

Abstract
Previous studies have shown that administration of ferristatin II to rats is associated with decreased serum iron, reduced transferrin saturation, and increased hepatic hepcidin expression. BMP and IL-6 signaling act via Smad and Stat3 transcription factors, respectively, to increase expression of hepcidin, the master regulator of iron metabolism. In this study, we aimed to explore the underlying mechanism of ferristatin II action on hepcidin production. We found that ferristatin II greatly increased hepcidin expression both in vivo and in vitro. In the rat liver, ferristatin II treatment decreased expression of Smad downstream targets Smad7 and Id1 and increased expression of Stat3 downstream targets α-2-macroglobulin, α-1-acid glycoprotein, and C-reactive peptide. Ferristatin II also increased Stat3 phosphorylation in the rat liver without affecting serum or hepatic IL-6 levels. It is unclear whether the Stat3 activation observed in vivo is a cause or a consequence to hepcidin induction. Reporter gene expression studies demonstrated that ferristatin II synergized with BMP6 and IL-6 to enhance hepcidin expression in vitro. However, this synergy was not due to activation of either Smad or Stat3 signaling, raising the possibility that ferristatin II may activate a novel pathway for hepcidin regulation.
Keywords: iron transport, hemochromatosis, BMP6, IL-6
hepcidin is the master regulator of iron metabolism (17, 25, 26). Secreted by the liver, it controls systemic iron homeostasis through the rapid degradation of the iron exporter ferroportin, which in turn decreases intestinal iron absorption and the mobilization of iron stores (21, 35). Hepcidin expression is transcriptionally regulated by various stimuli including iron (26), hypoxia (22), erythropoiesis (22), and inflammation (20). Many diseases of iron overload arise upon deregulation of hepcidin production. Mutations in hemojuvelin (HJV), transferrin receptor 2 (TFR2), hepcidin (HAMP), and hemochromatosis (HFE) genes all diminish hepatic hepcidin production. Inappropriately low hepcidin results in chronic iron accumulation and eventually the development of hemochromatosis and other pathologies (16, 23, 27, 32).
Bone morphogenetic protein (BMP) signaling plays an essential role in hepcidin regulation (1, 33). BMPs are members of the TGF-β superfamily and are responsible for a variety of biological functions. BMP6 appears to be a primary regulator of hepcidin in vivo (1). BMP6 mRNA is detected in both hepatocytes and nonparenchymal liver cells and its expression is changed by low- and high-iron diets, correlating with liver iron stores, but does not appear to be affected by isolated changes in serum iron or transferrin saturation (8, 15, 36, 37). BMP6 binds its coreceptor hemojuvelin as well as type I and type II serine-threonine kinase receptors to form signaling complexes. These complexes have been reported to include HFE and TFR2 where they could act as iron sensors through interaction with transferrin (9). Activated BMP receptors phosphorylate Smad1, Smad5, and Smad8, which oligomerize with Smad4 and translocate to the nucleus to activate transcription. Consistent with this model, conditional liver-specific Smad4 knockout mice display decreased basal hepcidin production as well as diminished responses to iron loading (33). Most hepcidin regulators identified recently in an RNAi screen were dependent on BMP/Smad signaling (19).
Hepcidin levels increase during inflammatory conditions and its actions promote anemia of inflammation (20). Anemia of inflammation is characterized by a rapid drop in serum iron levels, decreased iron absorption, and iron accumulation in macrophages (8, 10, 14). Interleukin-6 (IL-6), a cytokine abundantly produced during inflammation, increases hepcidin production through Jak2/Stat3 activation (21, 34). However, the activation of Stat3 is not sufficient to induce hepcidin transcription alone but also requires Smad signaling. For example, disruption of BMP signaling either with Smad4 knockout in vivo or with BMP antagonists in vitro blunts hepcidin induction by IL-6 despite Stat3 activation (2, 33). The interaction between these signaling pathways is also evident in the ability of inflammatory mediators to induce activin B, another member of the TGF-β superfamily, which is capable of activating Smad1/5/8 (4).
In addition to BMP/Smad and IL-6/Stat3 pathways, other pathways have been reported to modulate hepcidin expression in a context-dependent manner. For example, endoplasmic reticulum (ER) stress can induce hepcidin production through the activation of the ER-stress-induced transcription factors CREBH or CHOP (24, 30). Estrogen is capable of inducing hepcidin through an estrogen response element in a BMP-dependent manner (13). On the other hand, testosterone and other growth factors have been shown to suppress the production of hepcidin (3, 11). Understanding the different regulatory pathways that control hepcidin is important for the development of potential therapeutics for iron disorders.
Through small molecule screening, our laboratory defined ferristatin I (NSC306711) and ferristatin II (NSC8679) as iron uptake inhibitors (5–7). These molecules were identified as structural homologs by using the Enhanced NCI Database Browser. It was determined that ferristatin I (5) and ferristatin II (7) interfered with transferrin-mediated iron delivery. Both were found to induce the degradation of transferrin receptor 1 (5, 6, 12). Mechanistically, the ferristatins promote internalization of transferrin receptor 1 by a lipid raft mechanism (7, 12). In this endocytic pathway, the receptor becomes degraded in lysosomal compartments, resulting in loss of iron uptake activity. Interestingly, degradation can be blocked by holo-transferrin and this effect requires ligand binding to the transferrin receptor 1 (7). This finding suggests that ferristatins may directly interact with the receptor, an idea that is supported by the observation that ferristatin II induces degradation of transferrin receptor-1 but not the close homolog transferrin receptor-2 (7). More recent experiments have shown that hepatic transferrin receptor 1 levels are reduced in rats injected with ferristatin II (7). These animals displayed hypoferremia associated with an unexpected induction of hepcidin, with reduced intestinal iron uptake and the failure to mobilize iron stores. We aimed in this study to delineate the pathways that regulate induction of hepcidin expression by ferristatin II using combined in vivo and in vitro approaches.
MATERIALS AND METHODS
Ferristatin II treatment.
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Harvard Medical Area Animal Care and Use Committee (Animal Experimentation Protocol AEP no. 04692). Sprague-Dawley rats (3-wk-old) were injected intravenously twice daily for 3 days with ferristatin II or vehicle control (saline). On day 4, rats were injected once, fasted for 6 h, and humanely killed for tissue collection. A total of amount of 40 mg/kg was injected over the course of the experiment. Ferristatin II was from Sigma-Aldrich (product no. C1144, lot no. 034K1279); HPLC analysis showed one major peak of ferristatin II that was visible across all wavelengths, with two minor contaminants. Systemic iron values and tissue nonheme iron levels were measured as previously described (7). IL-6 protein levels were measured by use of rat IL-6 ELISA (Signosis).
Cell culture conditions.
HepG2 cells stably transfected with a pGLA4.17 containing 3 kb of the human hepcidin promoter upstream of the firefly luciferase reporter gene [HepG2/Luc, a kind gift from Dr. Paula Fraenkel (38)] were maintained in α-MEM supplemented with 10% certified endotoxin-free FBS, and 50 μg/ml streptomycin at 37°C, 5% CO2. HeLa and McA-RH7777 cells were grown in DMEM containing 50 U/ml penicillin, 50 μg/ml streptomycin, l-glutamine, and 10% FBS.
In vitro ferristatin II treatment and luciferase assay.
HepG2/Luc cells were seeded in a white opaque 96-well plate (8 × 104 cells per well). Cells were allowed to recover in 10% FBS-containing medium, which was replaced with serum-free medium for 48 h prior to experiments; shorter times of serum deprivation did not show consistent ferristatin II effects. Cells were treated for 6 h with 1 μM ferristatin II (Sigma-Aldrich product no. C1144), 100 ng/ml BMP6 (R&D Systems), and/or 10 ng/ml IL-6 (R&D Systems) unless otherwise stated in the figure legends. Vehicle controls contained equivalent volumes of DMSO. To assay induction of the hepcidin promoter after treatments, the ONE-Glo Luciferase Assay System (Promega) was used according to the manufacturer's instructions. Protein quantification by Bradford assay confirmed that cellular content was uniform in wells under different experimental conditions used in our studies (134.77 ± 0.25 μg/well).
For ER stress experiments, HeLa cells were first washed three times with phosphate-buffered saline containing 1 mM MgCl2 and 0.1 mM CaCl2 (PBS++) and then washed once with serum-free medium. Cells were then treated with ferristatin II (50 μM), tunicamycin (1 μg/ml), or thapsigargin (1 μM).
Western blot analysis.
Cells were washed in ice-cold PBS and lysed in Nonidet P-40 (NP-40) lysis buffer (10 mM Tris pH 7.4, 40 mM HEPES pH 7.4, 1 mM EDTA, 5% glycerol, 1% NP-40) containing phosphatase inhibitors (10 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 50 mM sodium fluoride, 0.5 mM sodium orthovanadate) and protease inhibitors (Protease Inhibitor Set III, Calbiochem) for 60 min on ice. Lysates were cleared at 14,000 rpm for 30 min at 4°C, and 20–50 μg of the supernatant were loaded on 4–20% SDS-polyacrylamide gels. Livers from rats treated with or without ferristatin II were homogenized in NP-40 lysis buffer. Samples (50–100 μg) were electrophoresed on 10% SDS-polyacrylamide gels.
After transfer to nitrocellulose membranes, Western blots were blocked with 5% nonfat milk (non-phosphoproteins) or bovine serum albumin (for phosphoproteins) and immunoblotted for rabbit monoclonal anti-phosphoSmad1/5 (clone 41D10, phosphoSer463/465, 1:1,000, Cell Signaling Technology no. 9516), rabbit polyclonal anti-Smad1 (1:1,000, Cell Signaling Technology no. 9743), mouse monoclonal anti-phosphoStat3 (clone 3E2 phosphoTyr705, 1:1,000, Cell Signaling Technology no. 9138), rabbit monoclonal anti-Stat3 (clone 79D7, 1:2,000, Cell Signaling Technology no. 4904), mouse anti-actin (clone C4, 1:10,000, MP Biomedicals no. MAB1501), mouse anti-GAPDH (1:4,000, Abcam no. ab8245), rabbit anti-lamin B1 (1:1000, Abcam no. ab133741), or rabbit polyclonal anti-Grp94 (clone 9G10, 1:1,000, Enzo Life Sciences no. ADI-SPA-850). IRDye800- or IRDye680-conjugated donkey anti-mouse or donkey anti-rabbit (1:5,000, LI-COR) were used to detect immunoreactivity via an Odyssey Infrared Imaging System (LI-COR). Relative intensities of protein bands were normalized to actin or total nonphosphorylated proteins by using Odyssey version 2.1 software.
Quantitative real-time PCR.
Liver total RNA was isolated by using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. An additional step with phenol/chloroform/8-quinolinol was used to further purify the RNA. One to 5 μg RNA were reverse transcribed by using SuperScript III First-Strand Synthesis System (Invitrogen) with random hexamers and oligo(dT)20 primers. Gene expression was analyzed by quantitative real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) on an Applied Biosystems 7300 Real-Time PCR System. The cDNA was diluted 1:8 to 1:40 and 6 μl were used as template in a 15-μl reaction volume. The conditions used were 40 cycles 95°C for 90 s, 60°C for 10 s, and 72°C for 30 s (hepcidin, BMP6, Smad7); 40 cycles 95°C for 15 s, 55°C for 30 s, and 72°C [C-reactive protein (CRP), α-2-macroglobulin (α2M), α-1-acid glycoprotein (AGP)]; 40 cycles 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s (Id1, unspliced XBP1, spliced XBP1). Analysis was performed in triplicate for each sample. A dissociation curve analysis was performed to detect nonspecific products. 36B4 was used as a reference gene. All primers were used at a final concentration of 200 nM. The primers used were rat hepcidin 5′-TGACAGTGCGCTGCTGATG-3′ (forward), 5′-GGAATTCTTACAGCATTTACAGCAGA-3′ (reverse); rat unspliced XBP1 5′-TCCGCAGCACTCAGACTAC-3′ (forward), 5′-AGTTCCTCCAGATTAGCAGAC-3′ (reverse); rat spliced XBP1 5′-CTGAGTCCGAATCAGGTGCAG-3′ (forward), 5′-ATCCATGGGAAGATGTTCTGG-3′; rat α2M 5′-CCTAACTGTCAGGGCCAAATAC-3′ (forward), 5′-GCCAAGCCTCTACATTCTCTTC-3′ (reverse); rat AGP 5′-CCTAACTGTCAGGGCCAAATAC-3′ (forward), 5′-GCCAAGCCTCTACATTCTCTTC-3′ (reverse); rat CRP 5′-GCTTTTGGTCATGAAGACATG (forward); TCACATCAGCGTGGGCATAG (reverse); rat Smad7 5′-CCAACTGCAGACTGTCCAGA-3′ (forward), 5′-TTCTCCTCCCAGTATGCCAC-3′ (reverse); rat BMP6 5′-GACAGCAGAGTCGCAATCG-3′ (forward), 5′-AGCTCACGTAAAGCTCATGC-3′ (reverse); rat Id1 5′-GCGAGATCAGTGCCTTGG-3′ (forward), 5′-TTTTCCTCTTGCCTCCTGAA-3′ (reverse); human hepcidin 5′-CTGCAACCCCAGGACAGAG-3′ (forward), 5′-GGAATAAATAAGGAAGGGAGGGG-3′ (Reverse). 36B4 5′-AGATGCAGCAGATCCGCAT-3′ (forward) and 5′-GTTCTTGCCCATCAGCACC-3′ (reverse). Calculations for relative quantification were done using the comparative CT method (ΔΔCT).
Statistical analysis.
Reported values are expressed as means ± SE. Statistical significance was evaluated by two-tailed Student's t-test or ANOVA with multiple comparisons when appropriate. Differences were considered significant at P < 0.05.
RESULTS
Previous studies from our laboratory demonstrated hepcidin induction in rats injected with ferristatin II (7). We aimed to confirm this effect and explore its underlying mechanism. In the present study, rats injected with ferristatin II had a fourfold increase in hepatic hepcidin mRNA levels compared with saline-injected controls (Fig. 1). Treatment with ferristatin II was associated with a significant decrease in serum iron and transferrin saturation as well as increased splenic iron levels (Table 1). These changes in systemic iron parameters are consistent with hepcidin upregulation and the subsequent decrease in iron mobilization and absorption.
Fig. 1.

Ferristatin II induces hepcidin expression in vivo. Transcript levels of hepcidin, Smad 1/5/8 downstream targets (Smad7, BMP6, and Id1), and the acute-phase reactants α-2-macroglobulin (α2M), α-1-acid glycoprotein (AGP), and C-reactive protein (CRP) in rats treated with or without ferristatin II. The fold change in transcript level compared with control was calculated by the comparative ΔΔCt method with 36B4 as the reference gene; n = 4, *P < 0.01.
Table 1.
Systemic iron parameters in animals treated with or without ferristatin II
| Control (Vehicle) | Ferristatin II (40 mg) | P Value | |
|---|---|---|---|
| Body weight, g | 69.2 ± 1.7 | 65.0 ± 0.8 | 0.0642 |
| Hematocrit, % | 34.0 ± 2.3 | 32.1 ± 0.3 | 0.4457 |
| Serum iron, μg/ml | 3.0 ± 0.4 | 0.9 ± 0.4 | 0.0093 |
| Unsaturated iron binding capacity, μg/ml | 1.2 ± 0.2 | 2.8 ± 0.2 | 0.0016 |
| Total iron binding capacity, μg/ml | 4.2 ± 0.3 | 3.7 ± 0.3 | 0.2576 |
| Transferrin saturation, % | 71.2 ± 5.7 | 23.1 ± 8.1 | 0.0028 |
| Liver nonheme iron, μg/g | 59.2 ± 7.6 | 91.3 ± 14.9 | 0.1036 |
| Spleen nonheme iron, μg/g | 30.7 ± 4.5 | 64.8 ± 9.5 | 0.0176 |
Although many different cell types produce hepcidin, it has been shown that the liver is the physiologically relevant site of synthesis (39). To determine signaling pathways that might be activated by ferristatin II to induce hepcidin expression in the liver, we first examined hepatic transcript levels of Smad downstream targets, Smad7 and Id1, by qPCR (15). Gene expression of both factors was downregulated. We also determined BMP6 mRNA levels, since its transcription is correlated to liver iron. Like Smad7 and Id1, BMP6 transcript levels were decreased in livers from rats treated with ferristatin II (Fig. 1). Thus hepatic Smad signaling was not upregulated in ferristatin II-treated animals, and expression of the major activator of this pathway, BMP6, was downregulated. The second major pathway that promotes hepcidin transcription is through Jak2/Stat3 activation and the inflammatory response (20, 31, 34). Hepatic transcript levels of the rat Stat3 targets α2M, AGP, and CRP were all significantly increased in livers of the ferristatin II-treated rats (Fig. 1), indicating that this pathway was upregulated. Finally, since it has been reported that ER stress in iron-loaded liver could induce hepcidin through CREBH or CHOP (24, 30), we also examined whether ferristatin II might induce the unfolded protein response (UPR). XBP1 splicing, an indicator of ER stress and the UPR, was unchanged in livers from ferristatin II-treated rats compared with controls; moreover, cells treated with ferristatin II did not induce the folding chaperone Grp94, unlike the ER stress inducers tunicamycin and thapsigargin, which were used as controls (Fig. 2).
Fig. 2.
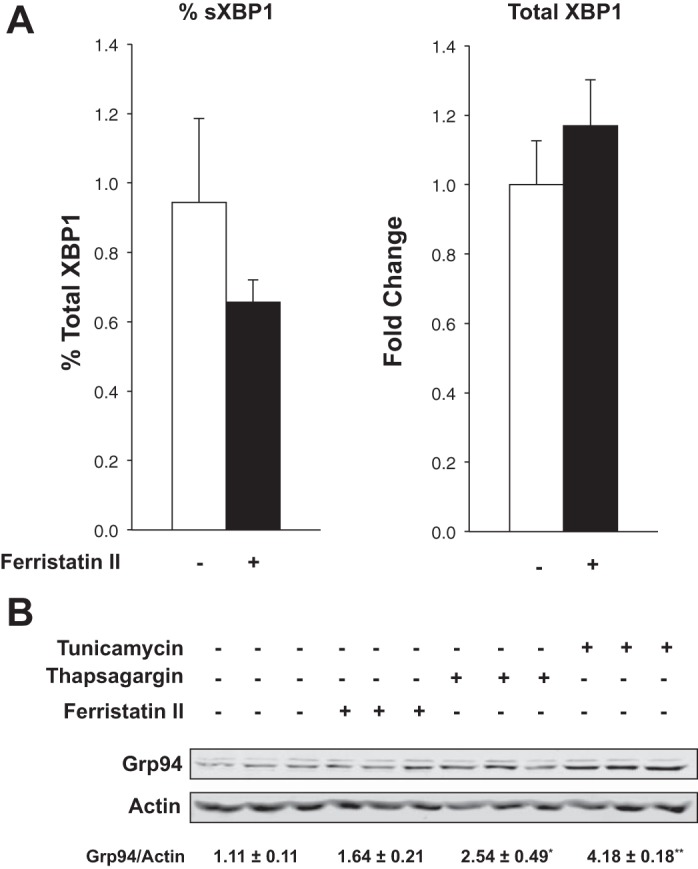
Ferristatin II does not induce endoplasmic reticulum (ER) stress in vivo or in vitro. A: XBP splicing is not induced in vivo. Levels of spliced, unspliced, and total XBP1 message in livers from rats treated with or without ferristatin II were determined by qPCR. Shown is the % spliced XBP1 (sXBP1) determined on the basis of total XBP1 and the fold difference in total determined for control and treated rats. B: Grp94 is not induced in vitro. HeLa cells were treated overnight with thapsigargin (1 μM) or tunicamycin (1 μg/ml) or treated 4 h with ferristatin II (50 μM). Cell lysates were immunoblotted to determine levels of Grp94. Actin was used as a loading control. Shown below are mean density ratios of Grp94/actin ± SE compared with vehicle controls (n = 3, *P < 0.05 and **P = 0.0001 determined by 2-tailed Student's t-test).
Since the transcriptional changes noted in Fig. 1 should reflect signal transduction pathways mediated through phosphorylation of Smads and Stats, we performed immunoblotting experiments. In our hands, Western blot analysis of Smad proteins was limited by available reagents, which did not reliably detect these factors in rat liver. Nonetheless, Western blots confirmed increased phosphorylation of Stat3 in liver lysates from ferristatin II-treated rats (Fig. 3), consistent with the induction of target genes associated with this pathway (Fig. 1). To investigate whether the phosphorylation of Stat3 reflected increased levels of the cytokine IL-6, which is known to activate the Jak/Stat3 pathway, we measured IL-6 protein in sera and liver lysates from treated and control rats. Ferristatin II treatments did not increase serum or liver IL-6 levels relative to saline-injected controls (Fig. 3).
Fig. 3.
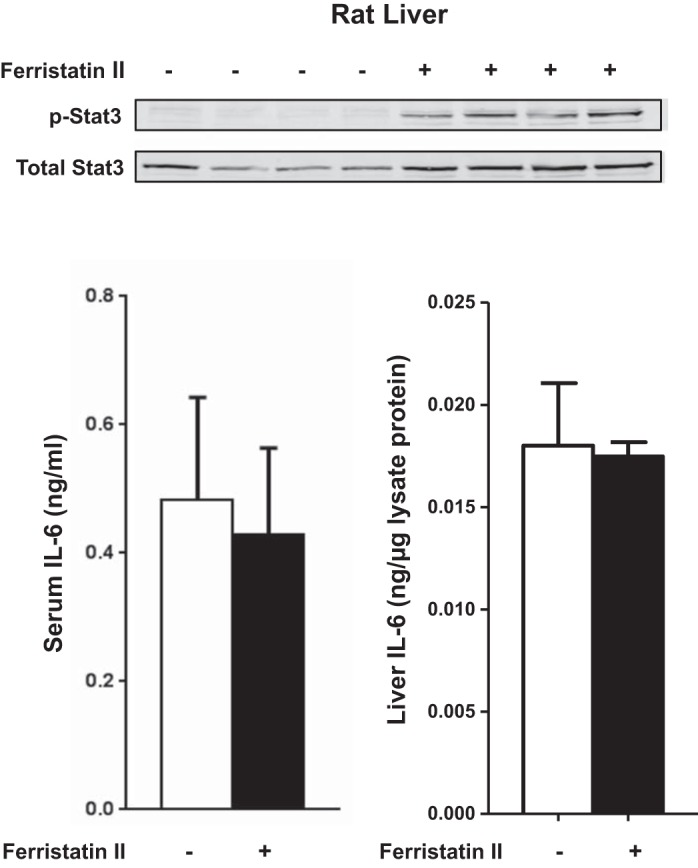
Ferristatin II increases Stat3 phosphorylation in vivo but liver and serum IL-6 protein levels do not change. Immunoblots of liver tissue lysates were probed for phospho-Stat3 and total Stat3. Serum and liver IL-6 levels were measured by ELISA.
We next turned to an in vitro model system using the human hepatocellular carcinoma cell line HepG2/Luc to further explore hepatic responses to ferristatin II. These cells are stably transfected with a construct containing 3 kb of the human hepcidin promoter upstream of the firefly luciferase reporter (38). When HepG2/Luc cells were treated with ferristatin II alone, the hepcidin promoter was not activated (Fig. 4). Since Smad activation is an essential component of hepcidin transcription (33), we also treated HepG2/Luc cells with BMP6 to induce hepcidin expression. Treatment with BMP6 alone increased luciferase activity greater than 10-fold compared with controls, but when combined with ferristatin II, an even greater increase was observed (>30-fold) (Fig. 4). Control experiments confirmed that comparable effects were observed on endogenous hepcidin transcript levels by qPCR methods (Fig. 4). These combined data demonstrate that ferristatin II synergizes with BMP6 to activate the hepcidin promoter.
Fig. 4.

Ferristatin II induces hepcidin expression in vitro. Hepcidin induction as measured by luciferase assay (A) or quantitative real-time RT-PCR (B) in HepG2/Luc cells treated with ferristatin II (1 μM) with or without BMP6 (100 ng/ml). Cells were serum starved for 48 h before the 6-h incubation, and luciferase activity was measured or RNA collected; n = 3 (left) and n = 4 (right) biological replicates, **P < 0.01.
We also studied activation of the Stat pathway in vitro. IL-6 did not induce luciferase activity, but when it was combined with BMP6 a greater than 200-fold increase in luciferase activity was observed in the HepG2/Luc cells (Fig. 5). These changes were not due to differences in cell proliferation since total protein levels were unchanged between various treatments. These results are consistent with others, demonstrating that Smad signaling is required for Stat3 activation of hepcidin expression (19, 33). As expected from results presented in Fig. 4, ferristatin II alone did not enhance luciferase activity but did display weak synergy with IL-6 to activate the hepcidin promoter (Fig. 5). Ferristatin II together with BMP6 and IL-6 produced a greater than 400-fold increase in luciferase activity (Fig. 5). The reporter gene experiments therefore indicate that ferristatin II not only influences BMP6-dependent hepcidin expression but also can enhance the effect of IL-6, which is also BMP6 dependent.
Fig. 5.
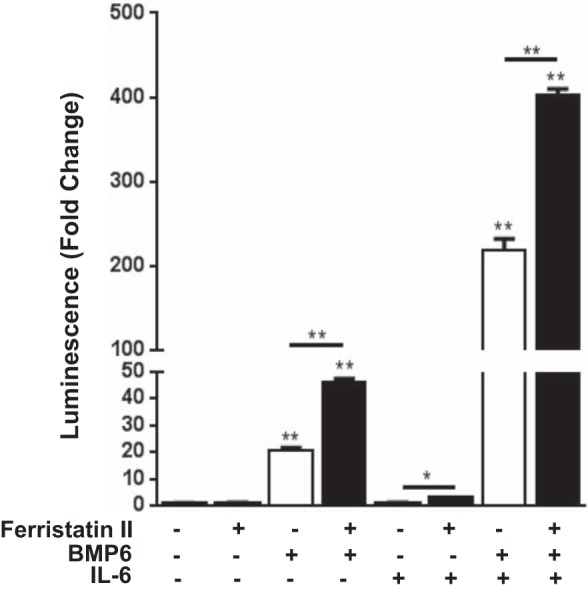
Ferristatin II synergizes with BMP6 and IL-6 to activate the hepcidin promoter. HepG2/Luc cells were serum starved for 48 h before incubation with or without ferristatin II (1 μM), BMP6 (100 ng/ml), and IL-6 (10 ng/ml) for 6 h; n = 4, *P < 0.05; **P < 0.01.
To determine whether expression of the reporter gene from the hepcidin promoter reflected changes in phosphorylation of Smads or Stat3, immunoblotting experiments were carried out. In contrast to the increased Stat3 phosphorylation observed in lysates from ferristatin II-treated rat livers (Fig. 3), phosphorylation of Stat3 in HepG2/Luc cells was unaffected by ferristatin II (Fig. 6). Notably, ferristatin II did not influence Stat3 phosphorylation induced by IL-6 with or without BMP6. Using anti-human Smad antibodies, we were able to detect phosphorylated Smad1/5 and total Smad1 in Western blots of HepG2/Luc cells (Fig. 6). Although BMP6-stimulated phosphorylation of Smad1/5 was observed, the extent of phosphorylation was unaffected by IL-6 or ferristatin II. Thus neither Stat3 nor Smad1/5 total phosphorylation was altered in hepatocytes treated with ferristatin II in vitro.
Fig. 6.
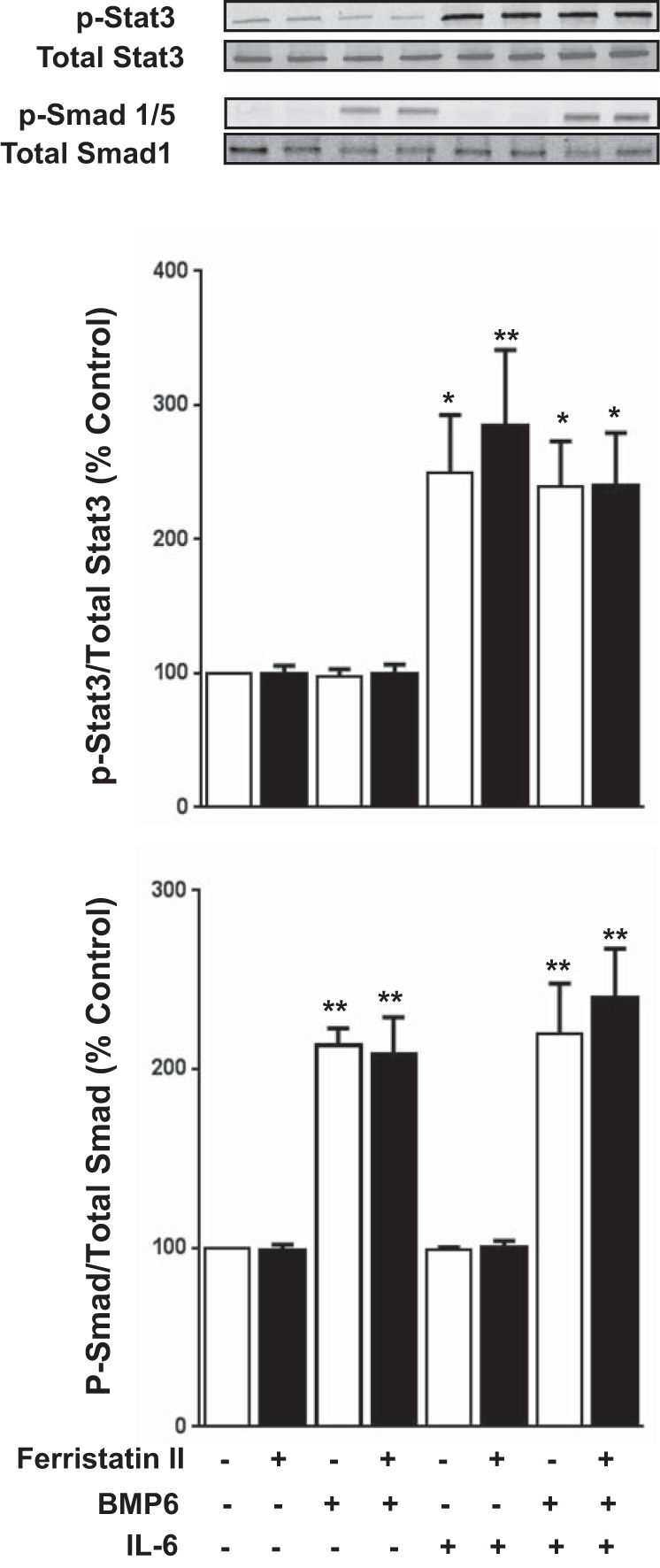
Ferristatin II does not increase total Stat3 or Smad 1/5 phosphorylation in vitro. HepG2/Luc cells were treated with or without ferristatin II (1 μM), BMP6 (100 ng/ml), and IL-6 (10 ng/ml) for 1 h. Representative immunoblots are shown in top panel. n = 3, *P < 0.05; **P < 0.01.
DISCUSSION
Ferristatin II (NSC8679) is a polysulfonated dye with structural similarity to the previously characterized small molecule iron transport inhibitor ferristatin (NSC306711). Both molecules have been shown to induce degradation of the transferrin receptor 1 in a clathrin-independent manner (7, 12). Importantly, ferristatin II does not degrade the upstream regulator of hepcidin, HFE (7). Under high-iron conditions, it is thought that iron-saturated transferrin binds to transferrin receptor 1 to displace HFE. HFE has been proposed to bind to a multimeric signaling complex to potentiate hepcidin synthesis (7, 28). Previous animal studies indicated that treatment with ferristatin II was associated with downregulation of transferrin receptor 1 and upregulation of hepcidin expression in liver (7). In preliminary cell culture experiments, we used siRNA instead of ferristatin II to reduce transferrin receptor 1 levels by 40%, but hepcidin expression was not affected (A. A. Alkhateeb and M. Wessling-Resnick, personal observations). We surmised that other pathways independent of transferrin receptor-1 degradation could be affected by ferristatin II and undertook this study to determine what upstream signaling pathways contribute to its ability to promote hepcidin transcription.
Hepcidin transcription is known to be regulated by Smad signaling (19, 33). Upon BMP activation, phospho-Smad1/5/8 associates with Smad4 to translocate into the nucleus to induce hepcidin transcription. Rats injected with ferristatin II showed increased liver hepcidin expression, along with reduced serum iron and splenic iron loading, consistent with our previous study (7). The latter responses reflect regulatory control of iron homeostasis by hepcidin associated with its increased synthesis. Because of reagent limitations, we were not able to directly assess the phosphorylation status of Smad proteins in rat liver. However, other Smad4 transcriptional targets (Id1 and Smad7) were downregulated in livers from rats treated with ferristatin II. These observations suggest that a downregulation of Smad signaling occurs with ferristatin II treatment. Interestingly, BMP6 mRNA levels were also reduced in rats treated with ferristatin II. BMP6 expression correlates with increased liver iron stores (8, 15, 36, 37), but nonheme iron levels were not reduced in livers from these animals (Table 1). Time course experiments will be required to study the temporal nature of ferristatin II activity and changes in iron status to discern how these are reflected in the drug's activity in vivo. It does appear, however, that increased hepcidin expression in livers of treated rats is not associated with activation of Smad signaling under our experimental conditions.
Stat3 signaling does appear to be activated in the liver of ferristatin II-treated animals. Through its receptor, the inflammatory cytokine IL-6 is known to increase hepcidin expression by activating the tyrosine kinase Jak2 to phosphorylate the Stat3 transcription factor, which translocates to the nucleus and associates with Stat3 response elements in the hepcidin promoter (31). Along with hepcidin, we observed that the Stat3 targets α2M, AGP, and CRP were induced by ferristatin II treatment. In addition, we detected phospho-Stat3 in livers of ferristatin II-treated rats but not control rats. However, neither serum nor hepatic IL-6 protein levels were increased in rats treated with ferristatin II, an observation that suggests that hepatic hepcidin induction is uncoupled to circulating or tissue levels of this particular cytokine. Since coculture experiments using macrophages and hepatocytes have shown that activated macrophages have the capacity to induce hepcidin in hepatocytes via an IL-6-independent mechanism (18), it is possible that other inflammatory factors play a role in vivo. The fact that α2M, AGP, and CRP are upregulated, along with the observed phosphorylation of Stat3, points to hepatic inflammatory response. In this regard, we cannot exclude the possibility that observed effects are provoked by a metabolite produced by hepatic clearance of the drug or to an impurity present in the compound preparation. We also considered ER stress as a possible xenobiotic response to the presence of the drug; however, in vitro experiments failed to support this idea.
To better understand the ferristatin II effects on hepatocytes, we characterized its actions using the hepatocellular carcinoma HepG2 cell line expressing a luciferase reporter under control of the hepcidin promoter (HepG2/Luc). Although ferristatin II alone did not induce luciferase expression, a strong synergistic increase was observed when the drug was combined with BMP6. Likewise, under serum-free conditions, IL-6 alone did not induce reporter gene expression, consistent with the idea that Stat3 activation of the hepcidin promoter requires activated Smad signaling (33). When BMP6, IL-6, and ferristatin II were combined, greater than 400-fold induction of hepcidin gene expression was observed. Unlike our in vivo studies, we did not observe an in vitro effect of ferristatin II on Stat3 phosphorylation, but using reporter gene expression we were able to show that that ferristatin II requires the presence of BMP6 to upregulate hepcidin expression and that it would act synergistically to enhance a Stat3-mediated response. It is important to note the divergence between in vivo and in vitro observations. It is possible these differences could be due to systemic metabolism as discussed above. Also, to observe synergistic effects of ferristatin II in the presence of BMP6 in vitro, HepG2/Luc cells required serum starvation for 48 h prior to the various treatments. The magnitude observed in the reporter gene expression studies most likely reflects minimal basal promoter activity and more complete activation when BMP6 is applied under these conditions. On the basis of our in vitro data, the Smad pathway is required for ferristatin II induction of hepcidin, but the fact that the Stat3 pathway is activated in vivo suggests that both this pathway and the Smad signaling pathway could possibly underlie the observed induction of hepcidin in ferristatin II-treated rats. Further study is required to determine how these regulatory pathways are involved, particularly since our studies in HepG2/Luc cells indicate that ferristatin II does not alter the phosphorylation of Stat3 or Smad 1/5.
Many forms of hereditary iron overload are characterized by inadequate hepcidin production due to the disruption of transcriptional regulation. Therefore, pharmacological induction of hepcidin has been proposed as an effective strategy for the treatment of hereditary iron overload (29). The observation that ferristatin II can synergize with BMP6 and IL-6 in vitro indicates that additional hepatocyte regulatory pathways could help enhance hepcidin synthesis.
GRANTS
Research reported in this publication was supported by NIDDK of the National Institutes of Health under award number R01DK064750 (M. Wessling-Resnick) and R01DK085250 (P. G. Fraenkel) and by NIGMS of the National Institutes of Health under award number R01GM083198 (P. G. Fraenkel).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.A.A., S.L.B., and M.W.-R. conception and design of research; A.A.A., P.D.B., A.M.G., and S.L.B. performed experiments; A.A.A., P.D.B., A.M.G., S.L.B., and M.W.-R. analyzed data; A.A.A., J.K., P.G.F., and M.W.-R. interpreted results of experiments; A.A.A., P.D.B., A.M.G., and S.L.B. prepared figures; A.A.A. drafted manuscript; A.A.A., P.D.B., J.K., S.L.B., P.G.F., and M.W.-R. edited and revised manuscript; A.A.A., P.D.B., A.M.G., J.K., S.L.B., P.G.F., and M.W.-R. approved final version of manuscript.
REFERENCES
- 1.Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, Babitt JL. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 41: 482–487, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest 117: 1933–1939, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachman E, Feng R, Travison T, Li M, Olbina G, Ostland V, Ulloor J, Zhang A, Basaria S, Ganz T, Westerman M, Bhasin S. Testosterone suppresses hepcidin in men: a potential mechanism for testosterone-induced erythrocytosis. J Clin Endocrinol Metab 95: 4743–4747, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besson-Fournier C, Latour C, Kautz L, Bertrand J, Ganz T, Roth MP, Coppin H. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 120: 431–439, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Brown JX, Buckett PD, Wessling-Resnick M. Identification of small molecule inhibitors that distinguish between non-transferrin bound iron uptake and transferrin-mediated iron transport. Chem Biol 11: 407–416, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Buckett PD, Wessling-Resnick M. Small molecule inhibitors of divalent metal transporter-1. Am J Physiol Gastrointest Liver Physiol 296: G798–G804, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrne SL, Buckett PD, Kim J, Luo F, Sanford J, Chen J, Enns C, Wessling-Resnick M. Ferristatin II promotes degradation of transferrin receptor-1 in vitro and in vivo. PloS One 8: e70199, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corradini E, Rozier M, Meynard D, Odhiambo A, Lin HY, Feng Q, Migas MC, Britton RS, Babitt JL, Fleming RE. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology 141: 1907–1914, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol 57: 1052–1060, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Ganz T. Systemic iron homeostasis. Physiol Rev 93: 1721–1741, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology 56: 291–299, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horonchik L, Wessling-Resnick M. The small-molecule iron transport inhibitor ferristatin/NSC306711 promotes degradation of the transferrin receptor. Chem Biol 15: 647–653, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, Tsuchiya K, Tamaki T. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PloS One 7: e40465, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurado RL. Iron, infections, and anemia of inflammation. Clin Infect Dis 25: 888–895, 1997. [DOI] [PubMed] [Google Scholar]
- 15.Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang RH, Deng C, Vaulont S, Mosser J, Coppin H, Roth MP. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 112: 1503–1509, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Kawabata H, Fleming RE, Gui D, Moon SY, Saitoh T, O'Kelly J, Umehara Y, Wano Y, Said JW, Koeffler HP. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 105: 376–381, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 480: 147–150, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Matak P, Chaston TB, Chung B, Srai SK, McKie AT, Sharp PA. Activated macrophages induce hepcidin expression in HuH7 hepatoma cells. Haematologica 94: 773–780, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mleczko-Sanecka K, Roche F, da Silva AR, Call D, D'Alessio F, Ragab A, Lapinski PE, Ummanni R, Korf U, Oakes C, Damm G, D'Alessandro LA, Klingmuller U, King PD, Boutros M, Hentze MW, Muckenthaler MU. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood 123: 1574–1585, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 113: 1271–1276, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 110: 1037–1044, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest 115: 2180–2186, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliveira SJ, Pinto JP, Picarote G, Costa VM, Carvalho F, Rangel M, de Sousa M, de Almeida SF. ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PloS One 4: e6618, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 276: 7806–7810, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 276: 7811–7819, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 33: 21–22, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 7: 205–214, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun CC, Vaja V, Babitt JL, Lin HY. Targeting the hepcidin-ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol 87: 392–400, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, Pietrangelo A. ER stress controls iron metabolism through induction of hepcidin. Science 325: 877–880, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109: 353–358, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Vujic Spasic M, Kiss J, Herrmann T, Galy B, Martinache S, Stolte J, Grone HJ, Stremmel W, Hentze MW, Muckenthaler MU. Hfe acts in hepatocytes to prevent hemochromatosis. Cell Metab 7: 173–178, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, Deng CX. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2: 399–409, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood 108: 3204–3209, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaji S, Sharp P, Ramesh B, Srai SK. Inhibition of iron transport across human intestinal epithelial cells by hepcidin. Blood 104: 2178–2180, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Zhang AS, Gao J, Koeberl DD, Enns CA. The role of hepatocyte hemojuvelin in the regulation of bone morphogenic protein-6 and hepcidin expression in vivo. J Biol Chem 285: 16416–16423, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Guo X, Herrera C, Tao Y, Wu Q, Wu A, Wang H, Bartnikas TB, Wang F. Bmp6 expression can be regulated independently of liver iron in mice. PloS One 9: e84906, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhen AW, Nguyen NH, Gibert Y, Motola S, Buckett P, Wessling-Resnick M, Fraenkel E, Fraenkel PG. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology 58: 1315–1325, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zumerle S, Mathieu JR, Delga S, Heinis M, Viatte L, Vaulont S, Peyssonnaux C. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 123: 3646–3650, 2014. [DOI] [PubMed] [Google Scholar]