

Abstract
Katanin is a heterodimeric enzyme that severs and disassembles microtubules. While the p60 subunit has the enzyme activity, the p80 subunit regulates the p60 activity. The microtubule-severing activity of katanin plays an essential role in axonal growth. However, the mechanisms by which neuronal cells regulate the expression of katanin-p60 remains unknown. Here we showed that USP47 and C terminus of Hsp70-interacting protein (CHIP) antagonistically regulate the stability of katanin-p60 and thereby axonal growth. USP47 was identified as a katanin-p60-specific deubiquitinating enzyme for its stabilization. We also identified CHIP as a ubiquitin E3 ligase that promotes proteasome-mediated degradation of katanin-p60. Moreover, USP47 promoted axonal growth of cultured rat hippocampal neurons, whereas CHIP inhibited it. Significantly, treatment with basic fibroblast growth factor (bFGF), an inducer of axonal growth, increased the levels of USP47 and katanin-p60, but not CHIP. Consistently, bFGF treatment resulted in a marked decrease in the level of ubiquitinated katanin-p60 and thereby in the promotion of axonal growth. On the other hand, the level of USP47, but not CHIP, decreased concurrently with that of katanin-p60 as axons reached their target cells. These results indicate that USP47 plays a crucial role in the control of axonal growth during neuronal development by antagonizing CHIP-mediated katanin-p60 degradation.
Introduction
Microtubules are assembled at the centrosome, released by the action of katanin, and transported to neurites, where they form bundles for the growth of axons and dendrites (Vale, 2003; Baas and Buster, 2004; Hirokawa and Noda, 2008). Katanin consists of two different subunits, p60 and p80. Of these, the p60 subunit (katanin-p60) is a member of the AAA ATPase superfamily and uses ATP as energy for its microtubule-severing activity (McNally and Vale, 1993; Hartman et al., 1998), which plays a key role in the growth of axons and dendrites, and in the formation of axonal branches. Significantly, the cellular level of katanin-p60 in the brain changes during development. The katanin-p60 level is elevated during axonal growth but falls as axons reach their target cells (Karabay et al., 2004). In addition, the level of katanin-p60 is dramatically increased in certain cellular regions, such as at the sites of axonal branch formation and within growth cones (Qiang et al., 2010). However, the mechanisms by which the neuronal cells regulate the protein level and thereby the activity of katanin-p60 remains unknown.
Ubiquitin-dependent proteolysis plays an essential role in the regulation of various cellular processes, including cell proliferation, differentiation, and apoptosis (Hochstrasser, 1996; Hershko and Ciechanover, 1998). Ubiquitin is covalently attached to target proteins by a cascade enzyme system consisting of ubiquitin activating (E1), conjugating (E2), and ligating (E3) enzymes, which in most, but not all, cases, causes degradation of ubiquitinated proteins by proteasome (Hershko and Ciechanover, 1998). Reversal of ubiquitination catalyzed by deubiquitinating enzymes also plays important roles in the control of numerous biological pathways through stabilization of regulatory proteins (Wilkinson, 1997; Kim et al., 2003).
The C terminus of Hsp70-interacting protein (CHIP) is a ubiquitin E3 ligase that has a U-box domain (Hatakeyama and Nakayama, 2003). It promotes ubiquitination of target proteins and escorts them to proteasome for degradation (McDonough and Patterson, 2003). CHIP also has a tetratricopeptide repeat (TPR) domain, which is involved in the interaction with numerous proteins, including HSP70 and HSP90 (Lamb et al., 1995; Ballinger et al., 1999).
USP47 is a deubiquitinating enzyme whose function is yet to be determined. Murine USP47 is expressed in neural precursor cells in prenatal phase and in olfactory bulb, cerebellum, hippocampus, and dentate gyrus of adult brain (National Center for Biotechnology Information GENESAT database). Both Ubp64E in Drosophila and a hypothetical protein T05H10.1 in Caenorhabditis elegans (Baek et al., 1997), which is expressed exclusively in neurons, have high homology with human USP47 in their amino acid sequences. Notably, Drosophila Ubp64E was identified as an interacting partner of Drosophila katanin-p60 (Giot et al., 2003).
Here, we showed that USP47 and CHIP inversely regulate the stability of katanin-p60 and thereby katanin-p60-mediated axonal growth. Moreover, basic fibroblast growth factor (bFGF) increased the expression of USP47, but not CHIP. On the other hand, the level of USP47, but not CHIP, decreased in parallel with that of katanin-p60 as axons reached their target cells. These findings revealed that antagonistic actions of USP47 and CHIP play a key role in the control of katanin-p60-mediated axonal growth during neuronal development.
Materials and Methods
Plasmids and antibodies.
Human USP47 cDNA was provided by Dr. K. Fujita (Biological Resource Center, National Institute of Technology and Evaluation, Chiba, Japan). pcDNA4-HisMax-USP47 was constructed by inserting the cDNA into KpnI and XhoI sites of pcDNA4-HisMax vector (Invitrogen). Katanin-p60 cDNA obtained from Korean UniGene Information was inserted into BglII and XbaI site of pFlag-CMV2 vector and pcDNA3-HA vector (Invitrogen). Other mammalian expression vectors used were pcDNA4.1-katanin-p60-V5-His, pcDNA3-Myc-CHIP, and pcDNA-HSP90. Site-directed mutagenesis was performed as recommended by the manufacturer's instructions (Stratagene).
Antibodies used were as follows: mouse monoclonal anti-Flag M2 (Sigma); mouse monoclonal anti-HA (Roche); rabbit polyclonal anti-HA Y-11; mouse monoclonal HSP90 α/β; mouse monoclonal HSP70; rabbit polyclonal CHIP H-231; goat polyclonal anti-katanin-p60 M-13; mouse monoclonal anti-β-actin (Santa Cruz Biotechnology); mouse monoclonal anti-USP47 (Abnova); and rabbit polyclonal anti-USP47 antibodies (Bethyl Laboratories). Polyclonal anti-katanin-p60 antibody was generated by injecting purified His-katanin-p60 (244–434 aa sequence) to mice. The purified katanin-p60 was obtained by using Ni2+-NTA agarose column as recommended by the manufacturer's instruction (Qiagen). Horseradish peroxidase-conjugated anti-rabbit IgG, anti-mouse IgG, FITC- and TRITC-conjugated anti-mouse and anti-rabbit IgG were used (Jackson ImmunoResearch).
Primary neuron culture.
Cultured cortical and hippocampal neurons were prepared from the brains of Sprague Dawley rats of either sex at embryonic day 18 (E18) (Chang and De Camilli, 2001). Dissociated neurons were plated in MEM containing 0.6% glucose, 1 mm pyruvate, 2 mm l-glutamine, 10% FBS, 100 μg/ml penicillin, and 100 μg/ml streptomycin for 4 h, and maintained in Neurobasal medium supplemented with B27, 0.5 mm l-glutamine, and 1 μg/ml gentamycin. All reagents used for the primary cell culture were purchased from Invitrogen, unless otherwise indicated.
Cell culture, transfection, and immunoprecipitation.
Neuro2A (a neuroblastoma cell line), HEK293T, and NIH3T3 cells were cultured at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% FBS, 2 mm l-glutamine, and 25 units/ml penicillin and streptomycin. Transfections of plasmids were performed using Metafectene reagent (Biontex). Transient transfection of plasmids to hippocampal neurons was performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). For immunoprecipitation, cell lysates were prepared in 50 mm Tris-HCl, pH 7.4, containing 150 mm NaCl, 1 mm EDTA, 1 mm N-ethylmaleimide (NEM), 1 mm sodium vanadate, 1 mm NaF, 1 mm PMSF, and 1× protease inhibitor cocktail (Roche) in the presence or absence of 0.5% (v/v) Triton X-100. Cell lysates were incubated with appropriate antibodies for 1 h at 4°C and then with protein A-conjugated Sepharose for the next 1 h.
Ubiquitination and deubiquitination assay.
HA-Ub and katanin-p60-V5-His were expressed in HEK293T cells with Myc-CHIP or Flag-USP47. Cell lysates were prepared by sonication in a lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% SDS, 0.5% Triton X-100, 1 mm EDTA, 1 mm PMSF, 1 mm NEM, and 1× protease inhibitor cocktail), followed by 10-fold dilution with the lysis buffer lacking SDS. After incubating precipitates with NTA resins at 4°C for 2 h, they were washed and subjected to pulldown with NTA resins followed by immunoblot with anti-HA antibody (Oh et al., 2011).
Immunocytochemistry of primary cultured neurons.
Cells grown on 0.1 mg/ml poly-d-lysine-coated coverslips were transfected with appropriate plasmids. After culturing for appropriate periods, they were fixed by incubation with 3.7% paraformaldehyde in PBS for 10 min; washed three times with 0.1% Triton X-100; permeabilized with 0.5% Triton X-100 for 5 min; and treated with 0.1% Triton, 10% goat serum, 1% BSA, and 1% gelatin. After incubation for 1 h with appropriate antibodies in 0.1% Triton X-100 and 3% BSA, they were treated for 1 h with FITC-labeled goat anti-mouse IgG and TRITC-labeled goat anti-rabbit IgG in 0.1% Triton X-100 and 3% BSA. Cells were then observed under a fluorescence microscope (Axioplan II, Zeiss) equipped with a 20×, 0.4 numerical aperture (NA) LD-achroplan objective lens. Images were captured by using a cooled charge-coupled device digital camera (Axio-Cam, Zeiss; see Fig. 7). Cells were also observed under a confocal microscope (Fluoview FV1000, Olympus) equipped with a 20×, 0.75 NA plan-apo objective lens (see Fig. 6).
Figure 7.
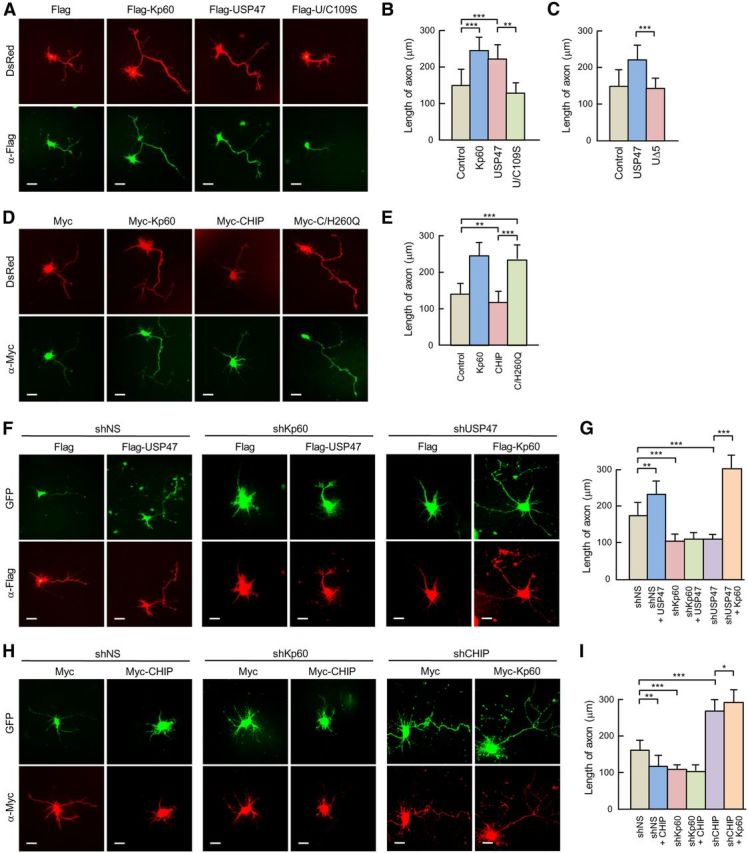
USP47 and CHIP inversely regulate axonal growth. A, DsRed was expressed in hippocampal neurons at 4 DIV with Flag only (by transfecting pFlag-CMV2), Flag-Kp60, Flag-USP47, or Flag-U/C109S. After culturing for 24 h, cells were fixed and stained with anti-Flag antibody (green). B, The length of axons was determined by using NeuronJ software. C, Flag, Flag-USP47, or Flag-UΔ5 was expressed in hippocampal neurons at 4 DIV. After culturing 24 h, cells were fixed and stained with anti-Flag antibody. The lengths of axons were determined as in B. Note that the stained cells were not shown. D, DsRed was expressed in hippocampal neurons with Myc (pcDNA3-Myc9), Myc-CHIP, or Myc-C/H260Q. After culturing for 24 h, cells were fixed and stained with anti-Myc antibody (green). E, The lengths of axons were determined as in B. F, GFP-shNS, GFP-shKp60, GFP-shUSP47, and GFP-shCHIP were expressed in hippocampal neurons at 3 DIV. After culturing for 24 h, cells were transfected with vectors expressing Flag, Flag-USP47, or Falg-Kp60. They were further cultured for the next 24 h. G, The length of axons was determined as in B. H, Experiments were performed as in F, except for the use of shCHIP and Myc-CHIP in place of shUSP47 and Flag-USP47, respectively. I, The lengths of axons were determined as in B. Statistical analysis was performed by using Student's t test. *p < 0.01; **p < 0.005; ***p < 0.001. Scale bars, 20 μm. Error bars indicate SD.
Figure 6.
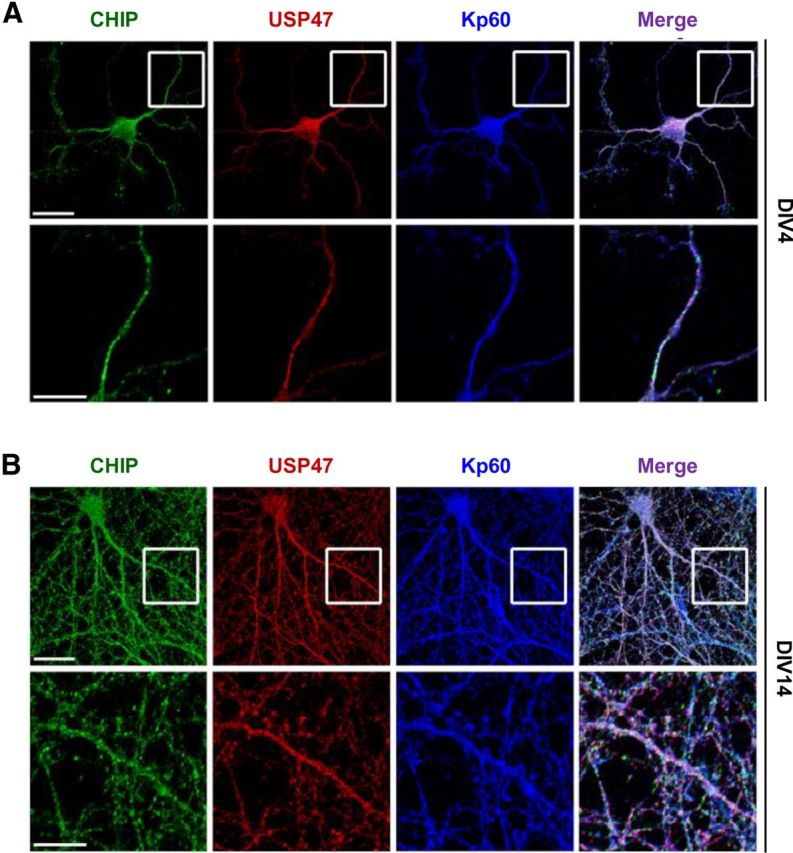
USP47 colocalizes with katanin-p60 and CHIP. A, B, Hippocampal neurons were cultured for 4 DIV (A) and 14 DIV (B) on slide glasses. Cells were then fixed; stained with anti-CHIP (green), anti-USP47 (red), and anti-katanin-p60 antibodies (blue); and observed under a fluorescent microscope. The bottom panels show the magnified views of the boxed regions of the top panels. Scale bars: top panels, 20 μm; bottom panels, 10 μm.
Quantitative analysis of neuronal morphology.
After image acquisition, images were cropped and processed using Adobe Photoshop. Image processing for morphometric analysis of neurons was performed using semimanual tracing with NeuronJ software (NIH). All morphometric experiments were repeated in at least three independent experiments with n > 12 experiments per condition in each experiment.
Results
USP47 interacts with katanin-p60
Since Drosophila Ubp64E, a homolog of human USP47, interacts with Drosophila katanin-p60 (Giot et al., 2003), we first examined whether human USP47 could interact with human katanin-p60. USP47 was expressed in HEK293T cells with and without katanin-p60. USP47 could be coimmunoprecipitated with katanin-p60 (Fig. 1A). Moreover, endogenous katanin-p60 could also be coimmunoprecipitated with endogenous USP47 in rat cortical neurons cultured for 7 d in vitro (DIV; Fig. 1B). These results indicate that katanin-p60 interacts with USP47 in mammalian cells.
Figure 1.

USP47 interacts with, deubiquitinates, and stabilizes katanin-p60. A, Flag-tagged katanin-p60 (Flag-Kp60) was expressed in HEK293T cells with HisMax-tagged USP47. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody or pulldown with NTA resins followed by immunoblot with anti-Xpress or anti-Flag antibody. Cell lysates were also directly probed with the same antibodies. The asterisk indicates IgG heavy chain. B, Lysates of cortical neurons at 7 DIV were immunoprecipitated with anti-USP47 (left) or anti-katanin-p60 antibody (right) followed by immunoblot with the same antibodies. C, HA-Ub and Kp60–V5-His were expressed in HEK293T cells with Flag-tagged USP47 or U/C109S. Cell lysates were subjected to pulldown with NTA resins followed by immunoblot with anti-HA or anti-V5 antibody. D, HA-Ub and Kp60–V5-His were expressed in HEK293T cells with Flag-tagged USP47, USP3, and USP10. Cell lysates were subjected to pulldown with NTA resins followed by immunoblot with anti-HA antibody. E, Neuro2A cells were transfected with shNS or shUSP47. After incubation for 48 h, cell lysates were subjected to immunoprecipitation with anti-Kp60 antibody followed by immunoblot with anti-Ub antibody. F, Flag-Kp60 was expressed in Neuro2A cells with HisMax-tagged USP47 or U/C109S. After culturing for 24 h, cells were treated with 100 μg/ml cycloheximide followed by immunoblot analysis. G, Neuro2A cells were transfected with shNS or shUSP47. After incubation for 48 h, they were treated with cycloheximide. H, Experiments were performed as in F, except expressing HisMax-UΔ5 in place of HisMax-U/C109S. I, Cortical neurons at 7 DIV were incubated with cycloheximide in the absence or presence of 10 μm MG132. Note that in A and C–E, cells were incubated with 10 μm MG132 for 4 h before the preparation of cell lysates.
To locate the katanin-p60-binding region within USP47, various deletions of USP47 were generated, expressed in HEK293T cells with katanin-p60, and subjected to immunoprecipitation analysis. Katanin-p60 bound to the middle region (amino acids 460–754) of USP47 (Fig. 2A). To determine the USP47-binding region within katanin-p60, deletions of katanin-p60 were generated and expressed in cells with USP47. USP47 bound to the AAA ATPase domain of katanin-p60 (Fig. 2B).
Figure 2.

Identification of the regions for binding between katanin-p60 and USP47. A, Deletions of USP47 (UΔ1–UΔ5) were tagged with HisMax to their N termini and expressed in HEK293T cells with and without Flag-Kp60 (top). Cell lysates were subjected to pulldown with NTA resins followed by immunoblot with anti-Flag or anti-Xpress antibody (bottom). B, Deletions of katanin-p60 (KΔ1-KΔ4) were tagged with V5-His to their C termini and expressed in cells with and without Flag-USP47 (top). Cell lysates were subjected to pulldown with Flag-M2-beads followed by immunoblot with anti-V5 or anti-Flag antibody (bottom). In the top panels, whether the proteins bind to each of the deletions or not was shown as “+” or “−,” and the binding regions were indicated by the arrows. The single and double asterisks indicate IgG heavy and light chains, respectively.
USP47 deubiquitinates and stabilizes katanin-p60
To determine whether USP47 can deubiquitinate katanin-p60, USP47 was expressed in HEK293T cells with katanin-p60 and ubiquitin. Ubiquitination of katanin-p60 could be reversed by USP47, but not by its catalytically inactive mutant (denoted U/C109S), in which the active site Cys109 was replaced by Ser (Fig. 1C). On the other hand, neither USP3 nor USP10 could remove ubiquitin from katanin-p60 (Fig. 1D), suggesting that USP47 specifically deubiquitinates katanin-p60. To confirm the ability of USP47 to deubiquitinate katanin-p60, a USP47-specific shRNA (shUSP47) was expressed in Neuro2A cells. Knockdown of endogenous USP47 by the shRNA led to a marked increase in the level of ubiquitinated katanin-p60, and this increase was further enhanced by treatment with MG132, a proteasome inhibitor (Fig. 1E). USP47 knockdown also resulted in a decrease in the level of katanin-p60 in cell lysates, and this decrease was abrogated by MG132 treatment. These results suggest that USP47 is responsible for deubiquitination and stabilization of katanin-p60.
To determine whether USP47-mediated deubiquitination indeed affects the stability of katanin-p60, USP47 or U/C109S was expressed in Neuro2A cells with katanin-p60 followed by incubation with cycloheximide, a protein synthesis inhibitor. USP47, but not U/C109S, could markedly increase the stability of katanin-p60 (Fig. 1F). Furthermore, the expression of shUSP47 significantly reduced the stability of endogenous katanin-p60, compared with that of a nonspecific shRNA (shNS; Fig. 1G). In addition, a mutant form of USP47 (UΔ5), in which the katanin-p60-binding region was deleted (Fig. 2A), no longer could stabilize katanin-p60 (Fig. 1H), indicating that the stability of katanin-p60 is at least in part controlled by USP47. Treatment with MG132 prevented the decrease in katanin-p60 stability, indicating that katanin-p60 is degraded by proteasome (Fig. 1I). Collectively, these results indicate that USP47 acts as a katanin-p60-specific deubiquitinating enzyme for its stabilization.
CHIP interacts with katanin-p60
To identify E3 ligases that are responsible for katanin-p60 ubiquitination, lysates prepared from cortical neurons at 7 DIV were immunoprecipitated with anti-katanin-p60 antibody. Precipitated proteins were subjected to SDS-PAGE followed by mass spectrometry (Fig. 3A). Identified katanin-p60-interacting proteins included STIP1, which contains a TPR domain (Fig. 3B), in addition to heat-shock proteins, such as HSP70 and HSP90, which are known to interact with the TPR domain. Since CHIP (also called STIP1 homologous and U-box-containing protein 1) also has a TPR domain and binds to HSP70 and HSP90, we suspected that CHIP is capable of interacting with katanin-p60. To test this possibility, CHIP and katanin-p60 were overexpressed in HEK293T cells. Reciprocal immunoprecipitation analysis showed that CHIP and katanin-p60 interact with each other (Fig. 4A). To confirm this finding, lysates from cortical neurons were subjected to immunoprecipitation. Endogenous CHIP could be coprecipitated with katanin-p60 as well as with HSP70 and HSP90 (Fig. 4B), suggesting that CHIP interacts with katanin-p60.
Figure 3.

Identification of katanin-p60-binding proteins. A, Lysates from cortical neurons at 7 DIV were subjected to immunoprecipitation with IgG or anti-katanin-p60 antibody. Precipitated proteins were subjected to SDS-PAGE followed by staining with Coomassie Brilliant Blue R250. They were also subjected to mass spectrometry for the identification of katanin-p60-binding proteins. The arrowheads indicate some of the proteins identified by the mass analysis. HC and LC indicate IgG heavy and light chains, respectively. M denotes size markers. B, List of proteins identified as katanin-p60-interacting proteins. Proteins bound to anti-katanin-p60 antibody were precipitated and analyzed by LC-MS/MS.
Figure 4.

CHIP interacts with, ubiquitinates, and destabilizes katanin-p60. A, Flag-Kp60 (top) or Kp60–V5-His (bottom) were expressed in HEK293T cells with Myc-CHIP. Cell lysates were subjected to immunoprecipitation with anti-Flag or anti-Myc antibody followed by immunoblot with anti-Myc or anti-Flag antibody, respectively. B, Lysates of cortical neurons at 7 DIV were immunoprecipitated with anti-CHIP (left) or anti-Kp60 antibody (right) followed by immunoblot with respective antibodies. C, Kp60–V5-His was expressed in HEK293T cells with Myc-tagged CHIP or C/K30A. Cell lysates were immunoprecipitated with anti-Myc antibody. D, Deletions of katanin-p60 (KΔ1–KΔ4), prepared as in Figure 2B, were expressed in HEK293T cells (top). Cell lysates were subjected to immunoprecipitation with anti-V5 antibody followed by immunoblot with anti-HSP90, anti-HSP70, or anti-V5 antibody (bottom). In the top panel, whether the proteins bind to each of the deletions or not was shown as “+” or “−,” and the binding region was indicated by the arrow. E, HA-Ub and Kp60–V5-His were expressed in HEK293T cells with Myc-tagged CHIP or C/H260Q. Cell lysates were pulled down with NTA resins. F, Neuro2A cells were transfected with shNS or shCHIP. After culturing for 48 h, cell lysates were immunoprecipitated with anti-Kp60 antibody. G, Kp60–V5-His was expressed in Neuro2A cells with Myc-tagged CHIP or C/H260Q. After culturing for 24 h, cells were treated with cycloheximide. H, Neuro2A cells were transfected with shNS or shCHIP. Cells were treated as in G. Note that in A, E, and F cells were incubated with 10 μm MG132 for 4 h before the preparation of cell lysates.
CHIP commonly transfers polyubiquitin chains to its target proteins through interaction with chaperone proteins, such as HSP90 and HSP70 (Ballinger et al., 1999). To determine whether the chaperone proteins are required for the interaction of CHIP with katanin-p60, we used a mutant form of CHIP (denoted C/K30A), in which Lys30 was replaced by Ala. It is known that C/K30A cannot bind the chaperone proteins (Jung et al., 2007). Immunoprecipitation analysis revealed that, unlike wild-type CHIP, C/K30A could interact with neither the chaperone proteins nor katanin-p60 (Fig. 4C), indicating that either HSP70 or HSP90 is required for the interaction of CHIP with katanin-p60. To locate the HSP90- and HSP70-binding region within katanin-p60, various deletions of katanin-p60 were generated, overexpressed in HEK293T cells, and subjected to immunoprecipitation analysis. Both HSP90 and HSP70 bound to the AAA domain of katanin-p60 (Fig. 4D). Collectively, these results indicate that HSP90 or HSP70 serves as an adaptor for the interaction between CHIP and katanin-p60.
CHIP ubiquitinates and destabilizes katanin-p60
To determine whether CHIP acts as a ubiquitin E3 ligase for katanin-p60, katanin-p60 was expressed in HEK293T cells with CHIP or its catalytically inactive mutant (denoted C/H260Q), in which the active site His260 was replaced by Gln. Expression of CHIP, but not C/H260Q, led to a significant increase in the level of ubiquitinated katanin-p60 (Fig. 4E), suggesting that CHIP is involved in the control of katanin-p60 stability. Of note was the finding that katanin-p60 was moderately ubiquitinated without CHIP overexpression and this modification could be strongly inhibited by C/H260Q, suggesting that the inactive mutant might act dominant negatively against endogenous CHIP. To determine the ability of endogenous CHIP to ubiquitinate katanin-p60, a CHIP-specific shRNA (shCHIP) was expressed in Neuro2A cells. Knock-down of CHIP led to a marked decrease in cellular levels of ubiquitinated katanin-p60 and this decrease was slightly compromised by MG132 treatment (Fig. 4F). CHIP knockdown also resulted in an increase in the level of katanin-p60 in cell lysates and this increase was further enhanced by MG132 treatment. These results suggest that CHIP is responsible for ubiquitination and destabilization of katanin-p60.
To determine whether CHIP-mediated ubiquitination indeed affects katanin-p60 stability, katanin-p60 was expressed in NIH3T3 cells with CHIP or C/H260Q followed by incubation with cycloheximide. Overexpression of CHIP significantly decreased katanin-p60 stability, but that of C/H260Q rather increased it (Fig. 4G), again suggesting that the mutant ligase acts dominant negatively against endogenous CHIP. Furthermore, the stability of endogenous katanin-p60 was increased by the expression of shCHIP, but not by that of shNS (Fig. 4H). These results indicate that CHIP could negatively regulate the stability of katanin-p60, in contrast to USP47, which could stabilize it.
Of note was the finding that both USP47 and HSP90 could bind to the AAA domain of katanin-p60 (Figs. 2B, 4D). Thus, it appeared possible that USP47 might compete with HSP90 and in turn with CHIP for binding to katanin-p60. To test this possibility, increasing amounts of USP47 or HSP90 were expressed in HEK293T cells with a constant amount of katanin-p60. Immunoprecipitation analysis revealed that the amount of USP47 coprecipitated with katanin-p60 is inversely correlated with that of CHIP as well as of HSP90 (Fig. 5A–D), suggesting that USP47 could compete with the chaperone proteins and in turn with CHIP for binding to katanin-p60. To determine whether the competitive binding of HSP90 to katanin-p60 against USP47 indeed influences katanin-p60 stability, Neuro2A cells transfected with pcDNA or pcDNA-HSP90 were incubated with cycloheximide. The stability of katanin-p60 was significantly reduced upon HSP90 expression (Fig. 5E). These results suggest that USP47 and CHIP could regulate the stability of katanin-p60 not only by their antagonistic catalytic functions, but also by competing for binding to the microtubule-severing enzyme.
Figure 5.

USP47 competes with HSP90 for binding to katanin-p60. A, Kp60–V5-His was expressed in HEK293T cells with and without increasing amounts of Flag-USP47. Cell lysates were subjected to immunoprecipitation with anti-V5 antibody followed by immunoblot with anti-HSP90, anti-V5, or anti-Flag antibody. B, The intensity of each protein band in A was scanned using a densitometer and was quantified using ImageJ software. The intensity of HSP90 or CHIP seen with an empty vector was expressed as 1.0, and the others were as its relative values. For USP47, the intensity seen upon transfection of 8 μg of USP47 cDNA was expressed as 1.0, and the others were expressed as its relative values. C, Myc-Kp60 was expressed with and without increasing amounts of HSP90–V5-His. Cell lysates were immunoprecipitated with anti-Myc antibody followed by immunoblot with anti-Myc, anti-V5, or anti-CHIP antibody. D, The intensity of each protein band in C was quantified as in B. The intensity of USP47 seen with an empty vector was expressed as 1.0, and the others were expressed as its relative values. For CHIP and HSP90, the intensity seen upon transfection of 8 μg of HSP90 cDNA was expressed as 1.0, and the others were expressed as its relative values. E, Neuro2A cells were transfected with pcDNA (Mock) or pcDNA-HSP90. After incubation for 24 h, cells were treated with cycloheximide. Note that in A and C, cells were incubated with 10 μm MG132 for 4 h before the preparation of cell lysates.
USP47 and CHIP antagonistically regulate axonal growth
Both CHIP and katanin-p60 are known to reside in most parts, if not all, of the neuron (Dickey et al., 2007; Yu et al., 2008; Ko et al., 2009). To determine the localization of USP47 and whether its localization may change during neuronal development, the rat hippocampal neurons at 4 DIV and 14 DIV were subjected to immunocytochemistry. Like katanin-p60, USP47 was found to reside in most parts of the neuron (Fig. 6). In addition, the localization of USP47 and CHIP remained unchanged during the culture periods examined, suggesting that the locality of both USP47 and CHIP does not change during neuronal development.
To examine whether USP47- and CHIP-mediated changes in katanin-p60 stability influence the growth of axons, we first overexpressed USP47, U/C109S, or katanin-p60 with DsRed in hippocampal neurons at 3 DIV. Immunocytochemical analysis revealed that USP47 overexpression, similar to that of katanin-p60, leads to a marked promotion of axonal growth, whereas U/C109S overexpression shows little or no effect (Fig. 7A). Quantitative analysis by using NeuronJ software confirmed the increase in axonal length by USP47, but not by U/C109S (Fig. 7B), suggesting that USP47 is involved in the control of axonal growth by stabilizing katanin-p60. To confirm this finding, the ability of USP47 to promote axonal growth was compared with that of UΔ5, a USP47 mutant that cannot bind to katanin-p60 (Fig. 2A). Unlike the wild-type enzyme, the UΔ5 mutant showed little or no effect on axonal growth (Fig. 7C), indicating that USP47 stimulates axonal growth by stabilizing katanin-p60. We next overexpressed CHIP, C/H260Q, or katanin-p60 with DsRed. In contrast to USP47, CHIP overexpression attenuated axonal growth (Fig. 7D,E). Remarkably, overexpression of C/H260Q promoted axonal growth nearly as well as that of katanin-p60, indicating that the inactive mutant acts as a dominant-negative inhibitor of endogenous CHIP. These results suggest that USP47 and CHIP are capable of altering axonal growth by antagonistically regulating the stability of katanin-p60.
To determine whether USP47 and CHIP are indeed involved in the control of axonal growth, USP47 and CHIP were expressed in hippocampal neurons with a katanin-p60-specific shRNA (shKp60) or shNS. Knockdown of katanin-p60 ablated the stimulatory effect of USP47 on axonal growth (Fig. 7F,G), whereas it showed little or no effect on CHIP-mediated inhibition of axonal growth (Fig. 7H,I). Furthermore, knockdown of USP47 by shUSP47 led to a decrease in axonal length, and this decrease could be reversed by katanin-p60 overexpression (Fig. 7F,G). On the other hand, depletion of CHIP by shCHIP resulted in a marked increase in axonal length, and this increase was slightly enhanced by katanin-p60 overexpression (Fig. 7H,I). Collectively, these results indicate that USP47 and CHIP inversely regulate axonal growth by controlling the stability of katanin-p60.
bFGF upregulates the expression of USP47 and katanin-p60
bFGF is a potent inducer of axonal growth (Thanos et al., 1989; Archer et al., 1999; Klimaschewski et al., 2004). It is also known to stabilize katanin-p60 (Qiang et al., 2010). Therefore, we examined whether bFGF might regulate the expression of USP47, CHIP, or both, and thereby the stability of katanin-p60. Treatment of cortical neurons with bFGF for increasing periods led to a gradual increase in the protein levels of USP47 and katanin-p60, but not of CHIP and HSP90 (Fig. 8A,B). On the other hand, other neurotrophins including EGF and BDNF showed little or no effect on the protein levels of USP47, HSP90, and katanin-p60 (data not shown). Furthermore, bFGF treatment caused a marked decrease in the level of ubiquitinated katanin-p60 (Fig. 8C), indicating that the ability of bFGF to induce the expression of USP47 is responsible for the stabilization of katanin-p60. We next examined the effect of USP47 knockdown on axonal growth of hippocampal neurons cultured with and without bFGF. As reported previously (Thanos et al., 1989; Archer et al., 1999; Klimaschewski et al., 2004), bFGF promoted axonal growth (Fig. 8D,E). Remarkably, the depletion of USP47 abrogated bFGF-mediated increase in axonal growth. These results demonstrate that bFGF-induced axonal growth is at least in part mediated by the increased expression of USP47.
Figure 8.

bFGF increases the expression of USP47 and katanin-p60. A, Cortical neurons at 3 DIV were treated with 60 ng/ml bFGF followed by immunoblot analysis. B, The band intensities seen in A were determined by using a densitometer. Similar data were obtained in three independent experiments. The open and closed symbols show without and with bFGF, respectively. C, Cortical neurons were incubated for 36 h with and without bFGF (60 ng/ml). Cell lysates were immunoprecipitated with anti-katanin-p60 antibody followed by immunoblot with anti-Ub or anti-Kp60 antibody. D, GFP-shNS and GFP-shUSP47 were expressed in hippocampal neurons at 3 DIV. After culturing them with and without bFGF (60 ng/ml) for 48 h, cells were fixed and observed. Statistical analysis was performed by using Student's t test. **p < 0.005; ***p < 0.001. Scale bars, 20 μm. E, The lengths of axons were determined as in Figure 7B. Error bars indicate SD. F, Cortical neurons were cultured and immunoblotted. G, The band intensities seen in F were determined by using a densitometer. Similar data were obtained in three independent experiments. H, A model for the control of axonal growth by USP47 and CHIP.
To determine whether the cellular levels of CHIP and USP47 change during neuronal differentiation, cortical neurons at 3–21 DIV were subjected to immunoblot analysis. During early period of the culture (up to ∼9 DIV), the levels of all proteins tested were unchanged (Fig. 8F,G). During later period of the culture (from ∼12 DIV); however, the level of USP47, but not of CHIP and HSP90, decreased concurrently with that of katanin-p60, suggesting that the decrease in USP47 level is responsible for the reduction in katanin-p60 level. Collectively, these results suggest that USP47 is involved in the control of katanin-p60 stability, and in turn the growth of axons during neuronal development, by antagonizing the ubiquitin E3 ligase function of CHIP.
Discussion
Based on the present findings, we propose a model for the control of axonal growth by antagonistic actions of the ubiquitin E3 ligase CHIP and the deubiquitinating enzyme USP47 (Fig. 8H). CHIP would ubiquitinate katanin-p60 for subsequent degradation by the 26S proteasome, leading to a decrease in the protein level of katanin-p60 in neuronal cells. On the other hand, USP47 would reverse the ubiquitination process by removing ubiquitin from katanin-p60, resulting in the stabilization and accumulation of katanin-p60. Thus, the changes in the level of USP47 relative to that of CHIP, and vice versa, would critically influence the stability and thereby the function of katanin-p60 in axonal growth. Consistently, CHIP overexpression or USP47 depletion in hippocampal neurons at 3 DIV led to a dramatic decrease in axonal growth, particularly its length, whereas USP47 overexpression or CHIP depletion showed an opposite effect, similar to katanin-p60 overexpression. Thus, the control of katanin-p60 stability by the antagonistic actions of CHIP and USP47 should be a critical mechanism for cells to keep katanin-p60 to appropriate levels at different stages of neuronal development.
bFGF, an inducer of axonal growth, upregulates the protein level of katanin, and the microtubule-severing activity of katanin-p60 critically contributes to the growth of axons and dendrites (Yu et al., 2005, 2008; Riano et al., 2009). However, it was unknown how bFGF increases the stability of katanin-p60. In this study, we showed that bFGF treatment leads to a marked increase in the protein level of USP47 with little or no effect on that of CHIP, resulting in a decrease in the level of ubiquitinated katanin-p60. Moreover, USP47 depletion was found to abrogate the stimulatory effect of bFGF on axonal growth. These findings provide a mechanical rationale for a bFGF-induced increase in the stability of katanin-p60 and in turn for katanin-mediated promotion of axonal growth during the early period of neuronal differentiation.
The cellular level of katanin-p60 in the brain is known to change during development. In addition, the level of katanin in cultured neurons is high when axons are avidly growing, but it declines as they stop growing upon reaching their targets (Karabay et al., 2004). However, the mechanism for the changes in the level of katanin during neuronal development was unknown. Here we showed that the protein levels of USP47, CHIP, and HSP90 as well as of katanin-p60 are kept constant during the early period of in vitro culture of hippocampal neurons (i.e., when axons are growing), suggesting that the maintenance of katanin-p60 to an optimal level by the balanced actions of CHIP and USP47 is required for axonal growth. On the other hand, the level of USP47 gradually decreased in parallel with that of katanin-p60 during the later periods of the culture (i.e., when axons reach their target cells). Since the levels of CHIP and HSP90 were not altered under the culture conditions, it is likely that overriding of CHIP level on that of USP47 is responsible for the decrease in katanin-p60 level. Thus, it appears that USP47 plays a crucial role in the control of axonal growth by antagonizing CHIP-mediated ubiquitination and destabilization of katanin-p60 during the early period of neuronal development.
So far, three different ubiquitin E3 ligases that control the stability of katanin-p60 have been identified. In C. elegans, katanin-p60 called Mei-1 is ubiquitinated by a Cul3/BTB E3 ligase complex (Pintard et al., 2003; Xu et al., 2003). In this system, Mel-26, a BTB domain-containing protein, serves as an adaptor protein that mediates the interaction of Mei-1 with Cul3 for its ubiquitination and subsequent degradation by proteasome. This E3 ligase-mediated degradation of Mei-1 is involved in the control of spindle formation during oocyte meiosis (Dow and Mains, 1998). In mammals, katanin-p60 is also ubiquitinated by a Cul3/BTB E3 ligase complex, in which the BTB domain-containing KLDHC5 (also called Cbt9) acts as the adaptor protein for the binding of katanin-p60 and Cul3 (Cummings et al., 2009). This E3 ligase-mediated katanin-p60 degradation is involved in the progression of mitosis most likely by regulating the spindle formation. An additional mammalian E3 ligase that ubiquitinates kapanin-p60 is the EDVP complex consisting of EDD, DDB1, and VPRBP (Maddika and Chen, 2009). Interestingly, DYRK2 is required not only for the assembly of the E3 complex, but also for the promotion of katanin-p60 ubiquitination by phosphorylating the substrate. Thus, like the Cul3/KLDHC5 E3 ligase, the DYRK2–EDVP complex appears to also play a crucial role in regulating normal mitotic progression. However, it remained unknown whether these E3 ligases also play a role in the control of katanin-p60 stability in neuronal cells, and thus in axonal growth.
Of note is a recent finding that the overexpression of DYRK2 inhibits the elongation of axon and dendritic branches (Slepak et al., 2012). This result suggests that the DYRK2–EDVP complex may also participate in ubiquitination and subsequent degradation of katanin-p60 in neuronal cells. In our study, knockdown of endogenous CHIP by shCHIP did not completely stabilize endogenous katanin-p60 (Fig. 4H), suggesting that additional ubiquitin E3 ligases may participate in the destabilization of katanin-p60. Together, these results suggest that both CHIP and DYRK2-EDVP may be involved in the control of katanin-p60 stability in neuronal cells, and thereby in axonal growth. However, it is also possible that CHIP and/or USP47 serve as downstream targets for phosphorylation by DYRK2 in the control of katanin-p60 stability.
Among deubiquitinating enzymes (USPs), relatively little is known about the biological function of USP47. The amino acid sequence of USP47 catalytic domain is significantly different from that of others. In addition, it was reported that USP47 is unable to cleave off ubiquitin from the ubiquitin-β-galactosidase fusion protein, when they were coexpressed in Escherichia coli (Quesada et al., 2004). However, it has recently been shown that USP47 is capable of deubiquitinating DNA polymerase-β, which plays a key role in base-excision repair, indicating that USP47 is involved in the control of cell viability in response to DNA damage (Parsons et al., 2011). In fact, DNA polymerase-β was the only substrate identified as a target of USP47. In this study, we identified katanin-p60 as a new target substrate of USP47 having an uncommon catalytic domain. Thus, determination of the x-ray crystallographic structure of USP47 and its complex with katanin-p60, which is under investigation, should provide the unique mechanism for the catalytic function of the protease.
Footnotes
This work was supported by National Research Foundation of Korea Grants NRF-2005-084-C00025 and M10533010001-05N3301 to C.H.C., and National Science Foundation Grant 0841245 to P.W.B., S.W.Y. and J.M.P. were the recipients of the BK21 Fellowship.
The authors declare no competing financial interests.
References
- Archer FR, Doherty P, Collins D, Bolsover SR. CAMs and FGF cause a local submembrane calcium signal promoting axon outgrowth without a rise in bulk calcium concentration. Eur J Neurosci. 1999;11:3565–3573. doi: 10.1046/j.1460-9568.1999.00773.x. [DOI] [PubMed] [Google Scholar]
- Baas PW, Buster DW. Slow axonal transport and the genesis of neuronal morphology. J Neurobiol. 2004;58:3–17. doi: 10.1002/neu.10281. [DOI] [PubMed] [Google Scholar]
- Baek SH, Choi KS, Yoo YJ, Cho JM, Baker RT, Tanaka K, Chung CH. Molecular cloning of a novel ubiquitin-specific protease, UBP41, with isopeptidase activity in chick skeletal muscle. J Biol Chem. 1997;272:25560–25565. doi: 10.1074/jbc.272.41.25560. [DOI] [PubMed] [Google Scholar]
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, De Camilli P. Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci. 2001;4:787–793. doi: 10.1038/90489. [DOI] [PubMed] [Google Scholar]
- Cummings CM, Bentley CA, Perdue SA, Baas PW, Singer JD. The Cul3/Klhdc5 E3 ligase regulates p60/katanin and is required for normal mitosis in mammalian cells. J Biol Chem. 2009;284:11663–11675. doi: 10.1074/jbc.M809374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Patterson C, Dickson D, Petrucelli L. Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol Med. 2007;13:32–38. doi: 10.1016/j.molmed.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Dow MR, Mains PE. Genetic and molecular characterization of the Caenorhabditis elegans gene, mel-26, a postmeiotic negative regulator of mei-1, a meiotic-specific spindle component. Genetics. 1998;150:119–128. doi: 10.1093/genetics/150.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, Vijayadamodar G, Pochart P, Machineni H, Welsh M, Kong Y, Zerhusen B, Malcolm R, Varrone Z, Collis A, Minto M, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Hartman JJ, Mahr J, McNally K, Okawa K, Iwamatsu A, Thomas S, Cheesman S, Heuser J, Vale RD, McNally FJ. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell. 1998;93:277–287. doi: 10.1016/S0092-8674(00)81578-0. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Nakayama KI. Ubiquitylation as a quality control system for intracellular proteins. J Biochem. 2003;134:1–8. doi: 10.1093/jb/mvg106. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y. Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev. 2008;88:1089–1118. doi: 10.1152/physrev.00023.2007. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Jung Y, Xu W, Kim H, Ha N, Neckers L. Curcumin-induced degradation of ErbB2: a role for the E3 ubiquitin ligase CHIP and the Michael reaction acceptor activity of curcumin. Biochim Biophys Acta. 2007;1773:383–390. doi: 10.1016/j.bbamcr.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Karabay A, Yu W, Solowska JM, Baird DH, Baas PW. Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J Neurosci. 2004;24:5778–5788. doi: 10.1523/JNEUROSCI.1382-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Park KC, Chung SS, Bang O, Chung CH. Deubiquitinating enzymes as cellular regulators. J Biochem. 2003;134:9–18. doi: 10.1093/jb/mvg107. [DOI] [PubMed] [Google Scholar]
- Klimaschewski L, Nindl W, Feurle J, Kavakebi P, Kostron H. Basic fibroblast growth factor isoforms promote axonal elongation and branching of adult sensory neurons in vitro. Neuroscience. 2004;126:347–353. doi: 10.1016/j.neuroscience.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI, Zhang YJ, Jiang H, Ross CA, Moore DJ, Patterson C, Petrucelli L, Dawson TM, Dawson VL. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci U S A. 2009;106:2897–2902. doi: 10.1073/pnas.0810123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci. 1995;20:257–259. doi: 10.1016/S0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- Maddika S, Chen J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat Cell Biol. 2009;11:409–419. doi: 10.1038/ncb1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:CALBTC>2.0.CO%3B2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally FJ, Vale RD. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 1993;75:419–429. doi: 10.1016/0092-8674(93)90377-3. [DOI] [PubMed] [Google Scholar]
- Oh KH, Yang SW, Park JM, Seol JH, Iemura S, Natsume T, Murata S, Tanaka K, Jeon YJ, Chung CH. Control of AIF-mediated cell death by antagonistic functions of CHIP ubiquitin E3 ligase and USP2 deubiquitinating enzyme. Cell Death Differ. 2011;18:1326–1336. doi: 10.1038/cdd.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JL, Dianova II, Khoronenkova SV, Edelmann MJ, Kessler BM, Dianov GL. USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase beta. Mol Cell. 2011;41:609–615. doi: 10.1016/j.molcel.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–316. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- Qiang L, Yu W, Liu M, Solowska JM, Baas PW. Basic fibroblast growth factor elicits formation of interstitial axonal branches via enhanced severing of microtubules. Mol Biol Cell. 2010;21:334–344. doi: 10.1091/mbc.E09-09-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Díaz-Perales A, Gutiérrez-Fernández A, Garabaya C, Cal S, López-Otín C. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem Biophys Res Commun. 2004;314:54–62. doi: 10.1016/j.bbrc.2003.12.050. [DOI] [PubMed] [Google Scholar]
- Riano E, Martignoni M, Mancuso G, Cartelli D, Crippa F, Toldo I, Siciliano G, Di Bella D, Taroni F, Bassi MT, Cappelletti G, Rugarli EI. Pleiotropic effects of spastin on neurite growth depending on expression levels. J Neurochem. 2009;108:1277–1288. doi: 10.1111/j.1471-4159.2009.05875.x. [DOI] [PubMed] [Google Scholar]
- Slepak TI, Salay LD, Lemmon VP, Bixby JL. Dyrk kinases regulate phosphorylation of doublecortin, cytoskeletal organization, and neuronal morphology. Cytoskeleton (Hoboken) 2012;69:514–527. doi: 10.1002/cm.21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos S, Bähr M, Barde YA, Vanselow J. Survival and axonal elongation of adult rat retinal ganglion cells. Eur J Neurosci. 1989;1:19–26. doi: 10.1111/j.1460-9568.1989.tb00770.x. [DOI] [PubMed] [Google Scholar]
- Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/S0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–321. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- Yu W, Solowska JM, Qiang L, Karabay A, Baird D, Baas PW. Regulation of microtubule severing by katanin subunits during neuronal development. J Neurosci. 2005;25:5573–5583. doi: 10.1523/JNEUROSCI.0834-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Qiang L, Solowska JM, Karabay A, Korulu S, Baas PW. The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol Biol Cell. 2008;19:1485–1498. doi: 10.1091/mbc.E07-09-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]