

Abstract
Although direct myocardial depression has been implicated in the lethal effects of Bacillus anthracis lethal toxin (LT), in hearts isolated from healthy rats and perfused under constant pressure, neither LT or edema toxin (ET) in typically lethal concentrations depressed myocardial function. In the present study, we challenged rats with LT and ET and performed in vivo and ex vivo heart measures. Sprague-Dawley rats infused over 24 h with LT (n = 94), ET (n = 99), or diluent (controls; n = 50) were studied at 8, 24, or 48 h. Compared with control rats (all survived), survival rates with LT (56.1%) and ET (37.3%) were reduced (P < 0.0001) similarly (P = 0.66 for LT vs. ET). LT decreased mean arterial blood pressure from 12 to 20 h (P ≤ 0.05), whereas ET decreased it progressively throughout (P < 0.05). On echocardiography, LT decreased left ventricular (LV) ejection fraction at 8 and 48 h but increased it at 24 h and decreased cardiac output (P ≤ 0.05 for the time interaction or averaged over time). ET decreased systolic and diastolic volumes and increased LV ejection fraction at 24 h (P ≤ 0.05). In isolated hearts perfused for 120 min under constant pressure, LT did not significantly alter LV systolic or developed pressures at any time point, whereas ET decreased both of these at 24 h (P < 0.0001 initially). ET but not LT progressively increased plasma creatine phosphokinase and cardiac troponin levels (P < 0.05). In conclusion, despite echocardiographic changes, in vivo lethal LT challenge did not produce evidence of myocardial depression in isolated rat hearts. While lethal ET challenge did depress isolated heart function, this may have resulted from prior hypotension and ischemia.
Keywords: anthrax, edema toxin, lethal toxin, heart function
anthrax (Bacillus anthracis) produces two binary toxins, lethal toxin (LT) and edema toxin (ET), which are both thought to play a role in hypotension and lethality during infection. Each toxin includes protective antigen (PA), which mediates cellular uptake of the toxins' toxigenic moieties: lethal factor, a zinc metalloprotease, for LT and edema factor, a molecule with calmodulin-dependent adenyl cyclase activity, for ET. The intracellular actions of lethal factor and edema factor differ. Lethal factor inhibits MAPKKs and stimulates inflammasome formation (6, 26). Edema factor increases intracellular cAMP levels (14, 15).
Evidence from in vivo models has raised the possibility that LT but not ET produces hypotension in part by depressing myocardial function (22). In canines, lethality and hypotension with a 24-h LT challenge was associated with gradual tachycardia and reductions in left ventricular (LV) ejection fraction (EF) on echocardiography, changes consistent with the effects of LT in some small animal models (21, 25, 28, 29). Different from LT, lethality and hypotension with 24-h lethal ET challenge in this canine model was associated with early and striking increases in heart rate (HR) and cardiac index but no significant changes in LVEF, alterations also noted by other groups (28, 29). These findings and others have suggested that although LT and ET both alter myocardial function, LT but not ET depresses it (22). However, since both toxins also decreased central venous pressure and systemic vascular resistance in the canine model, it was unclear whether their myocardial effects measured in vivo were direct ones or instead related to changes in preload or afterload.
We have previously used a constant pressure Langendorff isolated perfused rat heart model to examine the effects of LT and ET on heart function independent of the toxins' potential preload and afterload effects (11). In hearts isolated from healthy Sprague-Dawley rats, typically lethal LT concentrations had little effect on heart function. In contrast, lethal ET concentrations produced increases in HR and actually augmented myocardial function and coronary blood flow, changes consistent with increased intracellular cAMP (11). To further investigate the myocardial effects of LT and ET, 8, 24, or 48 h after the initiation of 24-h toxin or PA only (control) infusions in Sprague-Dawley rats, hearts were isolated and myocardial function was investigated in the constant pressure perfusion system. Immediately before heart isolation, animals had blood pressure and HR measures followed by in vivo echocardiography and blood sampling for cardiac markers. In other experiments, animals were euthanized at these same time points for cardiac electron microscopy.
MATERIALS AND METHODS
Animal care.
The protocol (ASP CCM09-03) used in the present study was approved by the Animal Care and Use Committee of the Clinical Center of the National Institutes of Health.
Study design.
Twenty-six weekly experiments were performed. Each week, Sprague-Dawley rats (200–230 g) with carotid arterial and jugular venous catheters were randomized to receive challenge with doses of B. anthracis ET (400 μg/kg edema factor + 800 μg/kg PA) or LT (50 μg/kg lethal factor + 100 μg/kg PA), which have been previously shown to produce ∼50% lethality rates (4), or with PA alone (control) as continuous 24-h infusions. A total of 243 animals were studied. Each experiment included at least one animal from each group. In one set of experiments (study 1), animals randomly selected at 8, 24, or 48 h had blood pressure and HR measured while awake via their indwelling carotid arterial catheters, after which they were lightly anesthetized with isoflurane (1–3%) and had echocardiography performed. After echocardiography, animals were more deeply anesthetized (3–5% isoflurane), and their hearts were excised and perfused in a constant pressure perfusion system. Immediately before heart excision, arterial blood was drawn for creatine phosphokinase (CPK), cardiac troponin I (cTnI), alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), creatinine, Na+, K+, arterial blood gas, and lactate measures at 8, 24, and 48 h. A subgroup of animals (n = 28) in study 1 had blood pressure and HR measured for 24 h, immediately after which their hearts were excised and perfused, and the perfusion effluent was collected for CPK, cTnI, and cAMP measures. The number of animals randomized and then studied in each experiment for study 1 at the three time points is shown in Table 1. In a second set of experiments (study 2), animals were randomly selected at 8, 24, or 48 h, anesthetized, and euthanized for cardiac electron microscopy experiments (Table 1). Before death, animals had blood pressure and HR measured and blood sampled as in study 1.
Table 1.
Number of animals randomized to 24-h infusions with diluent and PA alone (control), LT, or ET and then sampled at 8, 24, or 48 h after the start of infusion in each of 16 experiments in study 1 for echocardiography and perfused heart measures and 10 experiments in study 2 for electron microscopy measures
| Control Group |
LT Group |
ET Group |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Randomized | Sampled at 8 h | Sampled at 24 h | Sampled at 48 h | Randomized | Sampled at 8 h | Sampled at 24 h | Sampled at 48 h | Randomized | Sampled at 8 h | Sampled at 24 h | Sampled at 48 h | |
| Study 1 | ||||||||||||
| Experiment 1 | 2 | 1 | 1 | 4 | 1 | 1 | 3 | 1 | 1 | |||
| Experiment 2 | 2 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 1 | |||
| Experiment 3 | 2 | 1 | 1 | 4 | 1 | 1 | 4 | 1 | 1 | |||
| Experiment 4 | 2 | 1 | 3 | 1 | 1 | 3 | 1 | 2 | ||||
| Experiment 5 | 2 | 1 | 1 | 4 | 1 | 4 | 1 | 1 | ||||
| Experiment 6 | 2 | 1 | 3 | 2 | 1 | 3 | 1 | |||||
| Experiment 7 | 2 | 1 | 1 | 4 | 1 | 1 | 3 | 1 | ||||
| Experiment 8 | 2 | 1 | 1 | 5 | 1 | 1 | 5 | 1 | 2 | |||
| Experiment 9 | 2 | 1 | 1 | 4 | 1 | 2 | 4 | 1 | 1 | |||
| Experiment 10 | 2 | 1 | 1 | 5 | 1 | 1 | 5 | 1 | 2 | |||
| Experiment 11 | 2 | 1 | 1 | 4 | 1 | 2 | 4 | 1 | 1 | |||
| Experiment 12 | 1 | 1 | 3 | 1 | 2 | 3 | 1 | 1 | ||||
| Experiment 13* | 1 | 1 | 3 | 2 | 2 | 2 | ||||||
| Experiment 14* | 2 | 2 | 3 | 2 | 2 | 2 | ||||||
| Experiment 15* | 2 | 2 | 4 | 2 | 2 | 2 | ||||||
| Experiment 16* | 3 | 3 | 3 | 1 | 2 | 2 | ||||||
| Study 2 | ||||||||||||
| Experiment 1 | 2 | 1 | 1 | 2 | 1 | 3 | 1 | 1 | ||||
| Experiment 2 | 1 | 1 | 2 | 1 | 3 | 1 | ||||||
| Experiment 3 | 1 | 1 | 1 | 1 | 3 | 1 | ||||||
| Experiment 4 | 2 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | |||
| Experiment 5 | 2 | 1 | 2 | 1 | 1 | 4 | 1 | 1 | ||||
| Experiment 6 | 1 | 1 | 2 | 1 | 1 | |||||||
| Experiment 7 | 1 | 1 | 3 | 1 | 1 | 2 | 1 | |||||
| Experiment 8 | 5 | 1 | 1 | 3 | 9 | 1 | 1 | 2 | 10 | 1 | 1 | |
| Experiment 9 | 4 | 1 | 1 | 2 | 9 | 1 | 1 | 2 | 11 | 1 | 1 | 2 |
| Experiment 10 | 1 | 1 | 4 | 11 | 3 | |||||||
PA, protective antigen; LT, lethal toxin; ET, edema toxin.
These experiments tested hemodynamics, perfused isolated heart function, and effluent myocardial biomarkers and cAMP only.
Echocardiography.
Animals were lightly anesthetized with isoflurane (1–3%). M-mode echocardiography was performed using a Vevo 770 (Visualsonics, Toronto, ON, Canada) with a frame rate of 300–500 frames/s and a 12-MHz linear transducer as previously described (23). Data representing the average of nine cardiac cycles from at least two separate scans were analyzed. HR was averaged over the number of beats measured. After tracing end-diastolic and end-systolic dimensions, manufacturer software computed end-diastolic and end-systolic volumes (in μl), stroke volume (in μl), cardiac output (in ml/min), and EF (in %). Results were interpreted without knowledge of the study group.
Langendorff model.
Rats were anesthetized with 3–5% isoflurane and anticoagulated with heparin (500 IU/kg) through the inferior vena cava as previously described (11). Hearts were rapidly excised and arrested in cold (4°C) Krebs-Henseleit buffer (10, 11, 24, 27). A cannula was inserted in the aorta, and the heart was perfused retrograde at constant pressure (95 cmH2O) with Krebs-Henseleit buffer gassed with 95% O2 and 5% CO2 at 37°C and filtered through a 5-μm filter. A water-filled latex balloon connected to a water column and pressure transducer was introduced into the LV. LV end-diastolic pressure (LVEDP) was adjusted to between 4 and 8 mmHg. Hearts were allowed to equilibrate for 10 min, and serial readings of LV systolic pressure (LVSP; in mmHg), LVEDP (in mmHg), and HR (in beats/min) were then measured continuously over a 120-min period (ML880P PowerLab 16/30 with LabChart Pro, AD Instruments, Colorado Springs, CO). After equilibration and at 30-min intervals thereafter, measures obtained over the 5-min period immediately preceding each designated time point (i.e., 0, 30, 60, 90, and 120 min) were averaged for analysis. Coronary flow (CF; in ml/min) was calculated based on coronary effluent measures every 30 min. Hearts were perfused with fresh Krebs-Henseleit buffer throughout. Two to four hearts, including both control and toxin subjects, were studied daily. Calculated data included LV developed pressure (LVDP, in mmHg; LVDP = LVSP − LVEDP), the rate-pressure product (RPP, in mmHg·beats·min−1; RPP = LVDP × HR), the maximal rate of change in LV pressure during contraction (dP/dtmax; in mmHg/s), and the minimal rate of change in LV pressure during relaxation (dP/dtmin; in mmHg/s). In a subgroup of animals at 24 h (see above), perfusion effluent was collected at 0, 30, 60, and 90 min for cAMP, CPK, and cTnI measures.
Electron microscopy.
A section (2 × 2 mm) of the LV anterior wall was double fixed in PBS-buffered glutaraldehyde (2.5%) and osmium tetroxide (0.5%), dehydrated, and embedded into Spurr's epoxy resin. Ultrathin sections (90 nm) were made, double stained with uranyl acetate and lead citrate, and viewed with a JEOL JEM 1010 transmission electron microscope. Myocardial injury was assessed on the ultrastructural level with a numeric scale (where 0 = no injury and 5 = most severe injury) by a pathologist blinded to the study groups. The ultrastructural assessment included changes in the cardiac muscle's endothelial cell membranes and nuclei, evidence of myocyte mitochondrial degeneration and swelling in the outer and inner membranes, as well as damage to myocyte sarcomeres and nuclei.
Hemodynamic and other laboratory measures.
Indwelling arterial and central venous catheters from each animal were connected via protected extension tubing to pressure transducers for mean blood pressure (MBP) and HR measures or to syringe pumps for toxin delivery, respectively (5, 16). Arterial blood gases were measured with Critical Care Xpress (Nova Biomedical, Waltham, MA). Plasma or effluent chemistries (CPK, ALT, AST, BUN, and creatinine) were measured with Alfa Wassermann (Diagnostic Technologies, West Caldwell, NJ). Plasma and effluent cTnI were measured using ELISA (Life Diagnostics, West Chester, PA). Effluent cAMP was measured with a cAMP Chemiluminescent Immunoassay Kit (Arbor Assays, Ann Arbor, MI).
Toxin and treatments.
All toxin components (PA, lethal factor, and edema factor) were recombinant proteins prepared from Escherichia coli and provided by Human Genome Sciences (formerly of Rockville, MD) (3, 9, 12). ET and LT were comprised of edema factor or lethal factor, respectively, with PA in ratios of 1:2 on the basis of weight (4). The control solution contained diluent and PA alone.
Statistical analysis.
A Wilcoxon rank test compared survival between control and either LT- or ET-challenged animals and between LT- and ET-challenged animals, accounting for survival time, death of animals at 8, 24, or 48 h, and death within the 48-h experimental period. Electron microscopy scores for myocardial cell mitochondrial and endothelial injury were not normally distributed and were ranked using PROC RANK for analysis. cTnI was log transformed due to its abnormal distribution. Two-way ANOVA and two-way repeated-measures ANOVA, where applicable, accounting for toxin (LT, ET, or control) and time of measurement as well as one-way ANOVA comparing different time points in controls and toxin versus controls at each time point were performed on all continuous or ranked data using PROC MIXED in SAS (version 9.2) software (SAS Institute, Cary, NC). Two-sided P values of 0.05 or less were considered significant. Multiple comparisons were not controlled for. For echocardiography, electron microscopy, and blood chemistry and blood gas measures, values for control animals are shown in Table 2, whereas figures show the effect of each toxin compared with controls, calculated by subtracting mean control values from mean toxin values. For blood pressure, HR, and perfused heart experiments, figures compare values for control and LT- and ET-challenged animals.
Table 2.
Mean (±SE) values for echocardiography, electron microscopy, chemistry, and arterial blood gas measures for control animals at 8, 24 and 48 h after the start of an infusion of diluent with Protective Antigen only
| Echocardiography |
||||||
|---|---|---|---|---|---|---|
| End-systolic volume, μl | End-diastolic volume, μl | Ejection fraction, % | Heart rate, beats/min | Stroke volume, μl | Cardiac output, ml/min | |
| 8 h | 59.2 ± 12.6 | 257.8 ± 24.8 | 77.9 ± 3.5 | 393 ± 22 | 198 ± 20 | 77.4 ± 7.5 |
| 24 h | 81.5 ± 12.6 | 309.7 ± 24.8 | 74.5 ± 3.5 | 356 ± 25 | 228 ± 20 | 83.2 ± 8.4 |
| 48 h | 77.1 ± 8.5 | 309.9 ± 16.7 | 75.1 ± 2.3 | 320 ± 15 | 233 ± 13 | 74.7 ± 5.1 |
| Electron Microscopy |
||||||
|---|---|---|---|---|---|---|
| Endothelial membrane | Endothelial nucleus | Myocyte mitochondria outer membrane | Myocyte mitochondria inner membrane | Myocyte sarcomere | Myocyte nucleus | |
| 8 h | 8.3 ± 1.9 | 7.8 ± 1.7 | 8.8 ± 2.0 | 6.8 ± 2.0 | 8.9 ± 2.1 | 7.9 ± 1.6 |
| 24 h | 6.6 ± 2.2 | 6.8 ± 2.5 | 9.4 ± 2.4 | 8.9 ± 2.2 | 9.1 ± 2.3 | 8.6 ± 1.8 |
| 48 h | 7.7 ± 1.6 | 7.2 ± 1.1 | 6.5 ± 1.2 | 8.1 ± 1.9 | 5.81.5 | 7.6 ± 1.5 |
| Plasma Chemistry |
||||||
|---|---|---|---|---|---|---|
| CPK, U/l | cTnI, ln(ng/ml) | ALT, U/l | AST, U/l | BUN, mg/dl | Creatinine, mg/dl | |
| 8 h | 431 ± 143 | 0.01 ± 0.07 | 52.0 ± 34.3 | 160 ± 101 | 16.6 ± 9.3 | 0.34 ± 0.09 |
| 24 h | 235 ± 120 | 0.10 ± 0.05 | 31.7 ± 28.7 | 128 ± 84 | 17.6 ± 7.8 | 0.31 ± 0.07 |
| 48 h | 299 ± 115 | 0.00 ± 0.06 | 44.6 ± 26.2 | 213 ± 77 | 21.8 ± 7.1 | 0.25 ± 0.07 |
| Arterial Blood Gas |
|||||
|---|---|---|---|---|---|
| pH | Lactate, mmol/l | ABE, mmol/l | Na+, mmol/l | K+, mmol/l | |
| 8 h | 7.47 ± 0.01 | 1.25 ± 0.27 | 2.52 ± 0.69 | 141.6 ± 0.6 | 3.41 ± 0.14 |
| 24 h | 7.47 ± 0.01 | 1.01 ± 0.30 | 1.05 ± 0.78 | 141.8 ± 0.7 | 3.43 ± 0.17 |
| 48 h | 7.47 ± 0.01 | 1.05 ± 0.40 | 1.98 ± 1.05 | 141.6 ± 0.8 | 3.28 ± 0.18 |
CPK, creatine phosphokinase; cTnI, cardiac troponin I; ALT, alanine aminotransferase; AST, aspartate aminotransferases; BUN, blood urea nitrogen; ABE, arterial base excess.
RESULTS
Survival.
All animals challenged with PA alone (controls; n = 50) survived to the time of death (8, 24, or 48 h; Fig. 1). Compared with control animals, in animals challenged with LT (n = 94) or ET (n = 99) and after accounting for those euthanized for experiments, survival rates were significantly reduced (56.1% and 37.3%, respectively, P < 0.0001 for each) but not different when toxins were compared (P = 0.66).
Fig. 1.
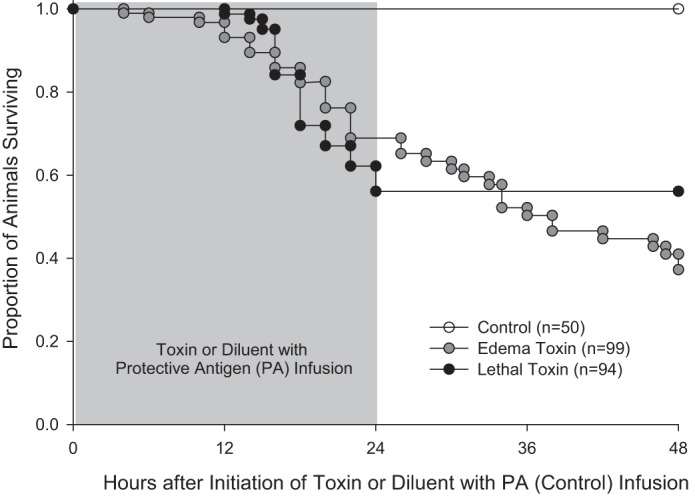
Proportion of animals surviving over time after the initiation of 24-h infusions of edema toxin (ET) or lethal toxin (LT) or diluent with protective antigen (PA) only (controls). Animals euthanized for experiments were considered to be survivors up until the time of death, at which time they were censored from further analysis. Both ET and LT resulted in progressive reductions in survival in patterns that were significantly different from control animals (P < 0.0001) but did not differ when the two toxins were compared (P = 0.66).
Mean arterial blood pressure and HR measures.
In control animals, compared with baseline MBP decreased over time (P < 0.0001 for the time interaction) but stayed between 100 and 120 mmHg. Compared with control animals, LT decreased MBP from 12 to 20 h (P ≤ 0.05) and HR from 14 to 22 h and at 44 h (P ≤ 0.05; Fig. 2). ET reduced MBP significantly at each time point (P ≤ 0.05) and in a pattern that increased over time (P < 0.0001 for the time interaction) and increased HR significantly from 2 to 18 h but decreased it at 30 and 32 h (P ≤ 0.05).
Fig. 2.

Serial mean ± SE mean arterial blood pressures (MBPs; A) and heart rates [HRs; in beats/min (bpm); B] measured for up to 48 h after the initiation of 24-h infusions of LT, ET, or diluent with PA only (controls).
Echocardiography measures.
On echocardiography in control animals, compared with 8 h, at 48 h, HR was lower (0.008) and end-diastolic volume was increased in a trend approaching significance (P = 0.08; Table 2). Compared with control animals, LT challenge decreased LVEF at 8 and 48 h but increased it at 24 h in patterns that differed over time (P = 0.049 for the time interaction; Fig. 3). LT also decreased cardiac output across the three time points in an overall pattern that was significant (P = 0.04 averaged over time). At 24 h, ET decreased both end-systolic and end-diastolic volumes but increased LVEF (P ≤ 0.05). However, stroke volume, possibly more informative than LVEF in the setting of reduced end-diastolic volume, was decreased at 24 h with ET, but not significantly. ET also increased HR at 48 h (P = 0.048).
Fig. 3.

Mean ± SE effects of LT or ET compared with control animals on echocardiography-determined left ventricular (LV) end-systolic volume (A), LV end-diastolic volume (B), LV ejection fraction (C), HR (D), stroke volume (E), and cardiac output (F) at either 8, 24, or 48 h after the initiation of 24-h infusions of toxin or diluent. Effects were calculated by subtracting control values from toxin values (see materials and methods). Data for control animals are shown in Table 2.
Blood cardiac markers.
Plasma CPK and cTnI measures did not change significantly over time in control animals (Table 2). Compared with control animals, LT challenge did not significantly alter blood CPK or cTnI levels at any of the three time points measured (8, 24, or 48 h; Fig. 4). ET challenge increased both measures significantly compared with control at 24 and 48 h (P ≤ 0.05).
Fig. 4.

Mean ± SE effects of LT or ET compared with control animals on blood creatine phosphokinase (CPK; A) and cardiac troponin I (cTnI; B) levels at either 8, 24, or 48 h after the initiation of 24-h infusions of toxin or diluent. Effects were calculated by subtracting control values from toxin values (see materials and methods). Data for control animals are shown in Table 2.
Isolated perfused cardiac measures.
Compared with control animals and despite its lethal and hypotensive effects, prior in vivo LT challenge did not significantly alter at any of the three time points (8, 24, or 48 h) LVEDP, LVSP, LVDP, RPP, HR, or dP/dtmin (Figs. 5 and 6). At 24 h, LT produced a small decrease in dP/dtmax (P = 0.054) at a single time point (60 min) and decreased CF from 30 to 120 min (P ≤ 0.05). However, even when the effects of LT on LVSP, LVDP, RPP, dP/dtmax, and dP/dtmin were analyzed across all perfusion time points (0, 30, 60, 90, and 120 min) at 24h, its effects did not differ over time (P = 0.19–0.49), and, when averaged, none of its overall effects were significantly different from control animals (P = 0.33–0.46). In perfusion effluent at 24 h, LT did not significantly alter CPK levels but did increase cTnI at 30 min (P = 0.02; Table 3). In contrast to LT, prior in vivo ET challenge had marked effects in isolated hearts. Compared with control animals, at 8 h, ET increased HR at 30 and 60 min of perfusion (P ≤ 0.05). Most notably, however, at 24 h, ET caused highly significant reductions in LVSP, LVDP, dP/dtmax, and RPP and increases in dP/dtmin evident immediately after equilibration at 24 h (P < 0.0001 for all except RPP, which was P = 0.0002). Decreases in LVSP and LVDP persisted and continued to be significant (P ≤ 0.05) for up to 90 and 60 min, respectively. ET also then increased RPP at 120 min (P < 0.05) at 24 h and increased it from 30 to 120 min at 48 h (P ≤ 0.001), possibly related to increases in HR with ET that were evident throughout perfusion at both 24 and 48 h (P ≤ 0.001). At 24 and 48 h, ET also increased CF throughout the perfusion period (P ≤ 0.001). Finally, at 48 h, ET increased dP/dtmax and decreased dP/dtmin from 60 to 120 min (P ≤ 0.05). ET was not associated with significant changes in CPK or cTnI in perfusion effluent. Also, while ET at 24 h was associated with increases in cAMP levels in perfusion effluent at 0, 30, and 120 min, these changes were not significant (Table 3).
Fig. 5.

Mean ± SE LV end-diastolic pressure (LVEDP), LV developed pressure (LVDP), LV systolic pressure (LVSP), and dP/dtmax in hearts excised from animals and perfused under constant pressure at either 8, 24, or 48 h after the initiation of 24-h infusions of LT, ET, or diluent with PA only (controls).
Fig. 6.

Mean ± SE dP/dtmin, HR, rate-pressure product (RPP), and coronary flow (CF) in hearts excised from animals and perfused under constant pressure at either 8, 24, or 48 h after the initiation of 24-h infusions of LT, ET, or diluent with PA only (controls).
Table 3.
Mean ± SE values for isolated perfused heart effluent CPK, cTnI, and cAMP levels at time 0 and 30, 60, and 90 min after perfusion
| Control Group | LT Group | ET | |
|---|---|---|---|
| CPK, U/l | |||
| Time 0 | 5.3 ± 0.5 | 5.9 ± 0.6 | 4.1 ± 0.5 |
| 30 min | 3.6 ± 0.5 | 3.2 ± 0.6 | 2.3 ± 0.6 |
| 60 min | 2.9 ± 0.5 | 2.1 ± 0.6 | 2.3 ± 0.6 |
| 90 min | 0.9 ± 0.5 | 2.1 ± 0.6 | 2.1 ± 0.5 |
| cTnI, ln(ng/ml) | |||
| Time 0 | 0.21 ± 0.02 | 0.27 ± 0.02 | 0.23 ± 0.02 |
| 30 min | 0.16 ± 0.02 | 0.24 ± 0.02* | 0.17 ± 0.02 |
| 60 min | 0.16 ± 0.02 | 0.20 ± 0.02 | 0.17 ± 0.02 |
| 90 min | 0.15 ± 0.02 | 0.19 ± 0.02 | 0.19 ± 0.02 |
| cAMP, pmol/ml | |||
| Time 0 | 8.0 ± 3.8 | 8.4 ± 4.1 | 12.3 ± 3.8 |
| 30 min | 7.2 ± 3.8 | 5.8 ± 4.1 | 14.9 ± 3.8 |
| 60 min | 7.1 ± 3.8 | 4.3 ± 4.1 | 6.8 ± 4.1 |
| 90 min | 10.3 ± 3.8 | 10.9 ± 4.1 | 15.6 ± 3.8 |
P = 0.02 compared with the control (diluent with PA alone) group.
Cardiac electron microscopy.
Electron microscopy revealed minimal myocardial injury from either LT or ET challenge. Compared with control hearts (Table 2), hearts from LT-challenged animals demonstrated myocyte sarcomere injury at 48 h (P = 0.05; Fig. 7). Hearts from ET-challenged animals demonstrated myocyte mitochondrial inner membrane and myocyte sarcomere injury at 24 h and sarcomere injury when averaged over the three time points (P ≤ 0.05).
Fig. 7.

Mean ± SE effects of LT or ET compared with control animals on ranked tissue injury scores determined with electron microscopy in sections taken from hearts of animals at either 8, 24, or 48 h after the initiation of 24-h infusions of toxin or diluent. Ranked injury scores were determined for endothelial membranes (A) and nuclei (B), myocyte mitochondrial outer (C) and inner (D) membranes, and myocyte sarcomeres (E) and nuclei (F). Effects were calculated by subtracting control values from toxin values (see materials and methods). Data for control animals are shown in Table 2.
Other blood measures.
Compared with control animals (Table 2), LT had no significant effects on chemistry and arterial blood gas measures (Fig. 8). In contrast, ET increased ALT and AST at 24 and 48 h and BUN at 8, 24, and 48 h and first decreased creatinine at 8 h and then increased it at 24 and 48 h (P ≤ 0.05 for each). ET also decreased pH at 24 h, increased lactate at 24 and 48 h, and decreased arterial base excess and Na+ at all time points, K+ at 8 h, and the alveolar-arterial gradient at 24 h (P ≤ 0.05 for each; alveolar-arterial gradient not shown). Changes with ET in ALT, BUN, creatinine, pH, arterial base excess, lactate, Na+, and K+ were greater at later time points (P ≤ 0.05 for the time interactions).
Fig. 8.

Mean ± SE effects of LT or ET compared with control animals on plasma or blood alanine aminotransferase (ALT; A), aspartate aminotransferase (AST; B), blood urea nitrogen (BUN; C), creatinine (D), arterial blood pH (E), lactate (F), arterial base excess (ABE; G), Na+ (H), and K+ (I) from animals at either 8, 24, or 48 h after the initiation of 24-h infusions of toxin or diluent. Effects were calculated by subtracting control values from toxin values (see materials and methods). Data for control animals are shown in Table 2.
DISCUSSION
In the present study in rats, a 24 h, B. anthracis LT challenge in doses causing significant reductions in survival and blood pressure did not produce evidence of myocardial depression in isolated perfused hearts at 8, 24, or 48 h, as reflected by measures of LVSP, LVDP, or RPP. None of these measures in LT-challenged hearts differed from control hearts over 120 min of observation at any of the three time points. Consistent with this, blood CPK and cTnI levels were also not altered in LT-challenged animals. Although a decrease in dP/dtmax was noted at 24 h, this change was small and only evident at a single time point (60 min). In fact, when data were analyzed over all perfusion time points at 24 h, the effects of LT on dP/dtmax, LVSP, LVDP, RPP, and dP/dtmin were not significantly different compared with control animals (P = 0.33–0.46).
These findings differ from other studies in mouse, rat, rabbit, and canine models in which LT challenge was noted on echocardiography to depress in vivo myocardial function, as reflected by changes in LV volumes, velocity of propagation, circumferential fiber shortening, or LVEF (13, 21, 25, 28). In these models, LT was also associated with increased cardiac enzymes in blood and myocardial injury on electron microscopy (13, 21). In studies that inhibited LT, PA-directed monoclonal antibody treatment in LT-challenged canines or selective deletion of anthrax toxin receptor-2 from cardiomyocytes in LT-challenged mice both increased LVEF as measured with echocardiography (1, 19). Possibly consistent with these other studies, in the present study, on echocardiography LT decreased LVEF at 8 and 48 h but not 24 h and reduced cardiac output when averaged over all time points. LT was also associated with modest changes on electron microscopy at 48 h but not in circulating CPK or cTnI levels.
Differing effects of LT on myocardial function in the present study versus studies like those noted above (13, 21, 24, 27) may reflect the techniques used to measure that function. Growing data suggest that LT decreases endothelial barrier and vascular smooth muscle function (8, 19). These changes could alter preload or afterload and secondarily affect myocardial performance measured in vivo with techniques like echocardiography (13, 21, 24, 27). Of note, in the prior canine study we conducted (24), although pulmonary artery occlusion pressure was not altered by LT, CVP was reduced and volume loading increased LVEF in some animals. Both of these latter findings suggest that reductions in preload related to LT may have influenced LVEF. Assessment of the effects of LT on myocardial function using measures less likely influenced by preload and afterload have been limited. On the one hand, a previously published abstract (2) reported that pressure-volume measures in four canines demonstrated reductions in stroke volume, end-systolic pressure, and LVEF and increased LVEDP in patterns consistent with heart failure. However, we have previously shown that perfusion with LT in concentrations typically producing high lethality rates in vivo did not alter the function of hearts isolated from healthy rats and tested for up to 240 min under constant pressure in a Langendorff system (11). In that study, LT only altered myocardial function when its concentration was increased to 10 times those producing lethality in vivo. Lack of an effect of LT (unless administered at very high concentrations) may have been because myocardial changes with ex vivo toxin administration required more time to develop than the model permitted. However, the present findings demonstrate that even when rat hearts are exposed in vivo to lethal LT doses over a 24-h period, once isolated, they do not demonstrate evidence of myocardial depression in a constant pressure perfusion system. Consistent with these findings in isolated hearts, LT was also not associated with increases in CPK or cTnI in blood, and its effects on cardiac electron microscopic findings were minimal. The basis for the reduction in CF with LT at 24 h is unclear. Since CF in this model is dependent on the perfusion pressure the system is exposed to, which was constant throughout, and the resistance of the coronary arteries, an effect of LT on the latter must be considered. Importantly, however, these reductions in CF were not associated with significant alterations in the pressure hearts were capable of generating.
It is possible that ex vivo measurement of heart function at later time points (e.g., 72 or 96 h) after the start of LT infusion might have demonstrated myocardial depression. However, lethality with LT appeared complete by 48 h in this model, and the relevance of later myocardial changes for lethality would be unclear. It is also possible that the myocardial effects of LT differ in the rat compared with other species. Notably, time to lethality with LT and the mechanisms underlying this lethality appear to differ in some rat and mouse models (18). However, we have found that the effects of either LT or ET on systemic hemodynamics have been comparable in species as different as the rat and canine (4, 25). Finally, LT challenge in doses more lethal or which had greater hypotensive effects than the doses used here may have resulted in myocardial depression in isolated hearts. Yet, LT in the present study produced highly significant lethality (P < 0.0001) approaching 40% and blood pressure reductions close to when hearts were isolated and measured. The reason why cTnI was increased in perfusion effluents at 24 h with LT challenge while these measures were unchanged in blood immediately before hearts were isolated is unclear. However, these cTnI levels are inconsistent with the absence of alterations in developed pressures or on electron microscopy in hearts exposed to LT.
Different from LT in the present study, hearts from rats exposed in vivo to a lethal ET challenge demonstrated both depressed and stimulated function. Most notably, hearts isolated 24 h after the start of ET, and when systemic blood pressure had reached its lowest point, had marked reductions in LVDP, LVSP, RPP, and dP/dtmax. There are several potential explanations for these changes. First, it is possible that ET directly depressed myocardial function. On the one hand, this appears unlikely, though, since acute increases in myocardial cAMP levels with ET would be expected to increase contractility (2, 11). As noted above, exposure of hearts isolated from healthy animals to ET in concentrations comparable with concentrations producing lethality in vivo increased LVDP and dP/dtmax as well as myocardial tissue and effluent cAMP levels. It is possible, though, that longer exposure to ET in vivo in the present study elicited mechanisms capable of producing myocardial depression. We have previously demonstrated that ET-induced arterial relaxation in an aortic ring model is partially endothelium dependent (15). These findings implicate endothelium-derived relaxant factors such as nitric oxide (NO) in the cardiovascular effects of ET. Studies have shown that cAMP can induce NO production (17, 30), whereas others have found an association between NO and cardiac dysfunction (7, 20). A second possibility, however, is that hypotension and ischemia resulting from the systemic vasodilatory effects of ET produced myocardial depression secondarily. Consistent with this, ET increased lactate and worsened liver and renal function (manifested by increased AST, ALT, BUN, and creatinine). In this regard, increases in CPK and cTnI in blood and changes on cardiac electron microscopy noted with ET may reflect myocardial ischemia as opposed to direct myocardial injury. Arguing against this possibility, though, is that while reductions in blood pressure with ET persisted until 48 h, evidence of myocardial depression had resolved by this later time point.
The depression in LV function in isolated hearts after ET challenge at 24 h contrasts with the increase in LVEF noted on echocardiography at this time point. However, reductions in afterload caused by hypotension with ET as well as relative differences in end-systolic and end-diastolic volumes may have increased LVEF on echocardiography.
While ET reduced LVDP, LVSP, and dP/dtmax at 24 h, it increased HR in isolated hearts at all three time points, dP/dtmax and RPP at 48 h, and CF at 24 and 48 h. These changes are very consistent with increased myocardial cAMP levels, and they are similar to changes we observed when healthy hearts were exposed to ET in the Langendorff system (11). Notably, though, whereas ET increased HR during perfusion in isolated hearts at all three time points, HR increases were only clearly evident in vivo on arterial tracings at 8 h and on echocardiography at 8 and 48 h. Thus, ongoing hypoperfusion or acidosis may have inhibited the chronotropic effects of increased cAMP in vivo. Also, although CF was increased when measured under constant pressure ex vivo, this increase may have been insufficient to maintain myocardial perfusion in the face of systemic hypotension with ET in vivo. Of note, increased HR and CF were still evident at 48 h in isolated hearts (i.e., 24 h after the cessation of ET challenge) and may have reflected the residual effects of toxin or were compensatory ones related to the prolonged systemic hypotension caused by ET.
In conclusion, the findings from the present study in a Langendorff constant pressure perfused rat heart model do not support a direct myocardial depressant effect of B. anthracis LT. Although ET challenge did depress myocardial function at 24 h, whether this was a direct effect of the toxin or was related to systemic hypotension and secondary ischemia is not clear. Further defining the mechanisms underlying the cardiovascular and lethal effects of these toxins will help improve the management of B. anthracis infection in the future.
GRANTS
This work was supported by the National Institutes of Health Intramural Program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.L., P.Q.E., and X.C. conception and design of research; Y.L., M.A.-A., J.S., P.Q., J.F., L.O., H.S.K., Y.F., P.Q.E., and X.C. performed experiments; Y.L., P.Q.E., and X.C. analyzed data; Y.L., M.A.-A., P.Q.E., and X.C. interpreted results of experiments; Y.L., P.Q.E., and X.C. prepared figures; Y.L., P.Q.E., and X.C. drafted manuscript; Y.L., M.A.-A., P.Q.E., and X.C. edited and revised manuscript; Y.L., P.Q.E., and X.C. approved final version of manuscript.
REFERENCES
- 1.Barochia AV, Cui X, Sun J, Li Y, Solomon SB, Migone TS, Subramanian GM, Bolmer SD, Eichacker PQ. Protective antigen antibody augments hemodynamic support in anthrax lethal toxin shock in canines. J Infect Dis 205: 818–829, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng CP, Masutani S, Cheng HJ, Cross M, Zhang CX, Zhou P, Cann J, Cline JM, Little WC, Kuo SR, Frankel AE. Progressive left ventricle, myocyte dysfunction, and heart failure in the lethality of anthrax toxin in conscious dogs. Circulation 116: 758–758, 2007. [Google Scholar]
- 3.Cooksey BA, Sampey GC, Pierre JL, Zhang X, Karwoski JD, Choi GH, Laird MW. Production of biologically active Bacillus anthracis edema factor in Escherichia coli. Biotechnol Prog 20: 1651–1659, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Cui X, Li Y, Li X, Laird MW, Subramanian M, Moayeri M, Leppla SH, Fitz Y, Su J, Sherer K, Eichacker PQ. Bacillus anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J Infect Dis 195: 572–580, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Cui X, Moayeri M, Li Y, Li X, Haley M, Fitz Y, Correa-Araujo R, Banks SM, Leppla SH, Eichacker PQ. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am J Physiol Regul Integr Comp Physiol 286: R699–R709, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280: 734–737, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Estan J, Ortiz MC, Lee SS. Nitric oxide and renal and cardiac dysfunction in cirrhosis. Clin Sci (Lond) 102: 213–222, 2002. [PubMed] [Google Scholar]
- 8.Guichard A, Nizet V, Bier E. New insights into the biological effects of anthrax toxins: linking cellular to organismal responses. Microbes Infect 14: 97–118, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gwinn W, Zhang M, Mon S, Sampey D, Zukauskas D, Kassebaum C, Zmuda JF, Tsai A, Laird MW. Scalable purification of Bacillus anthracis protective antigen from Escherichia coli. Protein Expr Purif 45: 30–36, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Hearse DJ, Sutherland FJ. Catecholamines and preconditioning: studies of contraction and function in isolated rat hearts. Am J Physiol Heart Circ Physiol 277: H136–H143, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Hicks CW, Li Y, Okugawa S, Solomon SB, Moayeri M, Leppla SH, Mohanty A, Subramanian GM, Mignone TS, Fitz Y, Cui X, Eichacker PQ. Anthrax edema toxin has cAMP-mediated stimulatory effects and high-dose lethal toxin has depressant effects in an isolated perfused rat heart model. Am J Physiol Heart Circ Physiol 300: H1108–H1118, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laird MW, Zukauskas D, Johnson K, Sampey GC, Olsen H, Garcia A, Karwoski JD, Cooksey BA, Choi GH, Askins J, Tsai A, Pierre J, Gwinn W. Production and purification of Bacillus anthracis protective antigen from Escherichia coli. Protein Expr Purif 38: 145–152, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence WS, Marshall JR, Zavala DL, Weaver LE, Baze WB, Moen ST, Whorton EB, Gourley RL, Peterson JW. Hemodynamic effects of anthrax toxins in the rabbit model and the cardiac pathology induced by lethal toxin. Toxins (Basel) 3: 721–736, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA 79: 3162–3166, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Cui X, Solomon SB, Remy K, Fitz Y, Eichacker PQ. B. anthracis edema toxin increases cAMP levels and inhibits phenylephrine-stimulated contraction in a rat aortic ring model. Am J Physiol Heart Circ Physiol 305: H238–H250, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Cui X, Su J, Haley M, Macarthur H, Sherer K, Moayeri M, Leppla SH, Fitz Y, Eichacker PQ. Norepinephrine increases blood pressure but not survival with anthrax lethal toxin in rats. Crit Care Med 37: 1348–1354, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu D, Homan LL, Dillon JS. Genistein acutely stimulates nitric oxide synthesis in vascular endothelial cells by a cyclic adenosine 5′-monophosphate-dependent mechanism. Endocrinology 145: 5532–5539, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Moayeri M, Leppla SH. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol 22: 317–325, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Zhang Y, Moayeri M, Liu J, Crown D, Fattah RJ, Wein AN, Yu ZX, Finkel T, Leppla SH. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 501: 63–68, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lv M, Liu K, Fu S, Li Z, Yu X. Pterostilbene attenuates the inflammatory reaction induced by ischemia/reperfusion in rat heart. Mol Med Rep 11: 724–728, 2015. [DOI] [PubMed] [Google Scholar]
- 21.Moayeri M, Crown D, Dorward DW, Gardner D, Ward JM, Li Y, Cui X, Eichacker P, Leppla SH. The heart is an early target of anthrax lethal toxin in mice: a protective role for neuronal nitric oxide synthase (nNOS). PLoS Pathog 5: e1000456, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remy KE, Qiu P, Li Y, Cui X, Eichacker PQ. B. anthracis associated cardiovascular dysfunction and shock: the potential contribution of both non-toxin and toxin components. BMC Med 11: 217, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su J, Cui X, Li Y, Mani H, Ferreyra GA, Danner RL, Hsu LL, Fitz Y, Eichacker PQ. SB203580, a p38 inhibitor, improved cardiac function but worsened lung injury and survival during Escherichia coli pneumonia in mice. J Trauma 68: 1317–1327, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutherland FJ, Hearse DJ. The isolated blood and perfusion fluid perfused heart. Pharmacol Res 41: 613–627, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Sweeney DA, Cui X, Solomon SB, Vitberg DA, Migone TS, Scher D, Danner RL, Natanson C, Subramanian GM, Eichacker PQ. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J Infect Dis 202: 1885–1896, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun 248: 706–711, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Wang QD, Tokuno S, Valen G, Sjoquist PO, Thoren P. Cyclic fluctuations in the cardiac performance of the isolated Langendorff-perfused mouse heart: pyruvate abolishes the fluctuations and has an anti-ischaemic effect. Acta Physiol Scand 175: 279–287, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Watson LE, Kuo SR, Katki K, Dang T, Park SK, Dostal DE, Tang WJ, Leppla SH, Frankel AE. Anthrax toxins induce shock in rats by depressed cardiac ventricular function. PLos One 2: e466, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watson LE, Mock J, Lal H, Lu G, Bourdeau RW, Tang WJ, Leppla SH, Dostal DE, Frankel AE. Lethal and edema toxins of anthrax induce distinct hemodynamic dysfunction. Front Biosci 12: 4670–4675, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Zhang XP, Hintze TH. cAMP signal transduction induces eNOS activation by promoting PKB phosphorylation. Am J Physiol Heart Circ Physiol 290: H2376–H2384, 2006. [DOI] [PubMed] [Google Scholar]