dependent mechanism that involved adipose angiotensin-converting enzyme 2 (ACE2). In this study, we hypothesized that provision of 17β-estradiol (E2) to ovariectomized (OVX) high-fat (HF)-fed female hypertensive mice would reverse obesity-hypertension through an ACE2-dependent mechanism. Pilot studies defined dose-dependent effects of E2 in OVX female mice on serum E2 concentrations and uterine weights. An E2 dose of 36 μg/ml restored normal serum E2 concentrations and uterine weights. Therefore, HF-fed OVX female Ace2+/+ and Ace2−/− mice were administered vehicle or E2 (36 μg/ml) for 16 wk. E2 administration significantly decreased body weights of HF-fed OVX female Ace2+/+ and Ace2−/− mice of either genotype. At 15 wk, E2 administration decreased systolic blood pressure (SBP) of OVX HF-fed Ace2+/+ but not Ace2−/− females during the light but not the dark cycle. E2-mediated reductions in SBP in Ace2+/+ females were associated with significant elevations in adipose ACE2 mRNA abundance and activity and reduced plasma ANG II concentrations. In contrast to females, E2 administration had no effect on any parameter quantified in HF-fed male hypertensive mice. In 3T3-L1 adipocytes, E2 promoted ACE2 mRNA abundance through effects at estrogen receptor-α (ERα) and resulted in ERα-mediated binding at the ACE2 promoter. These results demonstrate that E2 administration to OVX females reduces obesity-induced elevations in SBP (light cycle) through an ACE2-dependent mechanism. Beneficial effects of E2 to decrease blood pressure in OVX obese females may result from stimulation of adipose ACE2.
Keywords: 17β-estradiol, blood pressure, ovariectomy, renin-angiotensin system
the prevalence of hypertension has consistently risen in the United States largely due to the increasing prevalence of obesity. Data from the most recent National Health and Nutrition Examination Survey (NHANES) demonstrate that the prevalence of hypertension is rising at a faster rate in females than in males (6). It is generally well accepted, based on data from cross-sectional studies, that adult males are at a higher risk to develop hypertension than females until after menopause, when female prevalence of both obesity and hypertension increases markedly (27, 28). Mechanisms for the role of menopause in the increasing prevalence of hypertension in females, or for the faster rise in hypertension prevalence in females compared with males are unclear.
An activated renin-angiotensin system (RAS) has been suggested to contribute to the development of experimental and human obesity-induced hypertension (2, 9, 12, 14). Indeed, recent studies from our laboratory demonstrate that adipose tissue serves as a predominant source for elevated systemic concentrations of angiotensin II (ANG II) and the development of hypertension in male obese mice (41). Adipocytes express several components of the RAS necessary to synthesize, respond to, or metabolize ANG II. In male mice with diet-induced obesity-hypertension associated with increased plasma and adipose ANG II concentrations, adipose expression of angiotensin-converting enzyme 2 (ACE2) (14), which metabolizes ANG II to form angiotensin-(1–7) [ANG-(1–7)], was reduced, and plasma ANG-(1–7) concentrations plummeted to levels of ACE2-deficient mice (15). In contrast, female mice made obese from consumption of a high-fat (HF) diet did not exhibit changes in plasma ANG II concentrations and were resistant to the development of obesity-induced hypertension (15). Moreover, protection against obesity-hypertension in females was associated with increased adipose ACE2 expression and elevated plasma concentrations of the vasodilator ANG-(1–7) (15). Ovariectomy of HF-fed female mice reduced adipose ACE2 expression and plasma ANG-(1–7) concentrations and converted female mice to an obesity-hypertension phenotype (15). These results suggest that sexual dimorphism of obesity-hypertension may be associated with ACE2-mediated regulation of the ANG II/ANG-(1–7) balance, and that adipose ACE2 may contribute significantly to regulation of the balance of these vasoactive peptides.
17β-Estradiol (E2) has been reported to regulate ACE2 expression both in vitro and in vivo (15, 20, 21). Moreover, results from our laboratory demonstrated that E2 increases ACE2 mRNA abundance in adipocytes (15). In this study, we hypothesized that ovariectomy promotes obesity-induced hypertension in females through an E2-mediated mechanism. In addition, we hypothesized that E2 administration to ovariectomized (OVX) HF-fed females protects against obesity-hypertension through an ACE2-dependent mechanism. To determine whether exogenous E2 can also prevent the development of obesity-hypertension in males, we administered E2 to HF-fed wild-type and ACE2-deficient male mice. Finally, to explore mechanisms of E2 regulation of adipose ACE2, we defined the estrogen receptor (ER) responsible for E2-mediated regulation of ACE2 mRNA abundance in 3T3-L1 adipocytes and demonstrated direct interaction between ERα and the ACE2 promoter in adipocytes exposed to E2.
MATERIALS AND METHODS
Animal handling.
All studies using mice were approved by an Institutional Animal Review Committee at the University of Kentucky and were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. In pilot studies to determine the optimal E2 dose to activate adipose ACE2, wild-type female mice (C57BL/6, 8 wk of age) underwent ovariectomy, and then 2 wk later (to clear endogenous sex hormones) mice were assigned to groups to receive E2 (0, 36, 200 or 600 μg/ml, n = 5 mice/group) via Silastic tubing capsules for 1 mo; a group of intact female mice (n = 5) without drug treatment was included for comparison. At study end point, mice were anesthetized with ketamine-xylazine (100/10 mg/kg ip) for exsanguination and tissue harvest.
In a separate study, female mice underwent ovariectomy as described previously (15), followed by a 2-wk interval to clear endogenous sex hormones prior to the onset of HF feeding (ad libitum; 60% kcal as fat; D12492, Research Diets). Male and female wild-type (Ace2+/y, n = 20 mice; Ace2+/+, n = 20 mice, respectively) or whole body ACE2-deficient mice (Ace2−/y, n = 20 mice; Ace2−/−, n = 20 mice, respectively) 2 mo of age; back-crossed 10× onto a C57BL/6 background, were fed the HF diet for 16 wk. Silastic tubing inserts were filled with vehicle [sesame oil (Sigma-Aldrich, St. Louis, MO) or E2 (36 μg/ml) (19), equivalent to a dose of 1.2 μg·kg−1·day−1 (Sigma)] and implanted subcutaneously into the interscapular space of anesthetized mice (vehicle group, n = 8 mice/genotype; E2 group, n = 12 mice/genotype) for sustained delivery at the onset of HF feeding. Each month, empty Silastic tubing capsules were removed from anesthetized mice and replaced with tubing containing fresh drug. At study end point, body composition was quantified in conscious mice by echo-MRI. At study end point, mice were anesthetized with ketamine-xylazine (100:10 mg/kg ip) for exsanguination and tissue harvest.
Quantification of plasma parameters.
Plasma concentrations of ANG II in HF-fed male and female mice were quantified as described previously (7). Plasma renin concentrations were quantified as described previously (41). Serum E2 concentrations were quantified using a commercial estradiol ELISA kit (Calbiotech, ES180S-100) following the manufacturer's instructions.
Quantification of blood pressure.
Blood pressure was quantified by radiotelemetry at week 16 of HF feeding as described previously (14). Briefly, at week 15 of HF feeding, anesthetized (isoflurane to effect) mice were implanted with left carotid artery catheters followed by a 1-wk recovery, and then blood pressure was recorded (sampling every 5 min) for 5 consecutive days.
Tissue RNA extraction and quantitative RT-PCR.
Tissue RNA was isolated using the SV Total RNA Isolation system (Promega, Madison, WI). RNA concentrations were then determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE); 400 ng of RNA was used for reverse transcription to make cDNA using a Retroscript kit. cDNAs were amplified using SYBR Green PCR Master Mix(Quanta, Gaithersburg, MD). Estimation of amplified gene products was normalized to 18S RNA. Primer sequences used were as follows: ACE2: forward 5′-ACGAGATGGGACACATCCA-3′, reverse 5′-GAAAATCGGATGGCAGAAGA-3′; 18S: forward 5′-AGTCGGCATCGTTTATGGTC-3′, reverse 5′-CGAAAGCATTTGCCAAGAAT-3′; ERα: forward 5′-TCTCTGGAAGAGAAGGACCACATC-3′, reverse 5′-TGCAGAGTCAGGCCAGCTTT-3′; ERβ: forward 5′-GACACCTCTCTCCTTTAG-3′, reverse 5′-CAGGGTCTCTCTGTTTAC-3′.
Quantification of tissue ACE2 activity.
Mca-APK(Dnp) (BML-P163-0001), a fluorogenic substrate for ACE2, was purchased from Enzo Life Sciences (Farmingdale, NY). Adipose tissue and kidney samples were homogenized in ACE2 buffer [0.1 M Tris·HCl, 0.3 M NaCl, 10 μM ZnCl2 Z-pro-prolinol with protease inhibitors (Roche, Complete ULTRA Tablets), 0.5% Triton X-100, pH 7.0] using a Geno/Grinder 2010 for protein extraction. Total protein extracts (kidney, 10 μg; adipose tissue, 100 μg) were incubated with ACE2 reaction buffer (75 mM Tris·HCl, 1 M NaCl, 0.5 mM ZnCl2, 100 μM Z-pro-prolinol, 10 μM captopril, pH 7.0) and the intramolecularly quenched synthetic ACE2-specific substrate Mca-APK(Dnp) in black 96-well plates (100 μl total volume). ACE2 activity was quantified by fluorescence (read at excitation of 320 nm and emission at 405 nm, auto-cutoff at 420 nm). For adipose tissue, protein extracts were incubated with 25 μM Mca-APK(Dnp) for 2.5 h at 37°C; for kidney, protein extracts were incubated with 10 μM Mca-APK(Dnp) for 1 h at 37°C. Preliminary studies defined conditions where the ACE2 substrate concentration and time of incubation were not limiting. Total fluorescence was corrected for protein content (in kidney and adipose tissue homogenates) after subtracting blank values. ACE2 activity is expressed as relative fluorescence units per microgram of protein per hour (RFU·μg−1·h−1).
In vitro effect of E2 on ACE2 mRNA abundance in 3T3-L1 adipocytes.
3T3-L1 cells from ATCC were cultured and differentiated according to the manufacturer's standard protocol, with modification. Briefly, cells were seeded and cultured in six-well plates in regular DMEM containing 10% new-born calf serum (NCS) and 1% Pen-Strep. At 2 days postconfluence, cells were incubated with an adipocyte differentiation cocktail (regular DMEM with 10% fetal bovine serum, 1% Pen-Strep, 0.2 μM insulin, 0.5 mM IBMX, and 1.0 μM dexamethasone) for 2 days. On day 3, cells were incubated with Adipocyte Maintenance Medium (regular DMEM with 10% FBS, 1% Pen-Strep, and 0.2 μM insulin) for 2 days. Cells were than maintained in phenol red-free DMEM with 10% regular FBS until treatment. On day 8, mature adipocytes were serum starved with 1% charcoal stripped FBS (Gibco, Life Technologies) and incubated with vehicle (0.001% DMSO, ATCC, Manassas, VA) or E2 (100 nM) with or without an ERα antagonist (MPPD, 100 nM, Tocris Bioscience, Bristol, UK) for 24 h. At study end point, cells were washed with ice-cold PBS and harvested in 0.5 ml of TRIzol (Ambion, NY) for RNA extraction.
Chromatin immunoprecipitation in 3T3-L1 adipocytes.
We performed chromatin immunoprecipitation (ChIP) in 3T3-L1 adipocytes by using a commercial Simple ChIP Enzymatic Chromatin IP kit (Cell Signaling Technology) following the manufacturer's instructions and using an ERα antibody (rabbit polyclonal, HC-20X, Santa Cruz Biotechnology). Briefly, 3T3-L1 cells were seeded and differentiated in 150-mm culture dishes and differentiated to mature adipocytes as described above. On day 8, adipocytes were serum starved with 1% charcoal stripped FBS (Gibco, Life Technologies) and incubated with vehicle (0.001% DMSO, ATCC) or E2 (100 nM) for 1 or 12 h. Cells (2–3 dishes, 4 × 107 cells for each experiment to achieve appropriate starting material) were cross-linked and harvested for nuclei preparation and chromatin digestion following the manufacturer's protocol. After chromatin digestion, we determined DNA fragment size and concentration to optimize the assay. ERα antibody (10 μg) was incubated with chromatin (containing 20 μg DNA) overnight at 4°C with rotation. The use of control antibodies was strictly according to the manufacturer's protocol. Input chromatin (2%) as well as immunoprecipitated chromatin were reverse cross-linked and purified for RT-PCR. The primer set used to amplify the putative ERE site (5′-AGGTCAAACTCTCTG-3′) of the ACE2 promotor region is ACE2P2: forward AGCATCAAGGTCAAACTCTCTG, reverse AAAGGACCCTCTAGAGATGGAG (product size, 201 bp). PCR signals were normalized to signals of 2% input sample and were expressed as signal relative to input.
Statistical analysis.
Data are presented as means ± SE. Statistical analyses were performed using SigmaStat (SPSS). For two-factor analysis, a two-way ANOVA (genotype and treatment as between group factors, time as a repeated measure when applicable) was used to analyze end point measurements followed by Holm-Sidac for post hoc analyses. Values of P < 0.05 were considered to be statistically significant.
RESULTS
A low dose of E2 administered to OVX female mice restores plasma E2 concentrations to levels of ovary-intact females.
To determine a dose of E2 that would restore plasma E2 concentrations to physiological levels in OVX females, E2 (36, 200, or 600 μg/ml, equivalent to 1.2, 6.7, or 20 μg·kg−1·day−1) was administered to female OVX mice for 1 mo. Normal physiological E2 concentrations were determined using serum samples from ovary-intact female mice. Plasma E2 concentrations were significantly decreased at 1 mo of ovariectomy compared with intact female controls (Fig. 1A; P < 0.05). The lowest dose of E2 (36 μg/ml) restored serum E2 concentrations to levels that were not significantly different from those of intact females (Fig. 1A). Higher E2 doses resulted in a modest dose-dependent increase in serum E2 concentrations. We quantified uterine weight as a second measure of physiological E2 concentrations, which was reduced significantly in OVX females compared with controls (Fig. 1B; P < 0.05). The lowest dose of E2 restored uterine weight to the levels of ovary-intact female controls, whereas higher E2 doses resulted in supraphysiological increases in uterine weight (Fig. 1B; P < 0.05). We also administered E2 at the highest dose (600 μg/ml) to intact male mice, which resulted in a significant reduction in male sex organ weights (control 0.63 ± 0.02 g, E2 0.57 ± 0.02 g, P < 0.05). On the basis of these results, we chose to administer E2 (36 μg/ml) to HF-fed OVX female and male mice.
Fig. 1.
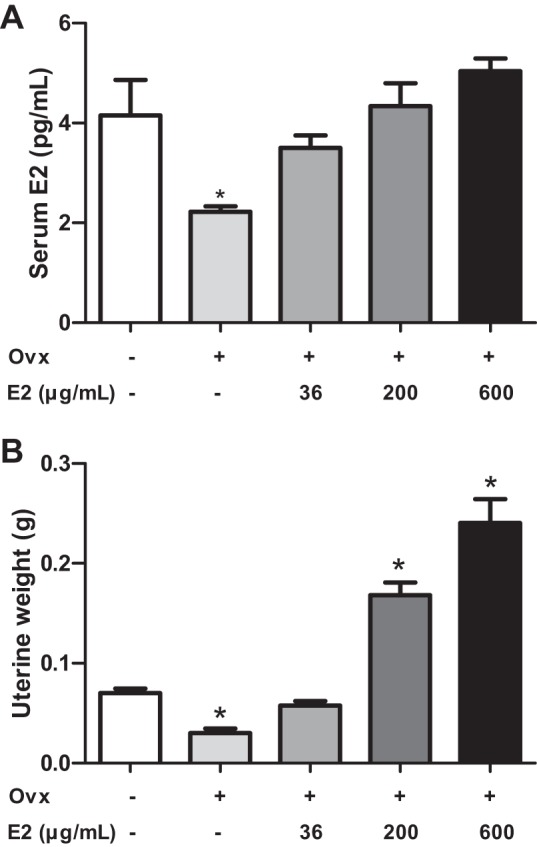
Effects of 17β-estradiol (E2) administration at various doses in ovariectomized (OVX) female mice fed standard mouse diet. A: serum E2 concentrations in intact vs. OVX females administered vehicle or different doses of E2 for 1 mo. B: uterine weights of mice in each group. Data are means ± SE from n = 5–10 mice/group. *P < 0.05 vs. intact female mice.
E2 administration reduces SBP during the light but not the dark cycle in Ace2+/+, but not in Ace2−/− OVX female mice, and has no effect on obesity-induced hypertension in male mice.
Female OVX mice exhibited significant increases in body weight with HF feeding, with no significant differences between genotypes (Fig. 2A; P > 0.05). E2 administration significantly decreased body weight (Fig. 2A; P < 0.05) and fat mass (Fig. 2B; P < 0.05) in Ace2+/+ and Ace2−/− female OVX mice, with no significant differences between genotypes. Moreover, E2 administration resulted in a significant increase in serum E2 concentrations in HF-fed OVX females of both genotypes (Table 1). Heart weight was significantly increased, whereas plasma renin concentrations were significantly decreased, in HF-fed Ace2−/− OVX females compared with Ace2+/+ controls, regardless of treatment group (Table 1; P < 0.05).
Fig. 2.
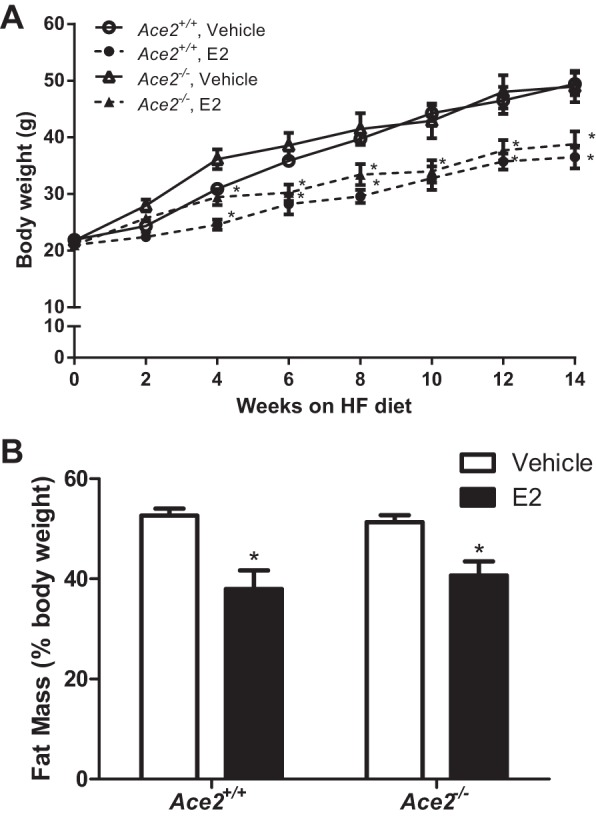
E2 administration significantly decreases body weight and fat mass of OVX HF-fed Ace2+/+ and Ace2−/− female mice. ACE2, angiotensin-converting enzyme 2. Body weights (A) and fat mass (%body weight; B) of HF-fed female Ace2+/+ or Ace2−/− mice administered vehicle or E2 (36 μg/ml). Data are means ± SE from n = 8–12 mice/group/genotype. *P < 0.05 vs. vehicle within genotype.
Table 1.
Characteristics and radiotelemetry parameters of HF-fed OVX female wild-type and ACE2-deficient mice administered vehicle or E2
| OVX Female Mice |
Ace2+/+ |
Ace2−/− |
||
|---|---|---|---|---|
| Vehicle | E2 | Vehicle | E2 | |
| Body weight, g | 49.4 ± 2.0 | 36.9 ± 2.0* | 48.9 ± 2.8 | 38.8 ± 2.3* |
| Total fat mass, g | 26 ± 1.2 | 15.1 ± 1.7* | 23.5 ± 2.8 | 16.8 ± 1.8* |
| Total lean mass, g | 21.5 ± 0.7 | 20.1 ± 0.5 | 20.9 ± 1.3 | 20.0 ± 0.4 |
| Heart weight, g | 0.16 ± 0.01 | 0.16 ± 0.01 | 0.18 ± 0.01† | 0.18 ± 0.01† |
| Uterine weight, g | 0.030 ± 0.004 | 0.055 ± 0.006* | 0.019 ± 0.001 | 0.058 ± 0.006* |
| Serum E2, pg/ml | 3.37 ± 0.14 | 4.75 ± 0.32* | 3.43 ± 0.48 | 4.03 ± 0.36* |
| Plasma ANG II, pg/ml | 464.0 ± 141.0 | 124.4 ± 49.3* | 158.4 ± 55.3† | 267.4 ± 143.1 |
| Plasma renin, pg/ml/30 min | 1.59 ± 0.37 | 2.08 ± 0.41 | 0.69 ± 0.24† | 0.97 ± 0.31† |
| Heart rate, bpm | 619 ± 11 | 665 ± 9* | 646 ± 15 | 661 ± 12 |
| Pulse pressure, mmHg | 37.3 ± 1.2 | 35.6 ± 1.0 | 43.2 ± 1.5 | 41.3 ± 1.3 |
| MAP, mmHg | ||||
| Light cycle | 113.1 ± 0.5 | 107.6 ± 1.5* | 110.3 ± 3.2 | 108.0 ± 1.6 |
| Dark cycle | 122.3 ± 1.3 | 121.2 ± 1.4 | 123.1 ± 4.6 | 121.3 ± 2.2 |
| 24 h | 118.1 ± 0.6 | 115.0 ± 1.5 | 116.7 ± 3.9 | 115.0 ± 1.9 |
| SBP, mmHg | ||||
| Light cycle | 130.1 ± 1.0 | 123.6 ± 1.1* | 130.0 ± 3.9 | 127.3 ± 2.0 |
| Dark cycle | 141.2 ± 2.1 | 139.1 ± 0.9 | 145.3 ± 5.5 | 142.6 ± 2.7 |
| 24 h | 136.0 ± 1.3 | 132.0 ± 1.1 | 137.7 ± 4.7 | 135.0 ± 2.3 |
| DBP, mmHg | ||||
| Light cycle | 94.6 ± 0.6 | 90.0 ± 1.9 | 89.2 ± 3.0 | 88.8 ± 1.5 |
| Dark cycle | 102.2 ± 0.7 | 101.7 ± 1.9 | 99.6 ± 3.9 | 98.4 ± 2.0 |
| 24 h | 98.7 ± 0.2 | 96.4 ± 2.0 | 94.4 ± 3.4 | 93.6 ± 1.7 |
| Activity, counts/min | ||||
| Light cycle | 3.3 ± 0.5 | 4.3 ± 0.4 | 4.2 ± 0.5 | 3.7 ± 0.3 |
| Dark cycle | 7.9 ± 0.8 | 14.0 ± 0.9* | 12.0 ± 2.6 | 11.5 ± 1.9 |
Data are means ± SE; n = 4–8 mice per treatment/genotype.
OVX, ovariectomized; E2, 17β-estradiol; ACE2, angiotensin-converting enzyme 2; MAP, mean arterial pressure; SBP, systolic BP; DBP, diastolic BP.
P < 0.05 vs. vehicle within genotype;
P < 0.05 vs. Ace2+/+ within treatment.
We quantified plasma ANG II concentrations, ACE2 activity, and ACE2 mRNA abundance in adipose tissue and kidneys from HF-fed female OVX Ace2+/+ and Ace2−/− mice. Plasma ANG II concentrations were significantly decreased in HF-fed Ace2−/− female OVX mice compared with Ace2+/+ controls (vehicle groups, Fig. 3A; P < 0.05). Moreover, E2 administration significantly decreased plasma ANG II concentrations in HF-fed Ace2+/+ but not in Ace2−/− OVX females (Fig. 3A; P < 0.05). Consistent with E2-mediated reductions in plasma ANG II concentrations in HF-fed OVX Ace2+/+ females, E2 administration significantly increased ACE2 mRNA abundance (Fig. 3B; P < 0.05) and ACE2 activity (Fig. 3C; P < 0.05) in adipose tissue from Ace2+/+ but not Ace2−/− females. In contrast to adipose tissue, E2 administration had no effect on ACE2 mRNA abundance (Fig. 4A) but significantly reduced ACE2 activity in kidneys from HF-fed Ace2+/+ OVX females (Fig. 4B; P < 0.05). There was reduced ACE2 mRNA abundance and activity in kidneys and adipose tissue from female ACE2-deficient mice (Figs. 3 and 4).
Fig. 3.
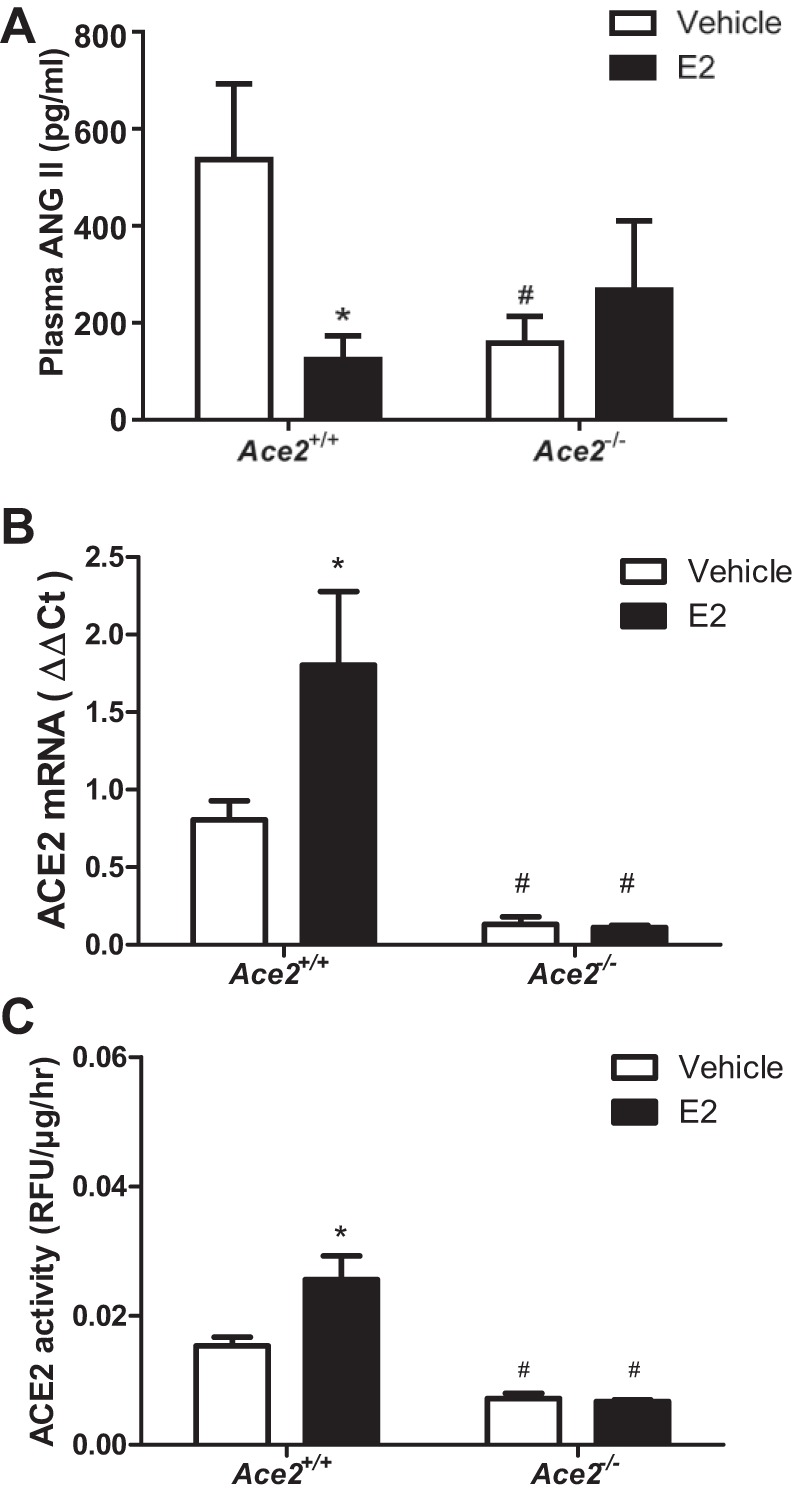
E2 administration reduces plasma ANG II concentrations and stimulates adipose ACE2 mRNA abundance and activity in HF-fed OVX Ace2+/+ but not in Ace2−/− female mice. A: plasma ANG II concentrations in HF-fed female Ace2+/+ and Ace2−/− OVX mice. Adipose ACE2 mRNA abundance (B) and ACE2 activity (C) in HF-fed female Ace2+/+ and Ace2−/− OVX mice. Data are mean ± SEM from n = 8–12 mice/treatment/genotype. *P < 0.05 compared with vehicle within genotype. #P < 0.05 compared with Ace2+/+ within treatment.
Fig. 4.
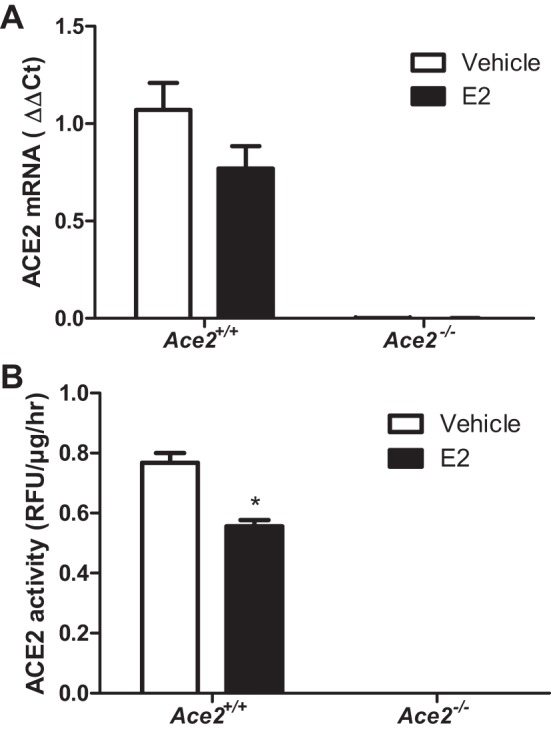
Effects of E2 administration on kidney ACE2 mRNA and enzymatic activity in HF-fed OVX female mice of each genotype. A: kidney ACE2 mRNA abundance in HF-fed OVX female mice. B: kidney ACE2 activity in HF-fed OVX female mice. Data are means ± SE from n = 5–10 mice/group/genotype. *P < 0.05 vs. vehicle within genotype.
E2 administration significantly decreased SBP during the light but not during the dark cycle in HF-fed Ace2+/+ OVX females (Fig. 5, A and B; P < 0.05). However, E2 administration had no effect on SBP (light or dark cycle) in Ace2−/− OVX females (Fig. 5, A and B; P > 0.05). When averaged over 24 h, E2 administration had no significant effect on SBP in HF-fed Ace2+/+ or Ace2−/− OVX females (Fig. 5C and Table 1; P > 0.05). In addition, E2 administration significantly reduced MAP during the light cycle but not the dark cycle in HF-fed Ace2+/+ but not in Ace2−/− OVX females (Table 1; P < 0.05). Interestingly, E2 administration significantly increased heart rate and physical activity (dark cycle) in HF-fed Ace2+/+ but not in Ace2−/− OVX females (Table 1; P < 0.05). In contrast to females, E2 administration had no effect on any parameter quantified in HF-fed Ace2+/y or Ace2−/y male mice (Figs. 6 and 7 and Table 2).
Fig. 5.
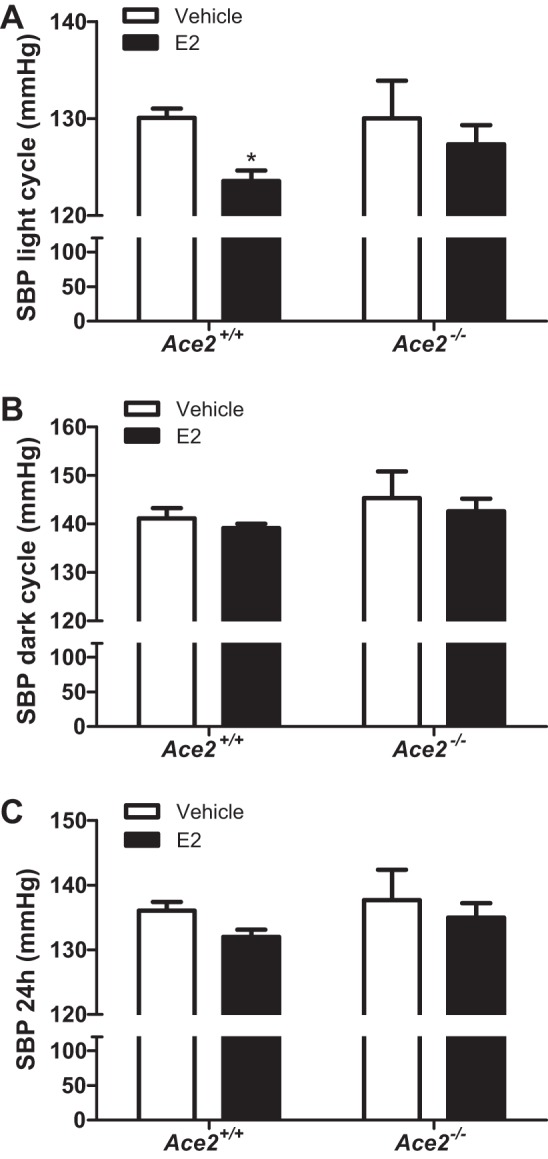
E2 administration lowers systolic blood pressure (SBP) of HF-fed Ace2+/+ but not Ace2−/− OVX female mice during the light but not the dark cycle. SBP during the light (A) or dark cycle (B) or 24-h average SBP (C) in HF-fed female Ace2+/+ and Ace2−/− OVX female mice. Data are means ± SE from n = 6–9 mice per group/genotype. *P < 0.05 vs. vehicle within genotype.
Fig. 6.

E2 administration has no effect on the development of obesity, tissue (adipose and kidney) ACE2 mRNA and enzymatic activity in HF-fed male mice of each genotype. Body weights (A), fat mass (%body weight; B), adipose ACE2 mRNA abundance (C), and adipose ACE2 activity (D) in HF-fed male Ace2+/y or Ace2−/y male mice administered vehicle or E2 (36 μg/ml). Kidney ACE2 mRNA abundance (E) and activity (F) in HF-fed male Ace2+/y or Ace2−/y mice administered vehicle or E2 (36 μg/ml). Data are means ± SE from n = 6–9 mice per group/genotype. *P < 0.05 vs. vehicle within genotype; #P < 0.05 vs. Ace2+/y within treatment.
Fig. 7.
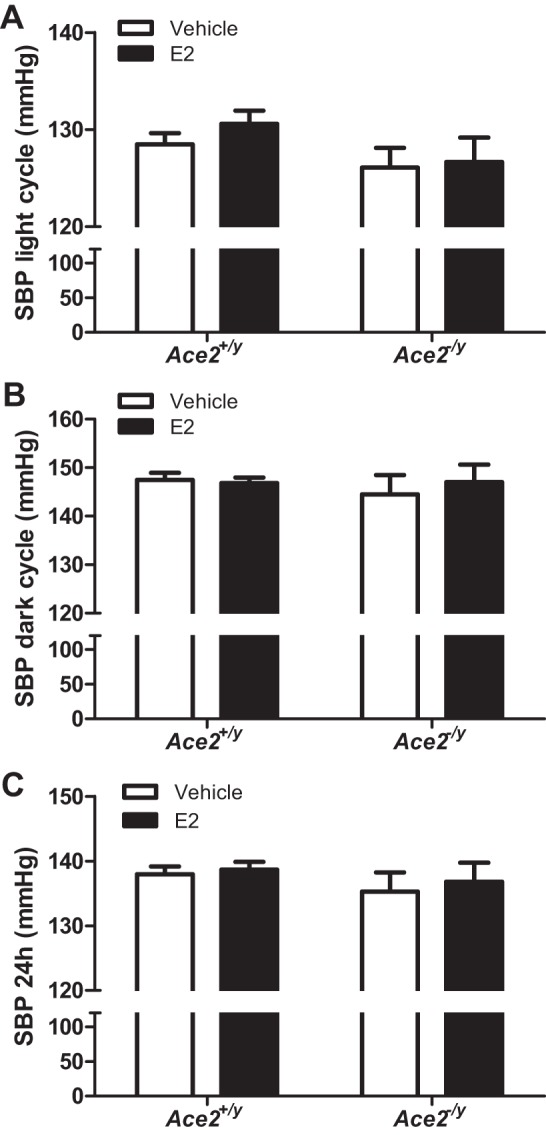
E2 administration had no effect on SBP of HF-fed male mice of either genotype. SBP during the light (A) or dark cycle (B) or 24-h average (C) in HF-fed male Ace2+/y and Ace2−/y mice. Data are means ± SE from n = 6–9 mice per group/genotype.
Table 2.
Characteristics and radiotelemetry parameters of HF-fed Ace2+/y and Ace2−/y male mice administered vehicle or E2
|
Ace2+/y |
Ace2−/y |
|||
|---|---|---|---|---|
| Male Mice | Vehicle | E2 | Vehicle | E2 |
| Body weight, g | 49.0 ± 2.0 | 48.8 ± 2.1 | 46.7 ± 1.5 | 45.2 ± 3.8 |
| Total fat mass, g | 19.4 ± 1.9 | 19.7 ± 1.2 | 17.9 ± 1.6 | 16.0 ± 2.5 |
| Total lean mass, g | 26.2 ± 0.7 | 25.5 ± 1.0 | 24.0 ± 0.7 | 24.8 ± 0.8 |
| Heart weight, g | 0.19 ± 0.01 | 0.18 ± 0.01 | 0.17 ± 0.01 | 0.17 ± 0.01 |
| Serum E2, pg/ml | 5.75 ± 0.78 | 5.72 ± 0.62 | 5.16 ± 0.61 | 5.19 ± 0.89 |
| Plasma ANG II, pg/ml | 117.4 ± 13.6 | 121.0 ± 23.0 | 112.3 ± 8.5 | 128.2 ± 18.6 |
| Plasma renin, pg/ml/30 min | 1.74 ± 0.51 | 1.45 ± 0.32 | 1.80 ± 0.40 | 1.25 ± 0.17 |
| Heart rate, bpm | 608 ± 6 | 611 ± 6 | 599 ± 6 | 612 ± 4 |
| Pulse pressure, mmHg | 36.5 ± 2.0 | 37.8 ± 0.9 | 37.2 ± 2.0 | 37.7 ± 2.3 |
| MAP, mmHg | ||||
| Light cycle | 112.0 ± 0.5 | 113.3 ± 1.2 | 109.5 ± 1.5 | 109.8 ± 1.8 |
| Dark cycle | 128.4 ± 1.2 | 127.3 ± 0.8 | 125.2 ± 2.9 | 127.2 ± 2.5 |
| 24 h | 120.2 ± 0.8 | 120.3 ± 1.0 | 117.3 ± 2.1 | 118.5 ± 2.0 |
| SBP, mmHg | ||||
| Light cycle | 128.5 ± 1.1 | 130.6 ± 1.4 | 126.1 ± 2.0 | 126.7 ± 2.5 |
| Dark cycle | 147.4 ± 1.4 | 146.8 ± 1.1 | 145.5 ± 3.9 | 147.0 ± 3.6 |
| 24 h | 138.0 ± 1.2 | 138.7 ± 1.2 | 135.3 ± 2.9 | 136.8 ± 3.0 |
| DBP, mmHg | ||||
| Light cycle | 94.5 ± 1.1 | 94.9 ± 1.0 | 91.5 ± 1.2 | 91.9 ± 1.5 |
| Dark cycle | 108.4 ± 1.9 | 106.8 ± 0.7 | 104.5 ± 2.0 | 106.3 ± 2.0 |
| 24 h | 101.5 ± 1.4 | 100.8 ± 0.8 | 98.0 ± 1.5 | 99.1 ± 1.7 |
| Activity, counts/min | ||||
| Light cycle | 2.0 ± 0.3 | 2.4 ± 0.4 | 1.9 ± 0.3 | 2.6 ± 0.6 |
| Dark cycle | 8.2 ± 0.8 | 8.6 ± 0.5 | 11.1 ± 2.3† | 12.8 ± 2.7† |
Data are means ± SE; n = 5–9 mice per treatment/genotype.
P < 0.05 vs. vehicle within genotype;
P < 0.05 vs. Ace2+/y within treatment group.
E2 promotes ACE2 mRNA abundance in 3T3-L1 adipocytes by eliciting ERα-mediated binding to the ACE2 promoter.
Administration of E2 to HF-fed OVX females promoted ACE2 mRNA abundance and activity in adipose tissue, but not in kidney, in a manner consistent with E2-mediated protection against obesity-hypertension. We quantified relative mRNA abundance of ERα and ERβ in adipose tissue from HF-fed Ace2+/+ OVX females administered vehicle. ERα mRNA abundance was >150-fold greater than that of ERβ in adipose tissue from obese females (Fig. 8A; P < 0.05). Moreover, E2 administration significantly increased ERα mRNA abundance in adipose tissue of HF-fed OVX females compared with vehicle controls in both genotypes (Fig. 8B; P < 0.05). We used 3T3-L1 adipocytes to define mechanisms for E2-mediated regulation of ACE2. As previously demonstrated (15), E2 (100 nM) stimulated ACE2 mRNA abundance in 3T3-L1 adipocytes, and these effects were abolished by an ERα antagonist (MPPD) (Fig. 9A; P < 0.05). To explore mechanisms for E2-mediated regulation of adipocyte ACE2, we performed a ChIP assay in 3T3-L1 adipocytes incubated with vehicle or E2 (100 nM) for 1 or 12 h followed by immunoprecipitation with an ERα antibody and RT-PCR. Following 12-h incubation with E2, there was a twofold enrichment of ERα binding to a putative estrogen response element (ERE, 5′-AGGTCAAACTCTCTG-3′) in the ACE2 promoter region (Fig. 9B), and PCR product size (201 bp) corresponded to the amplified promoter region of ACE2 (Fig. 9C).
Fig. 8.
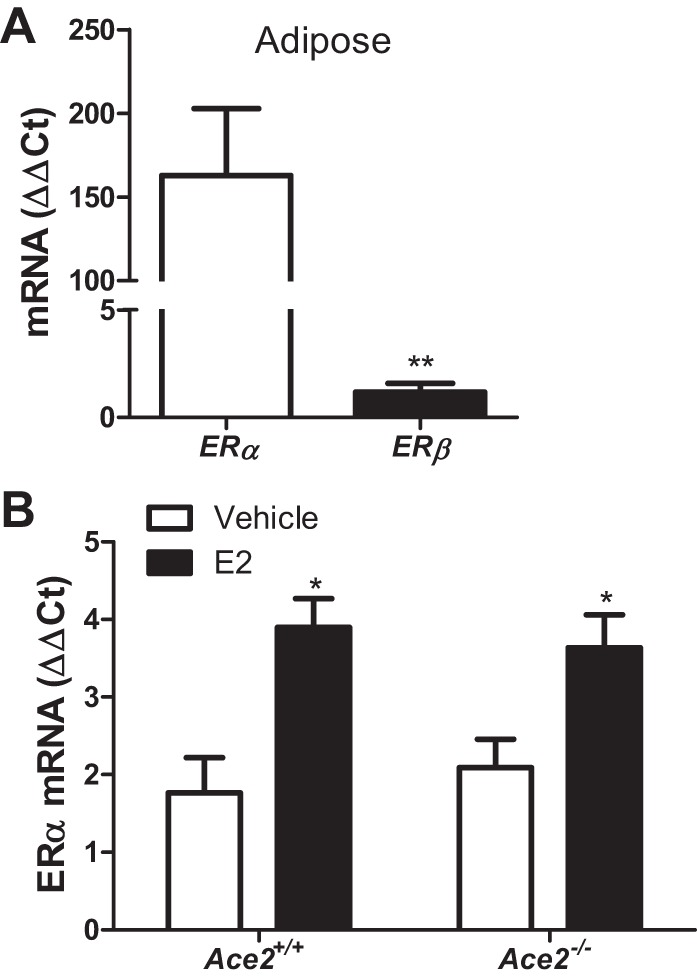
ERα is abundantly expressed in adipose tissue of female mice and increases upon E2 administration. A: mRNA abundance of ERα vs. ERβ in adipose tissue of HF-fed female Ace2+/+ OVX mice. B: ERα mRNA abundance in adipose tissue from HF-fed OVX female mice of each genotype administered vehicle or E2. Data are means ± SE from n = 5–10 mice per group/genotype. *P < 0.05 vs. vehicle within genotype, **P < 0.01 vs. ERα (by two-tailed t-test).
Fig. 9.

E2 binds to the ACE2 promoter to stimulate ACE2 expression in 3T3-L1 adipocytes. A: incubation of 3T3-L1 adipocytes with E2 results in an increase in ACE2 mRNA expression in 3T3L1 adipocytes that is abolished by an ERα antagonist (MPPD, 100 nM). B: incubation of 3T3-L1 adipocytes with E2 (12 h) as assessed by ChIP results in enrichment (twofold) of ERα to a putative estrogen response element (ERE) on the ACE2 promoter. C: PCR products (from B) illustrated by electrophoresis on a 2% agarose gel. Arrow denotes predicted ACE2 promoter product size (201 bp). Data (A, B) are means ± SE from n = 3 independent experiments. *P < 0.05 vs. vehicle; ##P < 0.01 vs. E2 treatment.
DISCUSSION
The results of this study demonstrate that E2 administration exhibits beneficial effects on the development of obesity-induced hypertension in female OVX mice through an ACE2-dependent mechanism. The major findings of the present study are 1) E2 administration stimulates adipose ACE2 mRNA abundance and activity in HF-fed OVX females; 2) E2 administration lowers plasma ANG II concentrations in HF-fed OVX females; 3) these effects most likely contribute to reductions in blood pressure of HF-fed OVX females administered E2; and 4) E2 administration reduces blood pressure during the light but not the dark cycle of HF-fed females. These effects were not observed in ACE2-deficient obese female mice, supporting an ACE2-dependent mechanism of E2 action. Moreover, beneficial effects of E2 administration at a dose that in preliminary studies physiologically restored serum E2 concentrations and uterine weights of OVX females to normal levels had no effect on blood pressure when administered to male obese mice. These results demonstrate that effects of ovariectomy to promote hypertension in obese female mice likely involve a lack of endogenous E2. Moreover, these results demonstrate that E2 exhibits protective effects to reduce SBP (light cycle) in females through an ACE2-dependent mechanism. Finally, effects of E2 to promote ACE2 mRNA abundance and activity were evident in adipose tissue, but not in kidneys, of obese female hypertensive OVX mice. In addition, E2 promoted ACE2 mRNA abundance through direct interaction at the ACE2 promoter in 3T3-L1 adipocytes. These results suggest that adipose tissue may be a target of E2 to stimulate ACE2 and protect females from obesity-hypertension.
Cross-sectional (11, 36), but not longitudinal, studies (39) indicate menopause as a hallmark of increased hypertension prevalence in adult females. Postmenopausal women not only have higher SBP (4–5 mmHg) than premenopausal women (37), but also the prevalence of hypertension and the rate of increase in postmenopausal women are higher than those observed in premenopausal women (6, 36). Observations performed during the menstrual cycle (10, 22) and early pregnancy (33) suggest an inverse correlation between endogenous E2 levels and female arterial blood pressure. It is generally well accepted that the prevalence of obesity also increases in postmenopausal women (36, 37), most likely due to reductions in local or systemic E2 concentrations. Our results extend previous findings related to the sexual dimorphism of hypertension to the setting of obesity and demonstrate that ovariectomy, a form of castration-induced menopause in mice, promotes the development of obesity-induced hypertension through an E2-dependent mechanism. An extension of these findings is that endogenous E2 may protect against the development of hypertension in premenopausal obese females, whereas a lack of E2 in postmenopausal females may contribute to the rising prevalence of both obesity and hypertension in this population.
Estrogen has been reported to exert beneficial effects to lower blood pressure through suppression of the expression of several components of the RAS necessary to synthesize or respond to ANG II (26, 32). In addition, studies have demonstrated that E2 positively regulates the expression of ACE2, an enzyme that also suppresses the RAS, and the effect of E2 may be attributed to ER action at two putative EREs on the ACE2 promoter (17). Indeed, we previously demonstrated that E2 promoted mRNA abundance of ACE2 in 3T3-L1 adipocytes (15). Results from this study extend previous findings by demonstrating that E2 stimulates ACE2 mRNA in 3T3-L1 adipocytes through ERα-mediated signaling, as demonstrated through blockade of E2-mediated regulation of ACE2 by an ERα antagonist. Moreover, ChIP results reveal the interaction of ERα with a putative ERE on the ACE2 promoter.
We (15) previously demonstrated that female mice do not exhibit an increase in blood pressure when fed a HF diet compared with low-fat-fed controls and that these effects were reversed by ovariectomy. Results from this study extend previous findings by demonstrating that effects of ovariectomy to promote an obesity-hypertension phenotype are at least partially reversed by E2 administration in wild-type but not in ACE2-deficient females. Interestingly, effects of E2 to stimulate ACE2 were present in adipose tissue but not in kidneys from obese females, suggesting, as has been previously described, that E2 regulates ACE2 in a tissue-specific manner (1). Moreover, since beneficial effects of E2 to lower blood pressure were absent in HF-fed ACE2-deficient females, these results demonstrate that E2 acts through an ACE2-dependent mechanism. It is unclear why the blood pressure lowering effect of E2 was seen during the light phase (resting phase), but not during the dark phase (active phase). However, E2 administration stimulated physical activity (25) and heart rate in Ace2+/+ obese OVX females during the dark but not during the light cycle. This stimulatory effect of E2 during the dark cycle may have masked its ability to decrease blood pressure. It is also unclear from the present studies if beneficial effects of E2 to promote adipose ACE2 expression are directly responsible for the observed reductions in plasma ANG II concentrations and blood pressure of female OVX mice. Future studies should address the role of adipocyte ACE2 in the development of obesity-induced hypertension in male and female mice.
An extension of these findings to the clinical setting would include potential administration of E2 to postmenopausal hypertensive females. However, the feasibility of using E2 therapeutically to treat hypertension in females (obese and/or postmenopausal) has been limited due to its carcinogenic properties as well as potential worsening cardiovascular outcomes reported in several controlled clinical trials (13, 30, 34). Indeed, several studies in humans and animal models assessing the effects of estrogen on blood pressure have yielded mixed results (3, 4, 18, 29, 31, 35, 40). Our results suggest that the dose of E2 may be a critical determinant of its efficacy to lower blood pressure. We used a dose of E2 that in preliminary studies restored physiological plasma E2 concentrations and uterine weights of OVX females, and that in obese OVX females resulted in a significant increase in plasma E2 concentrations. Moreover, this dose of E2 activated adipose ACE2 and reduced plasma ANG II concentrations in obese OVX females, most likely contributing to observed reductions in obesity-induced hypertension. An interesting finding of the present study was the lack of effect of E2 administration to protect male mice from obesity-induced hypertension. These results suggest that male mice do not express sufficient levels of ERs in relevant tissues to respond to exogenous E2 (e.g., adipose) or may require increased doses of E2 to lower blood pressure. We did not pursue administration of higher E2 doses to obese male mice, as this would not be a viable therapeutic option in humans due to a variety of ancillary properties of the sex hormone.
While administration of E2 to OVX HF-fed females reduced SBP (light cycle) in the present study, it also resulted in less weight gain, which could have contributed to the efficacy of E2 to prevent obesity-induced hypertension. Indeed, administration of E2 to HF-fed female mice has been previously reported to blunt the development of obesity (24, 42). In this study, E2-mediated reductions in body weight were observed in OVX HF-fed wild-type females as well as in females with whole body ACE2 deficiency. However, only the wild-type OVX females exhibited reductions in blood pressure following E2 administration. Thus, it is unlikely that E2-mediated reductions in body weight serve as the primary mechanism contributing to E2-mediated reductions in SBP in OVX females.
In conclusion, results from this study demonstrate that administration of E2 to OVX females reduced SBP during the light but not the dark cycle, blunting the development of obesity-induced hypertension. Moreover, as effects of E2 to activate adipose ACE2, reduce plasma ANG II concentrations, and decrease blood pressure were not evident in ACE2-deficient obese females, these results suggest that E2-mediated reductions in SBP are ACE2 dependent. Finally, our results demonstrate direct effects of E2 to promote ACE2 expression in adipocytes through interactions with ERE on the ACE2 promoter. These results suggest that low-dose E2 may afford protection against obesity-hypertension in postmenopausal females.
GRANTS
This work was supported by National Institutes of Health Grant HL-073085 (to L. A. Cassis), through Cores supported by National Institutes of Health Grant GM-103527 (L. A. Cassis), and from an American Heart Association Grant (14PRE20380163).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.W. and L.A.C. conception and design of research; Y.W. and V.L.E. performed experiments; Y.W. analyzed data; Y.W. and L.A.C. interpreted results of experiments; Y.W. prepared figures; Y.W. and L.A.C. drafted manuscript; Y.W., R.S., S.E.T., F.B.-Y., and L.A.C. edited and revised manuscript; Y.W., R.S., S.E.T., F.B.-Y., V.L.E., and L.A.C. approved final version of manuscript.
REFERENCES
- 1.Bharadwaj MS, Strawn WB, Groban L, Yamaleyeva LM, Chappell MC, Horta C, Atkins K, Firmes L, Gurley SB, Brosnihan KB. Angiotensin-converting enzyme 2 deficiency is associated with impaired gestational weight gain and fetal growth restriction. Hypertension 58: 852–858, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boustany CM, Bharadwaj K, Daugherty A, Brown DR, Randall DC, Cassis LA. Activation of the systemic and adipose renin-angiotensin system in rats with diet-induced obesity and hypertension. Am J Physiol Regul Integr Comp Physiol 287: R943–R949, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Cacciatore B, Paakkari I, Hasselblatt R, Nieminen MS, Toivonen J, Tikkanen MI, Ylikorkala O. Randomized comparison between orally and transdermally administered hormone replacement therapy regimens of long-term effects on 24-hour ambulatory blood pressure in postmenopausal women. Am J Obstet Gynecol 184: 904–909, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Campos C, Sartorio CL, Casali KR, Fernandes RO, Llesuy S, da Rosa Araujo AS, Bello-Klein A, Rigatto KV. Low-dose estrogen is as effective as high-dose treatment in rats with postmenopausal hypertension. J Cardiovasc Pharmacol 63: 144–151, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Compton DR, Sheng S, Carlson KE, Rebacz NA, Lee IY, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J Med Chem 47: 5872–5893, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Cutler JA, Sorlie PD, Wolz M, Thom T, Fields LE, Roccella EJ. Trends in hypertension prevalence, awareness, treatment, and control rates in United States adults between 1988–1994 and 1999–2004. Hypertension 52: 818–827, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation 110: 3849–3857, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Dieudonne MN, Leneveu MC, Giudicelli Y, Pecquery R. Evidence for functional estrogen receptors alpha and beta in human adipose cells: regional specificities and regulation by estrogens. Am J Physiol Cell Physiol 286: C655–C661, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Dobrian AD, Davies MJ, Prewitt RL, Lauterio TJ. Development of hypertension in a rat model of diet-induced obesity. Hypertension 35: 1009–1015, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Dunne FP, Barry DG, Ferriss JB, Grealy G, Murphy D. Changes in blood pressure during the normal menstrual cycle. Clin Sci 81: 515–518, 1991. [DOI] [PubMed] [Google Scholar]
- 11.Eferakeya AE, Imasuen JE. Relationship of menopause to serum cholesterol and arterial blood pressure in some Nigerian women. Public Health 100: 28–32, 1986. [DOI] [PubMed] [Google Scholar]
- 12.Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, Luft FC, Sharma AM. Weight loss and the renin-angiotensin-aldosterone system. Hypertension 45: 356–362, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, Hulley S, Herd A, Khan S, Newby LK, Waters D, Vittinghoff E, Wenger N, Group HR. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA 288: 49–57, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Gupte M, Boustany-Kari CM, Bharadwaj K, Police S, Thatcher S, Gong MC, English VL, Cassis LA. ACE2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol 295: R781–R788, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, Karounos M, Cassis LA. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol 32: 1392–1399, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol 206: 13–22, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 87: 677–682, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez I, Delgado JL, Diaz J, Quesada T, Teruel MJ, Llanos MC, Carbonell LF. 17β-Estradiol prevents oxidative stress and decreasesblood pressure in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol 279: R1599–R1605, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Ingberg E, Theodorsson A, Theodorsson E, Strom JO. Methods for long-term 17beta-estradiol administration to mice. Gen Comp Endocrinol 175: 188–193, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Ji H, Menini S, Zheng W, Pesce C, Wu X, Sandberg K. Role of angiotensin-converting enzyme 2 and angiotensin(1–7) in 17beta-oestradiol regulation of renal pathology in renal wrap hypertension in rats. Exp Physiol 93: 648–657, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Joyner J, Neves LA, Granger JP, Alexander BT, Merrill DC, Chappell MC, Ferrario CM, Davis WP, Brosnihan KB. Temporal-spatial expression of ANG-(1–7) and angiotensin-converting enzyme 2 in the kidney of normal and hypertensive pregnant rats. Am J Physiol Regul Integr Comp Physiol 293: R169–R177, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Karpanou EA, Vyssoulis GP, Georgoudi DG, Toutouza MG, Toutouzas PK. Ambulatory blood pressure changes in the menstrual cycle of hypertensive women. Significance of plasma renin activity values. Am J Hypertens 6: 654–659, 1993. [DOI] [PubMed] [Google Scholar]
- 23.Lundholm L, Moverare S, Steffensen KR, Nilsson M, Otsuki M, Ohlsson C, Gustafsson JA, Dahlman-Wright K. Gene expression profiling identifies liver X receptor alpha as an estrogen-regulated gene in mouse adipose tissue. J Mol Endocrinol 32: 879–892, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Matyskova R, Zelezna B, Maixnerova J, Koutova D, Haluzik M, Maletinska L. Estradiol supplementation helps overcome central leptin resistance of ovariectomized mice on a high fat diet. Horm Metab Res 42: 182–186, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Morgan MA, Pfaff DW. Effects of estrogen on activity and fear-related behaviors in mice. Horm Behav 40: 472–482, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Nickenig G, Baumer AT, Grohe C, Kahlert S, Strehlow K, Rosenkranz S, Stablein A, Beckers F, Smits JF, Daemen MJ, Vetter H, Bohm M. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation 97: 2197–2201, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Ong KL, Tso AW, Lam KS, Cheung BM. Gender difference in blood pressure control and cardiovascular risk factors in Americans with diagnosed hypertension. Hypertension 51: 1142–1148, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Lopez FR, Chedraui P, Gilbert JJ, Perez-Roncero G. Cardiovascular risk in menopausal women and prevalent related co-morbid conditions: facing the post-Women's Health Initiative era. Fertil Steril 92: 1171–1186, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Pripp U, Hall G, Csemiczky G, Eksborg S, Landgren BM, Schenck-Gustafsson K. A randomized trial on effects of hormone therapy on ambulatory blood pressure and lipoprotein levels in women with coronary artery disease. J Hypertens 17: 1379–1386, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, and Writing Group for the Women's Health Initiative I. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA 288: 321–333, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S, Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation 95: 39–45, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Shenoy V, Grobe JL, Qi Y, Ferreira AJ, Fraga-Silva RA, Collamat G, Bruce E, Katovich MJ. 17β-Estradiol modulates local cardiac renin-angiotensin system to prevent cardiac remodeling in the DOCA-salt model of hypertension in rats. Peptides 30: 2309–2315, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Siamopoulos KC, Papanikolaou S, Elisaf M, Theodorou J, Pappas H, Papanikolaou N. Ambulatory blood pressure monitoring in normotensive pregnant women. J Hum Hypertens 10, Suppl 3: S51–S54, 1996. [PubMed] [Google Scholar]
- 34.Simon JA, Hsia J, Cauley JA, Richards C, Harris F, Fong J, Barrett-Connor E, Hulley SB. Postmenopausal hormone therapy and risk of stroke: the Heart and Estrogen-Progestin Replacement Study (HERS). Circulation 103: 638–642, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Sorensen MB, Rasmussen V, Jensen G, Ottesen B. Temporal changes in clinic and ambulatory blood pressure during cyclic post-menopausal hormone replacement therapy. J Hypertens 18: 1387–1391, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Staessen J, Bulpitt CJ, Fagard R, Lijnen P, Amery A. The influence of menopause on blood pressure. J Hum Hypertens 3: 427–433, 1989. [PubMed] [Google Scholar]
- 37.Staessen JA, Ginocchio G, Thijs L, Fagard R. Conventional and ambulatory blood pressure and menopause in a prospective population study. J Hum Hypertens 11: 507–514, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS. Antagonists selective for estrogen receptor alpha. Endocrinology 143: 941–947, 2002. [DOI] [PubMed] [Google Scholar]
- 39.van Beresteyn EC, van t Hof MA, De Waard H. Contributions of ovarian failure and aging to blood pressure in normotensive perimenopausal women: a mixed longitudinal study. Am J Epidemiol 129: 947–955, 1989. [DOI] [PubMed] [Google Scholar]
- 40.Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK. Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am J Physiol Heart Circ Physiol 307: H191–H198, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, Cassis LA. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension 60: 1524–1530, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu L, Brown WC, Cai Q, Krust A, Chambon P, McGuinness OP, Stafford JM. Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway-selective insulin resistance. Diabetes 62: 424–434, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]