

Abstract
Pulmonary hypertension (PH) and right ventricular hypertrophy (RVH) affect 25–35% of premature infants with significant bronchopulmonary dysplasia (BPD), increasing morbidity and mortality. We sought to determine the role of phosphodiesterase 5 (PDE5) in the right ventricle (RV) and left ventricle (LV) in a hyperoxia-induced neonatal mouse model of PH and RVH. After birth, C57BL/6 mice were placed in room air (RA) or 75% O2 (CH) for 14 days to induce PH and RVH. Mice were euthanized at 14 days or recovered in RA for 14 days or 42 days prior to euthanasia at 28 or 56 days of age. Some pups received sildenafil or vehicle (3 mg·kg−1·dose−1 sc) every other day from P0. RVH was assessed by Fulton's index [RV wt/(LV + septum) wt]. PDE5 protein expression was analyzed via Western blot, PDE5 activity was measured by commercially available assay, and cGMP was measured by enzyme-linked immunoassay. Hyperoxia induced RVH in mice after 14 days, and RVH did not resolve until 56 days of age. Hyperoxia increased PDE5 expression and activity in RV, but not LV + S, after 14 days. PDE5 expression normalized by 28 days of age, but PDE5 activity did not normalize until 56 days of age. Sildenafil given during hyperoxia prevented RVH, decreased RV PDE5 activity, and increased RV cGMP levels. Mice with cardiac-specific overexpression of PDE5 had increased RVH in RA. These findings suggest normal RV PDE5 function is disrupted by hyperoxia, and elevated PDE5 contributes to RVH and remodeling. Therefore, in addition to impacting the pulmonary vasculature, sildenafil also targets PDE5 in the neonatal mouse RV and decreases RVH.
Keywords: PDE5, pulmonary hypertension, right ventricle, cyclic nucleotides
bronchopulmonary dysplasia (BPD) is a common complication of prematurity, affecting approximately one-third of extremely low birthweight infants (13, 46). Despite modest success in the prevention of BPD with therapies including inhaled nitric oxide, caffeine, and vitamin A, it still remains a significant problem in preterm infants, and there are 10,000 new cases annually in the United States (5, 9, 22, 34, 41). Approximately 25–35% of infants with moderate to severe BPD develop further complications of pulmonary hypertension (PH), right ventricular hypertrophy (RVH), and RV failure that increase morbidity and mortality (1, 4, 12, 21). Little is known about how to best treat BPD-associated PH acutely or chronically. Mortality for infants with BPD-associated PH and RV failure is reported to be as high as 50% by 2 years of age, and these infants have a 4.6-fold risk of death compared with matched patients with BPD alone (21, 36). Furthermore, pulmonary hypertension is an independent risk factor for predicting death or need for tracheostomy prior to discharge (29). Thus the presence of PH, RVH, and RV failure in BPD infants negatively influences patient outcomes.
There is a gap in knowledge of the molecular mechanisms and pathophysiology leading to the development of BPD-associated PH and its ultimate endpoint of RVH and RV failure. BPD is associated with abnormal lung and pulmonary vasculature development. Histological hallmarks of BPD include alveolar simplification, failure of angiogenesis, vascular remodeling, and increased vascular tone leading to increased pulmonary vascular resistance (PVR) (16, 38). This increased PVR increases afterload and strain on the right ventricle (RV) (10). In response to the pressure-overload, the right ventricle increases ventricular wall thickness. While this is initially a compensatory mechanism, it ultimately can lead to RV dilation and failure (40). While increased afterload is commonly thought to trigger adaptation and remodeling in the RV, neurohumoral signaling, oxidative stress, inflammation, ischemia, and cell death may all contribute to RV dilatation and failure (10).
cGMP is an important intracellular second messenger involved in the regulation of cardiovascular homeostasis. Its role is multifaceted, and it achieves multiple different effects through compartmentalization within the cardiac myocyte (17, 26, 28). cGMP negatively modulates contractility and accelerates relaxation of cardiomyocytes, affecting lusitropy and inotropy (39). cGMP is primarily produced through two separate pathways in the heart: BNP-stimulated production of particulate guanylate cyclase (pGC) and nitric oxide synthase (NOS)-stimulated production of soluble guanylate cyclase (sGC). cGMP has been shown to play a role and help protect against the hypertrophic maladaptive responses and remodeling in the myocardium (39, 40).
Phosphodiesterases are a family of eleven enzymes that are important downstream regulators of cyclic nucleotide signaling (17). In cardiac myocytes, PDE1, PDE2, PDE3, and PDE5 are known to regulate cGMP and cAMP. PDE5 is a cGMP-specific PDE that plays a significant role in the hydrolysis of cGMP in the cardiac myocyte of the failing heart (20, 23, 35). PDE5 has been shown to be upregulated in adult mouse models and human samples of RV and LV failure (30, 42, 43). In addition, PDE5 inhibitors increase RV inotropy in animal models of heart failure and in human clinical data (30).
To date, all of the data regarding phosphodiesterase and cyclic nucleotide signaling in the cardiac myocyte have been derived from adult animal models of RV and LV overload and/or failure as well as adult human heart samples. It is unknown whether PDE5 and cGMP play a similar role in the neonatal heart in general or in the neonatal RV specifically in response to PH. In the present study, we sought to determine the role of PDE5 in the RV and LV of neonatal mice with hyperoxia-induced PH and RVH. We further examined the time course of the heart's recovery after ending the hyperoxia exposure.
MATERIALS AND METHODS
Animal protocols.
The animal protocol used for this study was submitted and approved by the Institutional Animal Care and Use Committee at Northwestern University. Newborn C57Bl/6 mouse pups of both sexes (Charles River, Wilmington, MA) from age-matched litters were exposed to 75% O2 (CH) in a Plexiglas chamber (Biospherix, Lacona, NY) or to room air (RA) within 24 h of birth. Exposure to hyperoxia was continuous for 14 days, with brief interruptions for animal care and to rotate the dams every 24 h (3). Some litters of mice, along with age-matched controls, were allowed to recover in room air for either 14 days or 42 days (Fig. 1). Some litters received sildenafil [Revatio (0.8 mg/ml sildenafil in 5% dextrose), Pfizer, New York] or vehicle (sterile 5% dextrose) injections (3 mg·kg−1·dose−1 sc) every other day starting from P0, for a total of 7 injections (25). After the completion of the assigned protocol, the pups were euthanized for tissue harvest. Finally, for some studies, we also utilized cardiomyocyte-specific PDE5 overexpressing mice (PDE5TG) on a Taconic C57Bl/6 background that were kindly provided by Dr. Stefan Janssens (KU Leuven, Leuven, Belgium) (32).
Fig. 1.
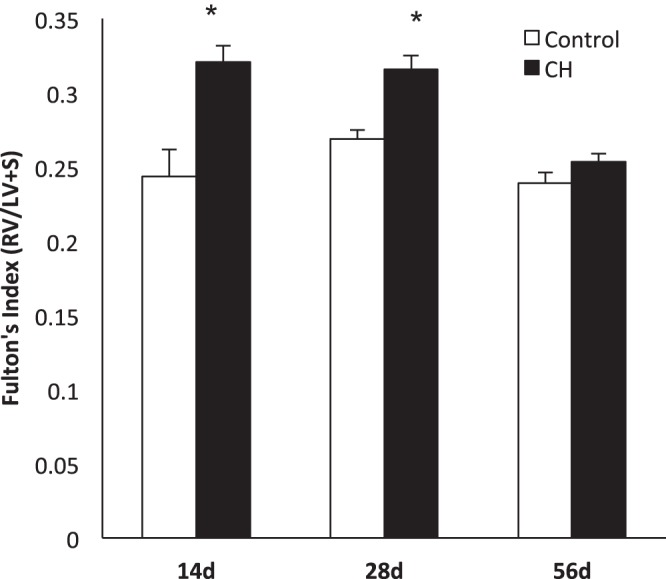
Right ventricular hypertrophy is present in chronic hyperoxia (CH) mice and slowly resolves with room air (RA) recovery: Fulton's index was measured at 3 time points in CH mice: 14 day mice (n = 6), 28 day mice (14 day exposure, 14 day recovery, n = 8), and 56 day mice (14 day exposure, 42 day recovery, n = 4). Comparisons were made with age-matched RA controls (n = 8, 13, and 4, respectively). Data are shown as means ± SE. RV, right ventricle; LV, left ventricle; S, septum. *P < 0.05 vs. age-matched controls.
Measurement of right ventricular hypertrophy (RVH).
Fulton's index, defined as the RV weight divided by the LV + septum weight, was used to assess RVH (3).
Western blot analysis.
Frozen RV tissue was pulverized, and total protein was collected and placed in lysis buffer (Millipore, Temecula, CA), supplemented with protease (Sigma-Aldrich, St. Louis, MO) and phosphatase inhibitors (EMD Biosciences, San Diego, CA) as previously described (14). Protein concentration was measured using the Bradford assay (11). PDE5 protein expression was assessed via Western blot, which was performed as previously described (14). Briefly, membranes were blocked at room temperature with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 (1× TBST) and incubated overnight at 4°C with primary antibody in 5% milk + 1× TBST at an appropriate dilution [1:750 for rabbit anti-PDE5a (Santa Cruz Biotechnology, Santa Cruz, CA) and 1:500 for β-actin (Sigma-Aldrich)] followed by secondary antibody at the appropriate dilution [anti-rabbit (1:750 Cell Signaling, Danvers, MA) and anti-mouse (1:1,000 Cell Signaling)]. Membranes were then washed and exposed via chemiluminescence (Pierce, Rockford, IL). Bands were analyzed using a Bio-Rad ChemiDoc XRS+ (Bio-Rad, Hercules, CA). Expression was normalized to β-actin. Data are shown as fold ± SE relative to control mice.
PDE5 activity assay.
Protein was prepared from snap-frozen RV tissue as described above and was purified over a Centri-Spin 10 column to remove any phosphate contamination (Princeton Separations, Adelphia, NJ). Protein concentration was then determined using the Bradford method. Total protein was assayed in duplicate for cGMP-hydrolytic activity using a commercially available colorimetric cyclic nucleotide phosphodiesterase assay kit (Enzo, Farmingdale, NY) with and without sildenafil (100 nM, Sigma-Aldrich) as previously described (24, 25). Results are shown as the sildenafil-inhibitable picomoles cGMP hydrolyzed per milligram total protein per minute.
cGMP enzyme immunoassay (EIA).
cGMP content in snap-frozen RV tissue was measured by EIA in duplicate, using a commercially available kit (Cayman Chemical, Ann Arbor, MI) as previously described (14). Results were measured using a Bio-Rad iMark automated plate reader (Bio-Rad) at 420 nm. Results are shown as picomoles cGMP per milliliter.
Measurement of medial wall thickness (MWT).
Small vessel remodeling was assessed by measuring MWT of small PAs. Mouse lungs were inflated and imaged using a Pixera microscope (40×); 6–8 images per animal were taken and analyzed in a blinded fashion. MWT was measured as the ratio of the area of small PA wall over the total PA area (25).
Measurement of alveolar area.
Lung sections were stained with hematoxylin for 16 h and lung morphometry images were taken on a Pixera microscope (20×); 6–8 images per animal were taken and analyzed in a blinded fashion. Mean alveolar area was measured using Scion software (Informer Technologies) (25).
Statistical analysis.
All data are expressed as means ± SE, with n representing the number of animals and with significance at P < 0.05. Results were analyzed by ANOVA with post hoc Bonferroni's analysis using Prism software (Graphpad Software, San Diego, CA).
RESULTS
RVH is present in CH mice and slowly resolves by 56 days of life (after 42 days of room air recovery).
Fulton's index was used to assess RVH in mice of all three age groups: 14 day exposure to CH, 28 days (14 day exposure + 14 day recovery), and 56 days (14 day exposure + 42 day recovery). The 14-day CH mice demonstrated significant RVH compared with RA controls (0.32 ± 0.02 vs. 0.24 ± 0.01, P < 0.05, Fig. 1). The 28-day CH mice continued to have RVH relative to age-matched RA controls (0.32 ± 0.01 vs. 0.26 ± 0.01, P < 0.05, Fig. 1). By 56 days, RVH was not present in the group of mice exposed to CH compared with age-matched RA controls (0.25 ± 0.01 vs. 0.24 ± 0.01, Fig. 1).
RV, but not LV + S, PDE5 expression is increased after exposure to chronic hyperoxia but normalizes with RA recovery.
As an increase in PDE5 protein expression has been shown in failing human hearts (30, 42, 43), we measured PDE5 expression in the RV of mice exposed to CH in all three age groups compared with age-matched RA controls. RV PDE5 expression was increased in 14-day CH mice compared with age-matched RA controls (2.7 ± 0.8-fold, P < 0.05, Fig. 2, A and B), However, it was not significantly different at 28 or 56 days compared with age-matched RA controls (Fig. 2, A and B). Meanwhile, there was no significant difference in LV PDE5 expression at any of the three time points in CH mice compared with age-matched RA controls (Table 1).
Fig. 2.

RV phosphodiesterase 5 (PDE5) expression is increased after 14 days of exposure to CH but normalizes with RA recovery. PDE5 expression was measured at 3 time points: 14 day mice, 28 day mice (14 day exposure and 14 day recovery), and 56 day mice (14 day exposure, 42 day recovery). A: RV PDE5 expression was measured by Western blot in CH mice (n = 7, 11, and 8, respectively) vs. age-matched RA controls (n = 11, 10, and 8, respectively). Data are shown as fold ± SE. *P < 0.05 vs. age-matched controls. B: representative Western blots of RV PDE5 expression were normalized to β-actin of CH vs. RA mice in the 3 different age groups.
Table 1.
LV PDE5/β-actin fold increase
| Control | Chronic Hyperoxia (CH) | |
|---|---|---|
| 14 day | 1 ± 0.29 (n = 5) | 1.18 ± 0.32 (n = 7) |
| 28 day | 1 ± 0.17 (n = 5) | 0.97 ± 0.18 (n = 7) |
| 56 day | 1 ± 0.24 (n = 6) | 1.30 ± 0.62 (n = 8) |
Values are means ± SE.
LV, left ventricle; PDE5, phosphodiesterase 5.
RV, but not LV + S, PDE5 activity is increased after CH and remains elevated after 14 days of RA recovery.
RV PDE5 activity was measured at the above-mentioned time points compared with age-matched RA controls. At 14 days, RV PDE5 activity was increased in CH mice compared with RA controls (402.9 ± 50.6 vs. 41.3 ± 79.6 pmol cGMP hydrolyzed·mg protein−1·min−1, P < 0.05, Fig. 3A). At 28 days, CH mice continued to have elevated RV PDE5 activity (196.5 ± 20.8 vs. 70.9 ± 23 pmol cGMP hydrolyzed·mg protein−1·min−1, P < 0.05, Fig. 3A). By 56 days, there was no significant difference in RV PDE5 activity found between the CH and RA control mice (Fig. 3A). Similar to the PDE5 expression data, there was no significant difference in LV PDE5 activity at any of the three time points in CH mice compared with age-matched RA controls (Table 2).
Fig. 3.
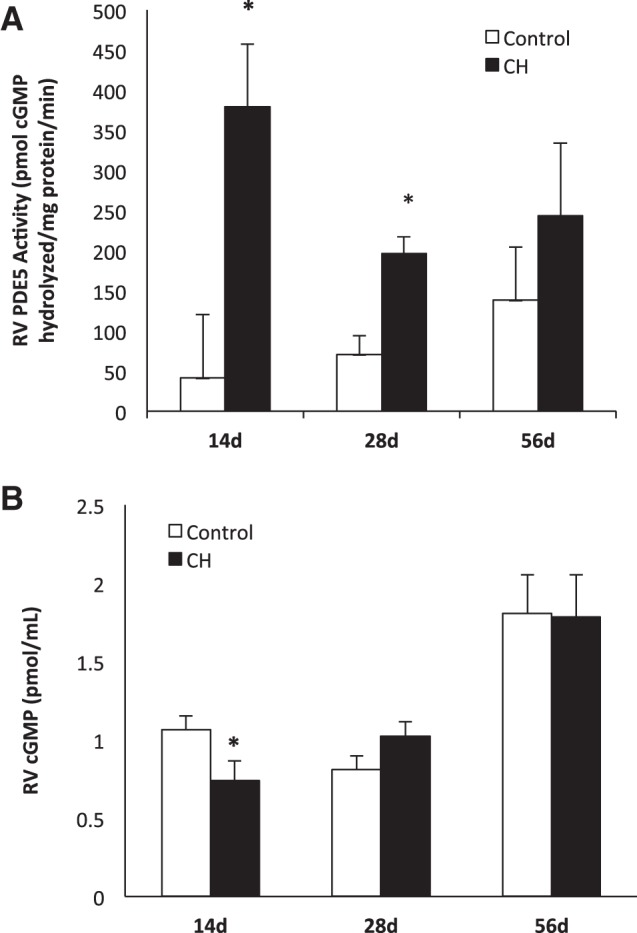
RV PDE5 activity is increased and cGMP decreased after CH but both normalize with RA recovery. A: RV PDE5 activity was measured at three time points: 14 day mice, 28 day mice (14 day exposure and 14 day recovery), and 56 day mice (14 day exposure, 42 day recovery). RV PDE5 activity was measured in CH mice (n = 7, 8, and 4, respectively) vs. age-matched RA controls (n = 10, 8, and 5) using a commercially available colorimetric cyclic nucleotide phosphodiesterase assay kit with and without sildenafil. Data are shown as means (pmol cGMP hydrolyzed·mg protein−1·min−1) ± SE. *P < 0.05 vs. age-matched controls. B: RV cGMP levels were measured in 14 day mice, 28 day mice, and 56 day mice by EIA in CH mice (n = 14, 8, and 4, respectively) vs. age-matched controls (n = 20, 8, and 3, respectively). Data are shown as means (pmol/ml) ± SE. *P < 0.05 vs. age-matched controls.
Table 2.
LV PDE5 activity
| Control | Chronic Hyperoxia (CH) | |
|---|---|---|
| 14 day | 231.9 ± 87.2 (n = 5) | 241.1 ± 202.5 (n = 5) |
| 28 day | 649.5 ± 209.5 (n = 8) | 586.8 ± 191.2 (n = 6) |
| 56 day | 412.6 ± 122.0 (n = 5) | 462.3 ± 212.5 (n = 6) |
Values are means ± SE in pmol cGMP hydrolyzed·mg protein−1·min−1.
RV cGMP levels are decreased after CH but normalized with RA recovery.
As cGMP is essential in lusitropy and contractility of the cardiac myocyte, cGMP levels were measured in the RV of mice exposed to CH, in all three age groups compared with age-matched RA controls. The 14-day CH mice demonstrated decreased cGMP levels in the RV (0.74 ± 0.13 vs. 1.06 ± 0.09 pmol cGMP/ml, P < 0.05, Fig. 3B). At 28 and 56 days, no significant differences were noted in cGMP levels in CH mice vs. age-matched controls (Fig. 3B).
Sildenafil treatment does not alter RV PDE5 protein expression.
There was no significant difference between RV PDE5 expression at 14 days in CH mice sildenafil compared with CH mice treated with vehicle (3.76 ± 2.25 vs 2.14 ± 1.3, Fig. 4A).
Fig. 4.
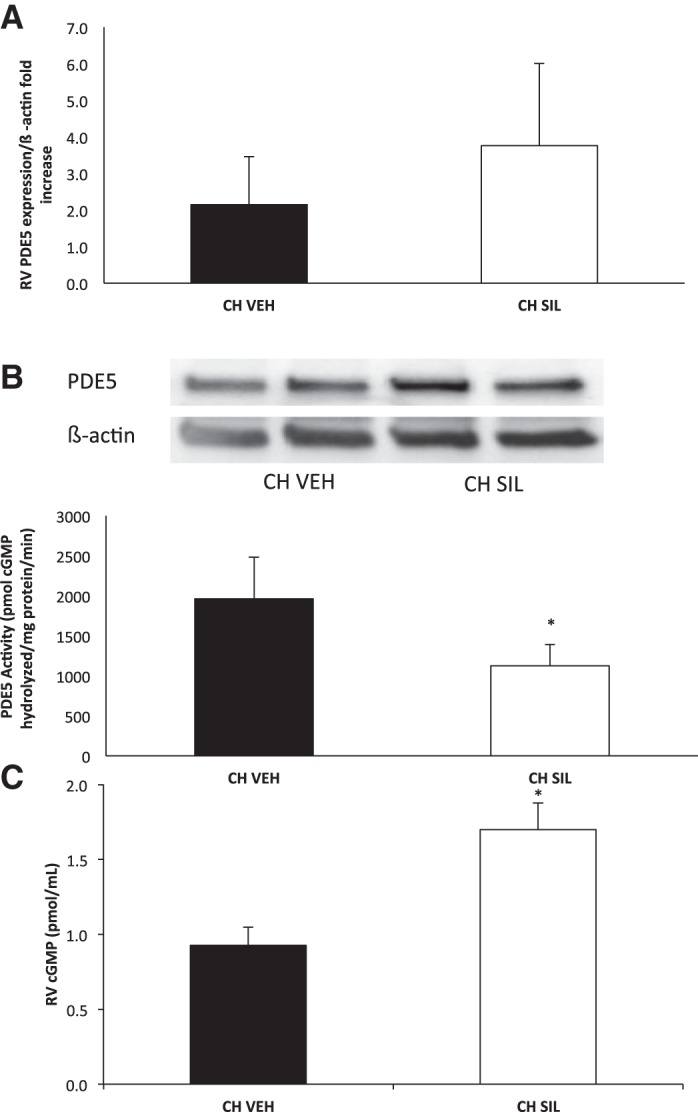
Sildenafil decreased RV PDE5 expression and activity and increased RV cGMP at 14 days in CH mice. A: RV PDE5 expression was measured in vehicle (VEH)- or sildenafil (SIL)-treated mice exposed to CH (n = 7 treated with vehicle, 8 treated with sildenafil). Data are shown as fold ± SE. B: RV PDE5 activity was measured in vehicle- or sildenafil-treated mice exposed to CH (n = 5 treated with vehicle, 4 treated with sildenafil) at 14 days using a commercially available colorimetric cyclic nucleotide phosphodiesterase assay kit with and without sildenafil. Data are shown as means (pmol cGMP hydrolyzed·mg protein−1·min−1) ± SE. *P < 0.05. C: RV cGMP levels were measured via EIA in vehicle- or sildenafil-treated mice exposed to CH (n = 5 treated with vehicle, n = 4 treated with sildenafil). Data are shown as means (pmol/ml) ± SE. *P < 0.05.
Sildenafil decreased PDE5 activity and increased cGMP in the RV in CH mice.
RV PDE5 activity was significantly decreased in 14-day CH mice receiving sildenafil compared with 14-day CH mice receiving vehicle (1,124.7 ± 265.7 vs 1,961.1 ± 520.9 pmol cGMP hydrolyzed·mg protein−1·min−1, P < 0.05, Fig. 4B). There was no difference in LV PDE5 activity in 14-day CH mice receiving sildenafil compared with CH mice receiving vehicle (data not shown). Consistent with the data on PDE5 activity, sildenafil treatment increased cGMP levels in the RV in 14-day CH mice receiving sildenafil compared with 14-day CH mice receiving vehicle (1.7 ± 0.18 vs. 0.93 ± 0.12 pmol cGMP/ml, P < 0.05, Fig. 4C).
Cardiac myocyte specific overexpression of PDE5 leads to RVH in RA at 14 days.
Mice with cardiac myocyte-specific PDE5 overexpression (PDE5TG) have previously been shown to be more susceptible to left-sided heart failure in the setting of pressure overload. Adult PDE5TG mice are reported to have nine times the expression of PDE5 relative to adult isogenic wild-type mice (32, 42). We verified that the PDE5TG RV had significant overexpression of PDE5 relative to the isogenic wild-type littermates at 14 days (Fig. 5A). Interestingly, while these mice have no functional defects detected by echocardiography at baseline as adults (32), we discovered that these mice have an increased Fulton's index indicative of RVH at baseline compared with controls in RA (0.32 ± 0.07 vs. 0.27 ± 0.04, P < 0.05, Fig. 5B). Since these mice were supposed to be cardiac-specific PDE5 overexpressers, we sought to determine if there were any changes in the medial wall thickness or alveolar area. As shown in Fig. 5, C and D, there is no change in the medial wall thickness of the PDE5 TG mice relative to their wild-type littermates. Neither is there any difference in alveolar area in the PDE5 TG mice relative to their wild-type littermates (Fig. 5, E and F). This confirms that the increase in Fulton's index seen in Fig. 5B is a result of the cardiac overexpression of PDE5.
Fig. 5.

PDE5-overexpressing mice have increased right ventricular hypertrophy in room air. A: representative Western blot of RV protein. M represents marker and PC represents PDE5 recombinant protein positive control. Lanes 1–4 show isogenic PDE5 wild-type mice, and lanes 5–8 show heterozygous PDE5-overexpressing (OE) (PDE5TG) mice. B: Fulton's index was measured at 14 days in heterozygous PDE5 wild-type (WT) mice (n = 14) and heterozygous PDE5-overexpressing mice (n = 12). Data are shown as means ± SE. *P < 0.05 vs. PDE5 wild-type mice. C: medial wall thickness was measured at 14 days in isogenic PDE5 wild-type mice (n = 6) and PDE5-overexpressing mice (n = 4). Data are shown as means ± SE. D: representative hematoxylin- and eosin-stained lung sections for PDE5WT and PDE5-overexpressing mice at 14 days in RA. E: mean alveolar area was measured at 14 days in isogenic PDE5 wild-type mice (n = 6) and PDE5-overexpressing mice (n = 4). Data are shown as means ± SE. F: representative hematoxylin-stained lung sections for PDE5WT and PDE5-overexpressing mice at 14 days in RA.
DISCUSSION
Phosphodiesterases, specifically PDE5, have been shown to be present in the cardiac myocyte, and limited data suggest PDE5 protein expression is increased in the failing or strained RV and LV. PDE5 is known to play a role in the cGMP hydrolysis and has been shown to be upregulated in models of heart failure (30, 32, 42). Until now, most data have been collected from adult animal models of pulmonary hypertension, pressure overload, and heart failure, which differs significantly from the neonatal model of hyperoxia-induced pulmonary hypertension. Furthermore, it is unknown if these pathways remain as critical at earlier time points in cardiac development. A recent study showed that perinatal inflammation combined with neonatal hyperoxia exposure resulted in early cardiac dysfunction (44). In the present study, we found that after 14 days of exposure to hyperoxia, mice had RVH compared with their age-matched RA controls. Moreover, this RVH continued to be present after recovery in room air, and only resolved after 56 days (Fig. 1). This suggests that hyperoxia-induced RVH is recoverable, but that it takes a prolonged period of time, as our 56 days represents a fully mature adult mouse.
We then sought to determine if cGMP and PDE5 signaling were impacted in this hyperoxia-induced model of RVH. As previous studies focused largely on failing hearts, it was unknown if we would detect cGMP signaling changes in our model where RVH precedes heart failure. In this study, we found that cGMP levels in the RV were decreased in 14 day mice exposed to hyperoxia. Decreased cGMP is likely to impair cardiac relaxation, contractility, and ion channel responsiveness. We found that, with room air recovery, these levels gradually normalized within 14 days of returning to RA.
To investigate the mechanisms responsible for the decrease in cGMP, we examined the expression and activity of PDE5 in the RV. In adult pulmonary arterial hypertension or heart failure in animals and humans, increased levels of RV PDE5 have previously been described (18, 30, 35). In 14 day mice exposed to CH, both RV PDE5 protein expression and activity are increased compared with RA controls. This suggests that upregulation of PDE5 in these mice causes increased hydrolysis of cGMP. There was no significant difference in either LV PDE5 protein expression and activity in mice exposed to CH vs. RA controls at 14 days, suggesting that hyperoxia-induced changes in PDE5 are specific to the RV and PDE5 is differentially regulated between the right and left ventricles of the heart. In addition, even though PDE5 expression returns to control levels after 14 days of room air recovery, PDE5 activity continued to remain elevated in those mice initially exposed to hyperoxia. We have previously described in this neonatal mouse model of pulmonary hypertension that PDE5 activity can be increased without increased PDE5 expression in the pulmonary vasculature (24). Our previous data suggest that PDE5 activity can be posttranslationally regulated by hyperoxia-induced ROS (15, 24, 25). Intriguingly, the timeline for the return of PDE5 activity to baseline mirrors that of the Fulton's index timeline, thereby suggesting that increased PDE5 activity may contribute to cardiac hypertrophy and remodeling. In support of that hypothesis, treatment with sildenafil, a PDE5 selective inhibitor, during hyperoxia led to decreased PDE5 activity and increased cGMP levels in the RV at 14 days. We have previously reported that the dose of sildenafil used here appears to be very selective for improving cGMP signaling in the cardiovascular system (25). We demonstrated that it can decrease RVH and pulmonary vascular remodeling; however, it did not improve lung morphometry in our neonatal mouse model of hyperoxia-induced lung disease and PH (25).
While sildenafil has been FDA-approved for the treatment of pulmonary arterial hypertension in adults and is currently under investigation in adults with heart failure (18, 28, 30, 35), use of sildenafil in the neonatal period is still experimental. There are two trials to suggest that sildenafil may be useful in term infants with pulmonary hypertension (6, 37). A recent single-center study in preterm infants with BPD-associated PH showed that short-term administration of sildenafil decreased PA pressures but did not significantly improve gas exchange (31). A retrospective review of patients with PH and chronic lung disease showed chronic use of sildenafil clinically improved PH (27). Our studies suggest that sildenafil prevents hyperoxia-induced RVH and attenuates hyperoxia-induced PDE5 activity in the mouse RV. Therefore, sildenafil in neonatal patients may target PDE5 in both the pulmonary vasculature and the right ventricle.
Since the sildenafil in our study was given systemically, it is difficult to determine whether the impact of sildenafil on the RV was due to a specific effect of PDE5-inhibition on the heart, pressure-unloading as a result of improvement in the pulmonary vasculature, or a combination of both pulmonary vascular and cardiac effects. Data evaluating the effect of sildenafil in the pressure overloaded RV is conflicting. Several studies using the monocrotaline-induced pulmonary hypertension adult animal model showed sildenafil reduced RVH and improved RV systolic function (19, 33), while studies using pulmonary artery banding models showed sildenafil did not impact RVH (2, 33), suggesting that the effects of sildenafil may vary in certain clinical situations. To date, no cardiac-specific PDE5 knockout mouse exists. However, there is a previously described mouse with cardiomyocyte-specific overexpression of PDE5 (32, 42). These mice have previously been described to have no hemodynamic changes in adulthood at baseline relative to isogenic wild-type mice as measured by echocardiography. Fulton's index is an indirect approximation of pulmonary hypertension. Unfortunately due to the size of the 14 day old mice (5–6 g on average) and the equipment available at our institution, we were unable to measure PA pressures or echocardiographic measures of pulmonary hypertension such as pulmonary artery acceleration time to right ventricular ejection time ratio (PAAT/ET) on echocardiogram. However, based on review of the literature, few have been successful to date with these measurements on such small pups. We do note that these mice have an increased Fulton's index, suggestive of RVH, at 14 days of age in room air. However, we have demonstrated that they do not have significant changes in either alveolar area or medial wall thickness (Fig. 5). While mild RVH is present, these mice survive into adulthood with few adverse events, suggesting that a second “hit” may be needed to expose the true impact of this PDE5 overexpression. Consistent with this phenomenon, PDE5TG mice have been described to have more pronounced LV hypertrophy when subjected to LV pressure overload (32, 42).
Taken together, our studies suggest a temporal relationship between RVH and increased PDE5 in the RV of mice exposed to CH for 14 days. These changes resolve with room air recovery, but they take a significant amount of time to do so. This represents an interesting model to study possible therapeutic interventions for human infants with BPD-associated pulmonary hypertension and RVH. We know that a subset of these infants will recover over time, like the mice, but others will develop progressive RVH, possibly leading to right heart failure and death. Thus, while our mice do not appear to develop the more malignant phenotype, the mouse model provides a platform to test potential therapies to speed the recovery of RVH in mice with established disease. More specifically, our data suggest that inhibition of PDE5 during hyperoxia exposure would likely normalize cGMP levels and PDE5 activity and thus contribute to either attenuation or prevention of RVH. It remains to be seen whether sildenafil given after hyperoxia-exposure can reverse established pulmonary vascular disease and RVH.
The clinical effects of long-term sildenafil use for BPD-associated pulmonary hypertension remain unknown. An extension phase of the STARTS trial reported that children ages 1–17 chronically taking a high dose of Revatio (> 6 mg·kg−1·day−1) had a higher risk of death than children taking lower doses (7, 8). However, this study did not include infants or children with PH associated with lung disease, so it is impossible to extrapolate these data to our BPD patient population of interest. Furthermore, we used subcutaneous injections of sildenafil at 3 mg·kg−1·dose−1 every other day, which falls below the high-dose group reported in the STARTS-2 trial, and closely mimics the doses given to term infants in clinical trials (37, 45). At this dosing range, we observed an improvement in RV PDE5 activity and cGMP, and we have previously published that this sildenafil dose improves RVH (25). Based on our previous studies where sildenafil did not improve alveolar area, low-dose sildenafil appears to be very selective for improving cGMP signaling in the cardiovascular system (25). Further clinical studies will be needed to determine whether sildenafil is safe in infants and what the optimal dose is to minimize side effects while maximizing cardiovascular gain. Finally additional studies will be needed in cells and animal models to determine if there might be drugs other than sildenafil with better safety profiles that can restore normal PDE5 activity and cGMP levels.
In conclusion, we have demonstrated that hyperoxia-induced RVH is significant and slow to recover after return to normoxia. Hyperoxia induces PDE5 expression and activity specifically in the RV, but not the LV + S. Sildenafil given during hyperoxia blunts hyperoxia-induced PDE5 activity and decreases in cGMP levels. Furthermore, mice overexpressing PDE5 in the cardiomyocytes develop RVH at baseline in room air. Together, these data suggest that PDE5 is a key modulator of right ventricle remodeling in the neonatal period. Future studies must further address both the mechanism(s) for PDE5 regulation in the RV as well as the evaluation of newer, potentially safer, pharmaceutical options to speed the regression of hyperoxia-induced RVH.
GRANTS
This study was supported by an American Academy of Pediatrics Marshall Klaus Perinatal Research Award (R. P. Heilman) and National Heart, Lung, and Blood Institute Grants HL-54705 (R. H. Steinhorn) and HL-109478 (K. N. Farrow).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.P.H., M.B.L., K.J.L., J.M.T., G.A.K., S.K.B., R.H.S., and K.N.F. conception and design of research; R.P.H., M.B.L., K.J.L., J.M.T., G.A.K., S.K.B., and K.N.F. performed experiments; R.P.H. and M.B.L. analyzed data; R.P.H., M.B.L., K.J.L., S.K.B., and K.N.F. interpreted results of experiments; R.P.H. and M.B.L. prepared figures; R.P.H. and M.B.L. drafted manuscript; R.P.H., M.B.L., K.J.L., J.M.T., G.A.K., S.K.B., R.H.S., and K.N.F. edited and revised manuscript; R.P.H., M.B.L., K.J.L., J.M.T., G.A.K., S.K.B., R.H.S., and K.N.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge technical support by J. Whitesides and critical reading of this manuscript by Dr. C. Epting. We thank Dr. S. Janssens for providing the cardiomyocyte-specific PDE5-overexpressing mice.
Present address for S. K. Berkelhamer: Univ. of Buffalo, Buffalo, NY.
REFERENCES
- 1.An HS, Bae EJ, Kim GB, Kwon BS, Beak JS, Kim EK, Kim HS, Choi JH, Noh CI, Yun YS. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Kor Circ J 40: 131–136, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen A, Nielsen JM, Peters CD, Schou UK, Sloth E, Nielsen-Kudsk JE. Effects of phosphodiesterase-5 inhibition by sildenafil in the pressure overloaded right heart. Eur J Heart Fail 10: 1158–1165, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, Kourembanas S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med 180: 1122–1130, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker CD, Abman SH, Mourani PM. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Pediatr Allergy Immunol Pulmonol 27: 8–16, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballard RA, Truog WE, Cnaan A, Martin RJ, Ballard PL, Merrill JD, Walsh MC, Durand DJ, Mayock DE, Eichenwald EC, Null DR, Hudak ML, Puri AR, Golombek SG, Courtney SE, Stewart DL, Welty SE, Phibbs RH, Hibbs AM, Luan X, Wadlinger SR, Asselin JM, Coburn CE NO. CLD Study Group. Inhaled nitric oxide in preterm infants undergoing mechanical ventilation. N Engl J Med 355: 343–353, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Baquero H, Soliz A, Neira F, Venegas ME, Sola A. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: a pilot randomized blinded study. Pediatrics 117: 1077–1083, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Barst RJ, Beghetti M, Pulido T, Layton G, Konourina I, Zhang M, Ivy DD, Investigators S. STARTS-2: long-term survival with oral Sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension. Circulation 129: 1914–1923, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, Sastry BK, Pulido T, Layton GR, Serdarevic-Pehar M, Wessel DL. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation 125: 324–334, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Berkelhamer SK, Mestan KK, Steinhorn RH. Pulmonary hypertension in bronchopulmonary dysplasia. Semin Perinatol 37: 124–131, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135: 794–804, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976. [DOI] [PubMed] [Google Scholar]
- 12.Check J, Gotteiner N, Liu X, Su E, Porta N, Steinhorn R, Mestan KK. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J Perinatol 33: 553–557, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fanaroff AA, Wright LL, Stevenson DK, Shankaran S, Donovan EF, Ehrenkrans RA, Younes N, Korones SB, Stoll BJ, Tyson JE, Bauer CR, Oh W, Lemons JA, Papile LA, Verter J. Very-Low-Birth-Weight Outcomes of the National-Institute-of-Child-Health-and-Human-Development Neonatal Research Network, May 1991 through December 1992. Am J Obstet Gynecol 173: 1423–1431, 1995. [DOI] [PubMed] [Google Scholar]
- 14.Farrow KN, Lakshminrusimha S, Czech L, Groh BS, Gugino SF, Davis JM, Russell JA, Steinhorn RH. SOD and inhaled nitric oxide normalize phosphodiesterase 5 expression and activity in neonatal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 299: L109–L116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farrow KN, Lee KJ, Perez M, Schriewer JM, Wedgwood S, Lakshminrusimha S, Smith CL, Steinhorn RH, Schumacker PT. Brief hyperoxia increases mitochondrial oxidation and increases phosphodiesterase 5 activity in fetal pulmonary artery smooth muscle cells. Antioxid Redox Signal 17: 460–470, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signaling. Am J Respir Crit Care Med 176: 1146–1153, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammond J, Balligand JL. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol 52: 330–340, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Hsu S, Nagayama T, Koitabashi N, Zhang M, Zhou L, Bedja D, Gabrielson KL, Molkentin JD, Kass DA, Takimoto E. Phosphodiesterase 5 inhibition blocks pressure overload-induced cardiac hypertrophy independent of the calcineurin pathway. Cardiovasc Res 81: 301–309, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janssen W, Schermuly RT, Kojonazarov B. The role of cGMP in the physiological and molecular responses of the right ventricle to pressure overload. Exp Physiol 98: 1274–1278, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Kass DA. Cardiac role of cyclic-GMP hydrolyzing phosphodiesterase type 5: from experimental models to clinical trials. Current Heart Fail Reports 9: 192–199, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV, Thomas KC, Mullen MP. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics 120: 1260–1269, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Kinsella JP, Cutter GR, Walsh WF, Gerstmann DR, Bose CL, Hart C, Sekar KC, Auten RL, Bhutani VK, Gerdes JS, George TN, Southgate WM, Carriedo H, Couser RJ, Mammel MC, Hall DC, Pappagallo M, Sardesai S, Strain JD, Baier M, Abman SH. Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N Engl J Med 355: 354–364, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Lee DI, Kass DA. Phosphodiesterases and cyclic GMP regulation in heart muscle. Physiology 27: 248–258, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Lee KJ, Berkelhamer SK, Kim GA, Taylor JM, O'Shea KM, Steinhorn RH, Farrow KN. Disrupted pulmonary artery cGMP signaling in mice with hyperoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 50: 369–378, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee KJ, Berkelhamer SK, Kim GA, Taylor JM, O'Shea KM, Steinhorn RH, Farrow KN. Disrupted pulmonary artery cyclic guanosine monophosphate signaling in mice with hyperoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 50: 369–378, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mokni W, Keravis T, Etienne-Selloum N, Walter A, Kane MO, Schini-Kerth VB, Lugnier C. Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PloS one 5: e14227, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr 154: 379–384, e371–372, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Movsesian MA, Kukreja RC. Phosphodiesterase inhibition in heart failure. Handbook Exp Pharmacol 237–249, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Murthy K, Savani RC, Lagatta JM, Zaniletti I, Wadhawan R, Truog W, Grover TR, Zhang H, Asselin JM, Durand DJ, Short BL, Pallotto EK, Padula MA, Dykes FD, Reber KM, Evans JR. Predicting death or tracheostomy placement in infants with severe bronchopulmonary dysplasia. J Perinatol 34: 543–548, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 116: 238–248, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Nyp M, Sandritter T, Poppinga N, Simon C, Truog WE. Sildenafil citrate, bronchopulmonary dysplasia and disordered pulmonary gas exchange: any benefits? J Perinatol 32: 64–69, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C, Marsboom G, Gheysens O, Vermeersch P, Biesmans L, Liu X, Gillijns H, Pellens M, Van Lommel A, Buys E, Schoonjans L, Vanhaecke J, Verbeken E, Sipido K, Herijgers P, Bloch KD, Janssens SP. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 119: 408–416, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schafer S, Ellinghaus P, Janssen W, Kramer F, Lustig K, Milting H, Kast R, Klein M. Chronic inhibition of phosphodiesterase 5 does not prevent pressure-overload-induced right-ventricular remodelling. Cardiovasc Res 82: 30–39, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt B, Roberts RS, Davis P, Doyle LW, Barrington KJ, Ohlsson A, Solimano A, Tin W, Caffeine for Apnea of Prematurity Trial Group. Caffeine therapy for apnea of prematurity. N Engl J Med 354: 2112–2121, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Shan X, Quaile MP, Monk JK, French B, Cappola TP, Margulies KB. Differential expression of PDE5 in failing and nonfailing human myocardium. Circ Heart Fail 5: 79–86, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slaughter JL, Pakrashi T, Jones DE, South AP, Shah TA. Echocardiographic detection of pulmonary hypertension in extremely low birth weight infants with bronchopulmonary dysplasia requiring prolonged positive pressure ventilation. J Perinatol 31: 635–640, 2011. [DOI] [PubMed] [Google Scholar]
- 37.Steinhorn RH, Kinsella JP, Pierce C, Butrous G, Dilleen M, Oakes M, Wessel DL. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr 155: 841–847, e841, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol 67: 623–661, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Takimoto E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ J 76: 1819–1825, 2012. [DOI] [PubMed] [Google Scholar]
- 40.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther 122: 216–238, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyson JE, Wright LL, Oh W, Kennedy KA, Mele L, Ehrenkranz RA, Stoll BJ, Lemons JA, Stevenson DK, Bauer CR, Korones SB, Fanaroff AA. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med 340: 1962–1968, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Vandenwijngaert S, Pokreisz P, Hermans H, Gillijns H, Pellens M, Bax NA, Coppiello G, Oosterlinck W, Balogh A, Papp Z, Bouten CV, Bartunek J, D'Hooge J, Luttun A, Verbeken E, Herregods MC, Herijgers P, Bloch KD, Janssens S. Increased cardiac myocyte PDE5 levels in human and murine pressure overload hypertrophy contribute to adverse LV remodeling. PloS one 8: e58841, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandeput F, Krall J, Ockaili R, Salloum FN, Florio V, Corbin JD, Francis SH, Kukreja RC, Movsesian MA. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther 330: 884–891, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Velten M, Gorr MW, Youtz DJ, Velten C, Rogers LK, Wold LE. Adverse perinatal environment contributes to altered cardiac development and function. Am J Physiol Heart Circ Physiol 306: H1334–H1340, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker DK, Ackland MJ, James GC, Muirhead GJ, Rance DJ, Wastall P, Wright PA. Pharmacokinetics and metabolism of sildenafil in mouse, rat, rabbit, dog and man. Xenobiotica 29: 297–310, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Walsh MC, Szefler S, Davis J, Allen M, Van Marter L, Abman S, Blackmon L, Jobe A. Summary proceedings from the bronchopulmonary dysplasia group. Pediatrics 117: S52–56, 2006. [DOI] [PubMed] [Google Scholar]