

Abstract
Dietary protein restriction has multiple benefits in kidney disease. Because protein intake is a major determinant of endogenous acid production, it is important that net acid excretion change in parallel during protein restriction. Ammonia is the primary component of net acid excretion, and inappropriate ammonia excretion can lead to negative nitrogen balance. Accordingly, we examined ammonia excretion in response to protein restriction and then we determined the molecular mechanism of the changes observed. Wild-type C57Bl/6 mice fed a 20% protein diet and then changed to 6% protein developed an 85% reduction in ammonia excretion within 2 days, which persisted during a 10-day study. The expression of multiple proteins involved in renal ammonia metabolism was altered, including the ammonia-generating enzymes phosphate-dependent glutaminase (PDG) and phosphoenolpyruvate carboxykinase (PEPCK) and the ammonia-metabolizing enzyme glutamine synthetase. Rhbg, an ammonia transporter, increased in expression in the inner stripe of outer medullary collecting duct intercalated cell (OMCDis-IC). However, collecting duct-specific Rhbg deletion did not alter the response to protein restriction. Rhcg deletion did not alter ammonia excretion in response to dietary protein restriction. These results indicate 1) dietary protein restriction decreases renal ammonia excretion through coordinated regulation of multiple components of ammonia metabolism; 2) increased Rhbg expression in the OMCDis-IC may indicate a biological role in addition to ammonia transport; and 3) Rhcg expression is not necessary to decrease ammonia excretion during dietary protein restriction.
Keywords: proximal tubule, protein restriction, collecting duct, ammonia, acid base
renal ammonia metabolism has an important role in both acid-base homeostasis and nitrogen metabolism (58, 60, 61).1 It is the primary component of basal net acid excretion, and changes in ammonia excretion are a primary component of changes in acid excretion in response to a wide variety of acid-base disorders. In addition, because ammonia excretion necessarily involves nitrogen excretion, changes in ammonia excretion contribute to nitrogen balance (57).
Dietary protein restriction is commonly used in the management of patients with chronic kidney disease because of beneficial changes in glomerular hemodynamics and subsequent changes in the rate of progression of loss of renal function (13, 38, 41, 45). Because protein metabolism is the primary determinant of endogenous acid production, dietary protein restriction also results in decreased endogenous acid loads (14, 48). To maintain acid-base homeostasis and to maintain nitrogen balance, dietary protein restriction demands an associated decrease in net acid and ammonia excretion.
The purpose of the current study was to determine the effect of dietary protein restriction on renal ammonia metabolism. We studied wild-type C57Bl/6 mice placed on isocaloric diets containing normal protein and with a 70% reduction in protein content. The reduced protein diet induced a rapid and substantial decrease in renal ammonia excretion. To determine the mechanism of this decrease in ammonia excretion, we examined changes in the expression of proteins involved in intrarenal ammonia metabolism and transport. Because dietary protein restriction resulted in a paradoxical increase in the expression of the ammonia transport protein Rh B glycoprotein (Rhbg) in the outer medullary collecting duct (OMCD) intercalated cell, we also examined the effect of collecting duct-specific Rhbg deletion. Finally, because the related ammonia-transporting protein Rh C glycoprotein (Rhcg) has a central role in renal ammonia transport (58, 59, 61, 62), we determined whether Rhcg deletion would alter ammonia excretion in mice with dietary protein restriction.
METHODS
Animals.
Wild-type C57Bl/6 mice were obtained from Harlan Sprague-Dawley (Indianapolis, IL). Mice with kidney collecting duct-specific deletion of Rhbg (CD-Rhbg-KO) were generated using Cre-loxP techniques. Briefly, we bred mice with floxed Rhbg alleles and expressing Cre-recombinase under control of the Ksp-cadherin promoter (Ksp-Cre) with mice homozygous for floxed Rhbg alleles but not expressing Ksp-Cre, as described previously (4, 5, 30, 34–37). Global Rhcg knockout mice were generated by breeding female Rhcg floxed mice with floxed Rhcg male mice expressing Cre-recombinase under control of the H+-ATPase B1 subunit promoter (B1-Cre). Mice in which Cre-recombinase expression in sperm resulted in heterozygous germline Rhcg gene recombination were crossbred until mice with homozygous Rhcg gene recombination were identified. These mice in which the B1-Cre transgene was not present were then used as global Rhcg knockout mice. We genotyped all mice used in these studies using tail-clip samples, as we have described previously (4, 5, 30, 34–37). Animal breeding was performed in the University of Florida College of Medicine Cancer and Genetics Transgenic Animal Core Facility by trained personnel. All animal studies were approved by the University of Florida College of Medicine and the North Florida/South Georgia Veterans Health System Institutional Animal Care and Use Committee.
Antibodies.
Affinity-purified antibodies to Rhbg and Rhcg generated in our laboratory have been characterized previously (5, 30, 34, 40, 53, 56). Norman Curthoys (Colorado State University) graciously provided antibodies to phosphate-dependent glutaminase (PDG), Fiona Karet (Cambridge Institute for Medical Research, Cambridge, England) graciously supplied antibodies to the a4 subunit of H+-ATPase, and H. Moo Kwon (Ulsan National Institute of Science and Technology, Ulsan, South Korea) graciously provided antibodies to NKCC2. Antibodies to phosphoenolpyruvate carboxykinase (PEPCK) were obtained from Cayman Chemical (Ann Arbor, MI), antibodies to glutamine synthetase (GS) were obtained from Chemicon (Temecula, CA), and antibodies to NHE3 were obtained from StressMarq Biosciences (Victoria, BC, Canada).
Protein diet.
Powdered, semisynthetic diets with either normal (20%) or low (6%) protein content were obtained from Harland Laboratories (TD.91352 and TD.90016, respectively). Mice were allowed to acclimate to metabolic cages (Tecniplast diuresis metabolic cage; Fisher Scientific) for 3 days while being fed a normal (20% protein) diet and were then randomized to normal or low-protein diets for the next 10 days. Daily food intake was measured. At all times, animals were provided ad libitum access to water. Daily urine was collected under mineral oil, and daily urine volume and pH were recorded. Urine samples were stored at −20°C until analyzed further.
Electrolyte measurements.
Urine ammonia was measured using a modification of a commercially available kit (A7553; Pointe Scientific, Canton, MI) as described previously (5, 34). Urine pH was measured using a micro-pH electrode (ROSS semi-micro pH; Orion 8115BN; Thermo Scientific). Serum bicarbonate was measured as total CO2 in 1-μl serum samples using a commercially available kit (Sigma-Aldrich, St. Louis, MO) modified for use in 1-μl samples. Plasma Na+ and K+ were measured using a flame photometer (Instrumentation Laboratory, Lexington, MA). Titratable acid was measured using standard techniques we have described previously (34).
Tissue preparation for immunolocalization.
Mice were anesthetized with inhalant isoflurane. The kidneys were preserved by in vivo cardiac perfusion with PBS (pH 7.4) followed by periodate-lysine-2% paraformaldehyde (PLP) and then cut transversely into several 2- to 3-mm-thick slices and immersed for 48 h at 4°C in the same fixative. Kidney samples from each animal were embedded in polyester wax made using polyethylene glycol 400 distearate (Polysciences, Warrington, PA) with 10% 1-hexadecanol, and 3-μm-thick sections were cut and mounted on gelatin-coated glass slides.
Immunohistochemistry.
Immunolocalization was accomplished using standard immunoperoxidase procedures. Briefly, sections were dewaxed in ethanol, rehydrated, and then rinsed in PBS. Endogenous peroxidase activity was blocked by incubating the sections in 3% H2O2 in distilled water for 45 min. The sections were blocked for 15 min with Serum-Free Protein Block (Dako Cytomation) and were then incubated at 4°C overnight with primary antibody. The sections were washed in PBS and incubated for 30 min with polymer-linked, peroxidase-conjugated goat anti-rabbit IgG (MACH2; Biocare Medical, Concord, CA), again washed with PBS, and then exposed to diaminobenzidine (DAB) for 5 min. The sections were washed in distilled water, dehydrated with xylene, mounted, and observed by light microscopy. Comparisons of labeling were made only between sections from the same immunohistochemistry experiment. Sections were examined on a Nikon E600 microscope equipped with DIC optics and photographed using a DXM1200F digital camera and ACT-1 software (Nikon). Color correction was performed using Adobe Photoshop CS2 software (Adobe Systems, San Jose, CA).
Double-immunolabeling procedure.
Double immunolabeling was done using sequential immunoperoxidase procedures as previously described (34). Briefly, tissue sections were labeled with the first primary antibody following the procedure described above. After the DAB reaction, sections were washed in PBS and then blocked using 3% H2O2 in methanol. The above procedure was repeated using a second primary antibody and Vector SG (Vector Laboratories, Burlingame, CA) for the peroxidase substrate, which produces a blue reaction product easily distinguished from the DAB brown reaction product. Sections were then washed with glass distilled water, dehydrated with xylene, mounted with Permount, and observed by light microscopy.
Protein preparation.
Animals were anesthetized with inhalant isoflurane, and the kidneys were rinsed by in vivo cardiac perfusion with PBS (pH 7.4). The right renal vasculature was clamped, the right kidney was rapidly removed, and the cortex and outer medulla were isolated rapidly under a dissecting microscope, snap-frozen in liquid nitrogen, and stored frozen at −70°C until used. The left kidney was perfused with PLP fixative for immunohistochemistry. Tissues were homogenized in T-PER Tissue Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL) using microtube pestles (USA Scientific, Ocala, FL), and protein was extracted according to the manufacturer's recommended procedures. An aliquot was obtained for protein determination using a BCA assay, and the remainder was stored frozen at −70°C until used.
Immunoblotting procedure.
Five to ten micrograms of renal protein were electrophoresed on 10% PAGE ReadyGel (Bio-Rad, Hercules, CA). Gels were then transferred electrophoretically to nitrocellulose membranes, blocked with 5 g/dl nonfat dry milk in Blotto buffer (50 mM Tris, 150 mM NaCl, 5 mM Na2EDTA, and 0.05% Tween 20, pH 7.6), and incubated at 4°C overnight with primary antibody diluted in nonfat dry milk. Loading and transfer equivalence were assessed with Ponceau S staining. After being washed, membranes were exposed to secondary antibody, goat anti-rabbit IgG (Millipore, Billerica, MA), conjugated to horseradish peroxidase at a dilution of 1:5,000. Sites of antibody-antigen reaction were visualized by using enhanced chemiluminescence (SuperSignal West Pico Substrate; Pierce) and a Kodak Image Station 440CF digital imaging system. Band density was quantified using Kodak 1D, version 5.0, software (Kodak Scientific Imaging, New Haven, CT). Band density was normalized such that mean density in the same region (cortex or outer medulla) in control tissues was 100. The absence of saturation was confirmed by examining pixel intensity distribution in all immunoblots.
Quantitative analysis of immunohistochemistry.
Quantitative immunohistochemistry was performed as we have described previously (54). Briefly, high-resolution digital micrographs were obtained of collecting duct segments in the inner stripe of the outer medulla by an observer blinded to the treatment status and genotype of the mouse. With the use of ImageJ software (version 1.34j; National Institutes of Health) and a Bamboo CTH-460 pen tablet (Wacom, Vancouver, WA), individual intercalated cells expressing Rhbg immunolabel were identified and circumscribed. Immunolabel intensity within the cell was assessed as the integrated pixel intensity, following subtraction of background intensity, within the outlined area using custom-written software executed in Microsoft Excel 2010.
Statistics.
Results are presented as means ± SE. Statistical analyses were performed using Student's t-test and P < 0.05 was taken as statistically significant; n refers to the numbers of animal studied.
RESULTS
Physiological parameters.
Wild-type mice were allowed to acclimate to metabolic cages for 3 days while being fed a 20% protein, semisynthetic diet and were then randomized to normal (20%) or low (6%) protein diets for the next 10 days. Mice were pair-fed such that daily food intake did not differ between the two treatment groups. Figure 1 shows that daily food intake and body weight were similar in the two treatment groups. Table 1 shows plasma electrolyte values in mice fed control and low-protein diets. Dietary protein restriction did not significantly alter serum sodium, potassium, or bicarbonate concentration.
Fig. 1.
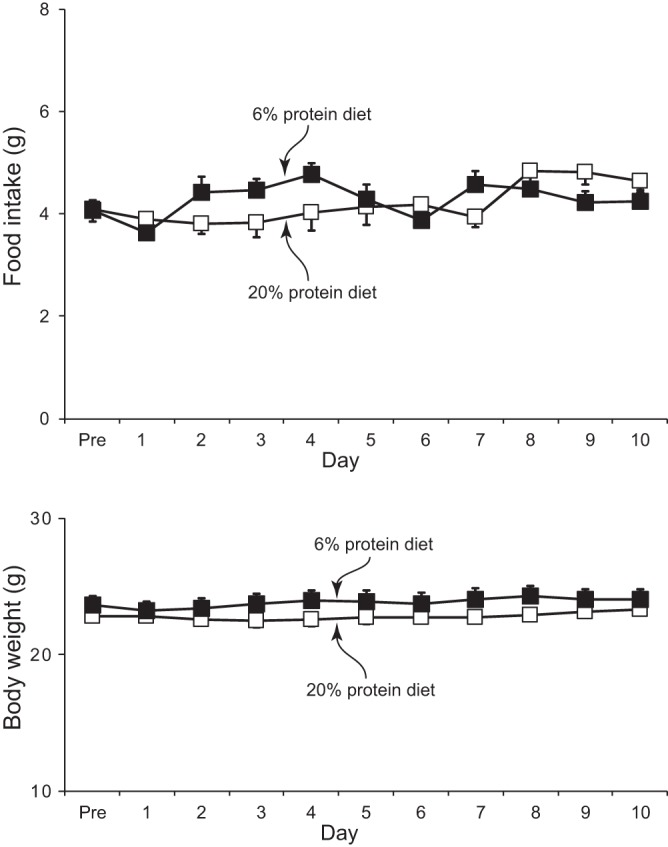
Effects of dietary protein restriction on food intake and growth. Top: food intake in mice on control, 20%, and low, 6%, protein diets. There was no significant difference between the two groups on any experimental day. Bottom: body weight during the experimental period; there was no significant effect protein intake on body weight. Values are means ± SE; n = 12/group.
Table 1.
Physiological parameters in mice fed 20 and 6% protein diets
| Parameters | 20% Protein Diet | 6% Protein Diet | P Value |
|---|---|---|---|
| Serum Na+, mmol/l | 149.3 ± 0.9 (5) | 151.8 ± 0.9 (5) | NS |
| Serum K+, mmol/l | 4.19 ± 0.24 (5) | 3.66 ± 0.12 (5) | NS |
| Serum HCO3−, mmol/l | 21.0 ± 2.0 (5) | 20.9 ± 1.1 (5) | NS |
Values are means ± SE. Numbers in parentheses are numbers of animals in each group.
NS, not significant.
Dietary protein restriction resulted in a rapid decrease in urinary ammonia excretion (Fig. 2). On the first day of the low-protein diet, urinary ammonia excretion decreased by an average of 64 ± 14% from the basal excretion rate, and on the second day, ammonia excretion had decreased an average of 85 ± 12%. There was no further decrease in urinary ammonia excretion on days 3–10. In the animals on the control protein diet, there were no significant time-dependent changes in urinary ammonia excretion. Thus, following dietary protein restriction, there is a rapid, significant, and persistent decrease in urinary ammonia excretion.
Fig. 2.
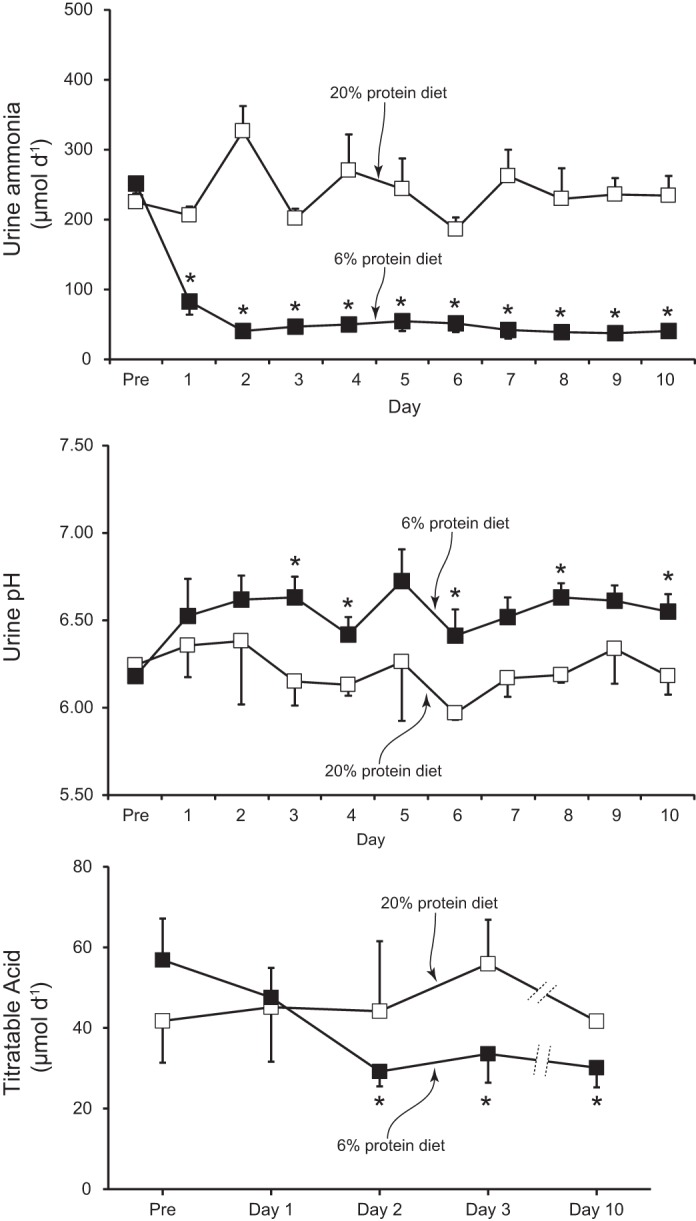
Effect of dietary protein restriction on urine ammonia, pH, and titratable acid. Top: changes in urinary ammonia excretion. Dietary protein restriction induced a rapid and sustained decrease in urinary ammonia excretion; there were no significant time-dependent changes in ammonia excretion in mice fed control, 20%, protein diet. *P < 0.05 vs. 20% protein diet; n = 11/group. Middle: changes in urine pH. Dietary protein restriction resulted in a slow, but gradual, alkalinization of urine pH. There were no time-dependent changes in urine pH in response to control, 20%, protein diet. Changes in urinary ammonia excretion were significant on the 1st day of dietary protein restriction, whereas change in urinary pH were not significant until the 3rd day. *P < 0.05 vs. 20% protein diet; n = 11/group. Bottom: titratable acid excretion. Dietary protein restriction did not change titratable acid excretion significantly on the 1st day, but it did on the 2nd day of dietary protein restriction, and there was no further change on either day 3 or day 10. *P < 0.05 vs. Pre; n = 5/group.
Dietary protein restriction also resulted in slow but significant urine alkalinization (Fig. 2). However, urine pH did not change significantly until the third day of dietary protein restriction, whereas urinary ammonia excretion changed significantly on the first day, with the maximal change on the second day. Mice fed a control protein diet had no significant changes in urine pH over 10 days. Thus urine alkalinization likely contributes to the decrease in urinary ammonia excretion in response to dietary protein restriction. The difference in the time course of urinary ammonia excretion and urinary pH suggests that changes in luminal pH are not the primary mechanism of the initial change in ammonia excretion.
Another component of net acid excretion is titratable acid excretion. In control animals continued on 20% protein diet, there were no time-dependent changes in titratable acid excretion. In mice given the 6% protein diet, titratable excretion did not change significantly on the first day, but it was significantly decreased starting on the second day, with no further change through day 10 (Fig. 2). The magnitude of the change, from 57 ± 10 to 30 ± 5 μmol/day, was substantially smaller than the change in ammonia excretion.
Effect of dietary protein restriction on proteins involved in ammoniagenesis.
Changes in urinary ammonia excretion could result from changes in ammonia production, intrarenal ammonia removal, or ammonia transport. To begin to differentiate these mechanisms, we examined the effects of dietary protein restriction on expression of proteins involved in renal ammoniagenesis. Two enzymes that play a central role in renal ammoniagenesis are PDG and PEPCK (59, 61). PDG catalyzes the initial enzymatic step in ammonia metabolism, conversion of glutamine to glutamate with release of NH4+, and PEPCK catalyzes the conversion of oxaloacetate to phosphoenolpyruvate, which not only releases HCO3− but also enhances the upstream production of ammonia (59, 61). Dietary protein restriction induced a small, ∼6%, but statistically significant, decrease in renal cortical PDG expression (Fig. 3). PEPCK expression, in contrast, exhibited a significantly larger, ∼60%, decrease in response to dietary protein restriction (Fig. 3).
Fig. 3.
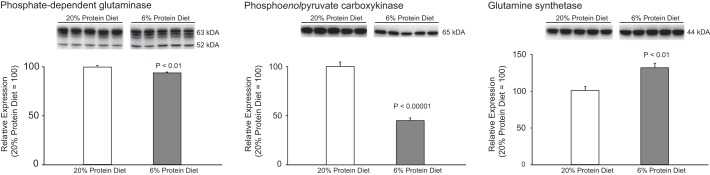
Effect of dietary protein restriction on proteins involved in ammoniagenesis. Left: renal cortical phosphate-dependent glutaminase (PDG) expression. Dietary protein restriction induced a small, but statistically significant, decrease in PDG expression. The multiple bands are due to kidney-specific endogenous proteinases (25). Middle: changes in renal cortical phosphoenolpyruvate carboxykinase (PEPCK) expression. Dietary protein restriction induced a substantial, statistically significant, decrease in PEPCK expression. Right: renal cortical glutamine synthetase expression. Glutamine synthetase catalyzes the regeneration of glutamine and decreases intrarenal ammonia availability. Dietary protein restriction resulted in a significant increase in glutamine synthetase expression as compared with 20% protein diet. The original blot for each of the proteins shown at right included tissues from mice with global Rhcg deletion provided either 20 or 6% protein diets and is shown in Fig. 8. Only the data from wild-type mice are shown at right. Values are means ± SE; n = 5/group.
The kidney also uses intracellular ammonia to regenerate glutamine via the reaction of ammonium with glutamate, a reaction catalyzed by the enzyme glutamine synthetase (12, 54). Dietary protein restriction resulted in a substantial and significant increase in renal cortical glutamine synthetase expression (Fig. 3).
Thus dietary protein restriction induces a significant decrease in urinary ammonia excretion. This appears to result, at least in part, from decreased ammoniagenesis via PDG and PEPCK and increased intrarenal ammonia utilization to regenerate glutamine via glutamine synthetase.
Effect of dietary protein restriction on ammonia transporters.
Renal ammonia excretion involves coordinated transport of NH3 and NH4+ by specific membrane proteins in specific renal epithelial cells (58, 61). Understanding the regulation of renal ammonia excretion requires evaluation of the role of ion transporters expressed in the proximal tubule, thick ascending limb of the loop of Henle, and collecting duct.
In the proximal tubule, the apical sodium hydrogen exchanger NHE3 is believed generally to secrete ammonium (27, 28, 32, 58). We observed that dietary protein restriction induced no significant change in renal cortical NHE3 expression (Fig. 4). Regulation of NHE3 expression does not appear to contribute to the decreased renal ammonia excretion.
Fig. 4.
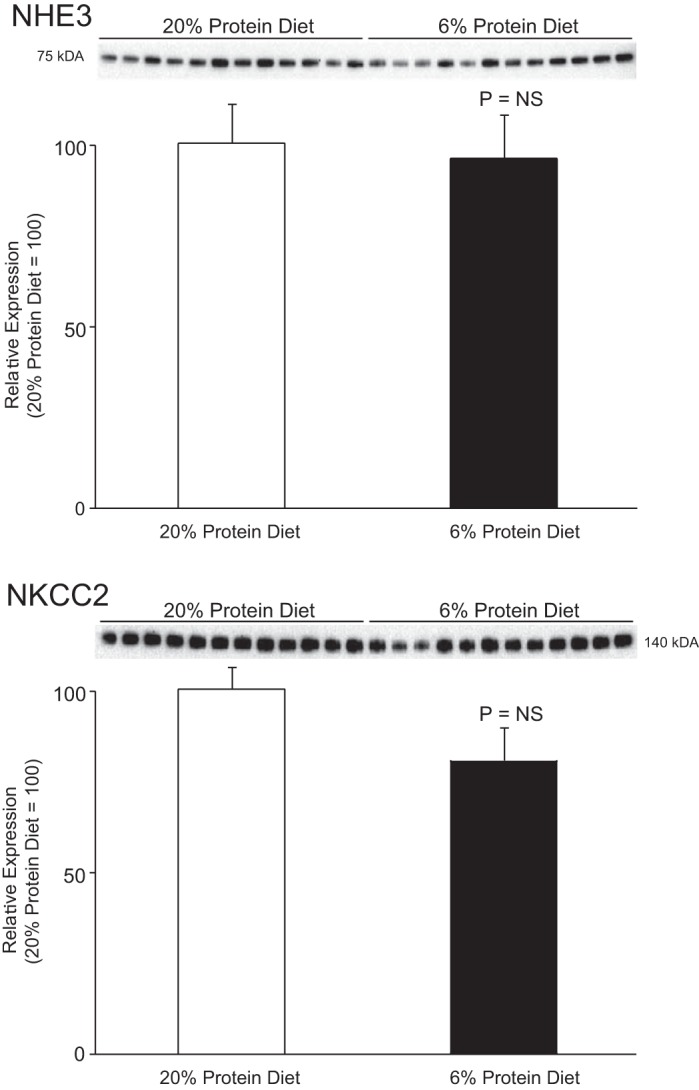
Effect of dietary protein restriction on NHE3 and NKCC2 expression. Top: effects of 6% (low-protein) and 20% (normal protein) diet on expression of the proximal tubule apical Na+/H+ exchanger NHE3. NHE3 is believed to be the major mechanism of proximal tubule ammonium secretion. There was no significant change in NHE3 expression in response to low-protein diet. Bottom: effects of dietary protein restriction on expression of NKCC2, the primary protein responsible for luminal ammonium reabsorption by the thick ascending limb. There was no significant change in NKCC2 expression in response to low-protein diet. Values are means ± SE; n = 12/group.
In the thick ascending limb of Henle, apical NKCC2 mediates ammonia reabsorption and contributes to subsequent medullary shunting (2, 3, 15, 27–29). Dietary protein restriction did not significantly alter NKCC2 expression (Fig. 4), suggesting that altered NKCC2 expression does not contribute to the decreased ammonia excretion.
The collecting duct secretes 60 to 80% of urinary ammonia, and the Rhesus glycoproteins Rhbg and Rhcg (21, 55) are the primary collecting duct ammonia-transporting proteins (58, 61, 62). In previous experimental models, changes in Rhbg and/or Rhcg expression have paralleled changes in total ammonia excretion (4, 5, 23, 24, 31, 34, 39, 50, 51). In contrast to these previous studies, dietary protein restriction, surprisingly, induced a significant increase in Rhbg expression in the outer medulla. (Fig. 5). Rhbg expression in the cortex and Rhcg expression in either the cortex or outer medulla were not significantly changed.
Fig. 5.

Effect of dietary protein restriction on Rhbg expression. Top left: effects of dietary protein restriction on steady-state Rhbg protein expression. There is no significant change in expression in the cortex, but dietary protein restriction induced a significant increase in expression in the outer medulla. Values are means ± SE; n = 6/group. Bottom left: immunolabel for Rhbg in the inner stripe of the outer medulla (ISOM) in control and low-protein diet-fed mice. Dietary protein restriction causes an increase in basolateral Rhbg immunolabel intensity in a subset of collecting duct cells. No expression is evident in noncollecting duct cells. Top right: colocalization of Rhbg (brown) with the intercalated cell marker H+-ATPase (blue) in the inner stripe of the outer medullary collecting duct. Rhbg expression in response to low-protein diet is restricted to cells identified as type A intercalated cells by the presence of apical H+-ATPase immunolabel. Bottom right: mean cellular Rhbg expression determined using quantitative immunohistochemistry. Dietary protein restriction resulted in a significant increase in mean Rhbg expression in the type A intercalated cell.
To determine whether increased outer medullary Rhbg expression reflected de novo expression in noncollecting duct cells or whether it was due to increased expression in specific collecting duct cell populations, we examined Rhbg expression using immunohistochemistry. In both control and dietary protein restriction mice, discrete basolateral Rhbg immunolabel was present in a subpopulation of collecting duct cells. Dietary protein restriction did not induce expression in cells outside of the collecting duct nor was there an evident change in subcellular distribution (Fig. 5). Instead, there appeared to be a significant increase in immunolabel intensity in a specific subpopulation of OMCD cells. These cells had a morphologic appearance consistent with intercalated cells, the cell-type known to be the primary Rhbg-expressing cell in the OMCD (22, 46, 53).
To confirm that the Rhbg-expressing cells in the low-protein diet kidney were intercalated cells, we performed double immunolabel experiments using antibodies to Rhbg and to the a4 subunit of H+-ATPase. Our results showed that the cells with strong basolateral Rhbg expression also exhibited apical H+-ATPase expression, thereby identifying the cells as type A intercalated cells (Fig. 5).
Finally, we used quantitative immunohistochemistry to confirm that dietary protein restriction increases type A intercalated cell Rhbg expression. Dietary protein restriction significantly increased Rhbg immunolabel expression in OMCD intercalated cells (Fig. 5). Thus, although dietary protein restriction decreased urinary ammonia excretion, there was increased expression of the ammonia-transporting protein Rhbg in type A intercalated cells in the OMCD.
Role of Rhbg in the renal response to dietary protein restriction.
The finding that dietary protein restriction increased Rhbg expression in OMCD type A intercalated cells while decreasing urinary ammonia excretion is not consistent with the current paradigm that Rhbg contributes to urinary ammonia excretion. To further examine Rhbg's role in ammonia excretion in dietary protein restriction, we examined the effect of collecting duct-specific Rhbg deletion on the renal response to this experimental maneuver. In preliminary studies, we observed that mice with collecting duct-specific Rhbg deletion grew normally and exhibited normal renal anatomy (data not shown).
We then compared the effect of a low-protein diet on urinary ammonia excretion in control and CD-Rhbg-KO mice. There was no significant difference in food intake or body weight in the different genotypes (data not shown). As seen in previous experiments, dietary protein restriction decreased ammonia excretion, but there was no significant difference in ammonia excretion between the two different genotypes, either under basal conditions or after initiation of dietary protein restriction (Fig. 6).
Fig. 6.
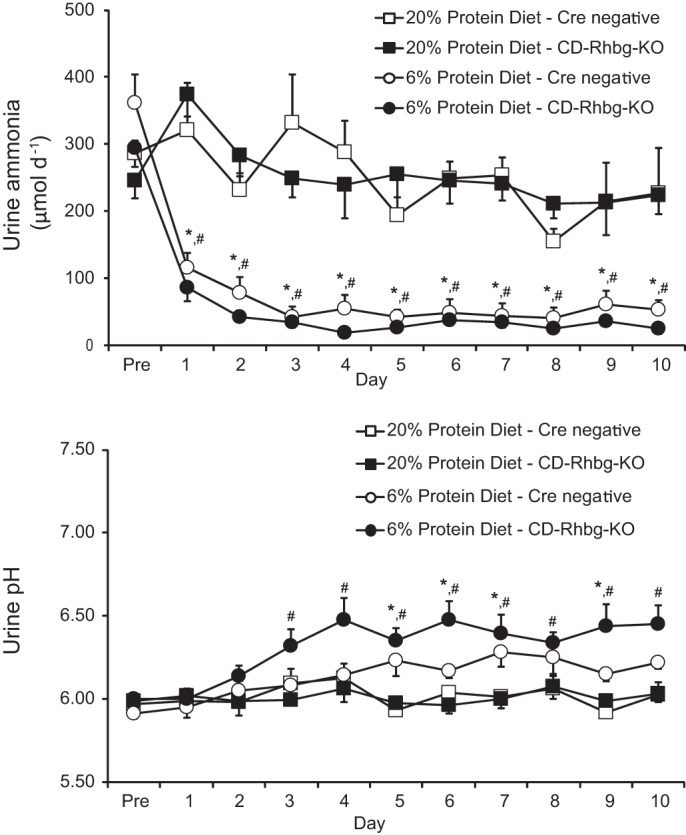
Effect of collecting duct-specific Rhbg deletion on response to dietary protein restriction. Top: urinary ammonia response. Dietary protein restriction induced a rapid decrease in urinary ammonia in mice with collecting duct-specific Rhbg deletion and in control, Cre-negative littermates. There was no significant difference in urinary ammonia excretion between control, Cre-negative littermates and mice with collecting duct-specific Rhbg deletion, either under basal conditions or following initiation of 6% protein diet. Bottom: urinary pH changes. There was no significant difference in urinary pH in the different genotypes, either under basal conditions or following initiation of dietary protein restriction. *P < 0.05, 20 vs. 6% protein diet in Cre negative mice; #P < 0.05, 20 vs. 6% protein diet in CD-Rhbg-knockout (KO) mice; n = 4/group on 20% protein and 6% protein diet on Cre-negative littermates and n = 6/group on 20% protein, n = 7/group on 6% protein diet on CD-Rhbg-KO mice.
Dietary protein restriction caused urine pH to increase in mice with collecting duct-specific Rhbg deletion and in control, Cre negative mice. However, there was no difference in urine pH between the two genotypes either under basal conditions or after initiation of the low-protein diet (Fig. 6).
Adaptive changes to the deletion of a protein by genetic manipulation can potentially compensate completely for its absence. However, there was no significant difference in expression of the key ammonia-metabolizing enzymes PDG, PEPCK, or glutamine synthetase (data not shown). Thus, during conditions of dietary protein restriction, adaptive changes in ammoniagenic enzyme protein expression do not appear to compensate for the absence of Rhbg.
Role of Rhcg in the response to dietary protein restriction.
In previous studies, we, and others, have shown that Rhcg has a critical role in renal ammonia excretion (6, 8, 34–36). Thus the absence of decreased Rhcg expression is unpredicted. Moreover, because Rhcg can be regulated by mechanisms other than steady-state protein expression (51), we were concerned that the lack of change in steady-state Rhcg protein in response to dietary protein restriction might not indicate that Rhcg has no role in the response to the stimulus. To examine this possibility, we examined mice with global Rhcg deletion.
First, we examined changes in urinary ammonia excretion. On a basal, 20%, protein diet, there was no difference in ammonia excretion between mice with intact or with global Rhcg deletion. After initiation of a 6% protein diet, urinary ammonia excretion decreased in both genotypes, and there was no significant difference in urinary ammonia excretion response in mice with global Rhcg deletion (Fig. 7). Dietary protein restriction also increased urine pH, and global Rhcg deletion significantly enhanced this response (Fig. 7). Food intake did not differ between wild-type and Rhcg deletion mice, either during 20 or 6% protein diets. Titratable acid excretion did not differ between wild-type and Rhcg deletion mice on 20% protein diet, but, consistent with the urine pH changes, on day 2 and 10 of dietary protein restriction it was slightly less in mice with Rhcg deletion than in mice with intact Rhcg expression (data not shown).
Fig. 7.
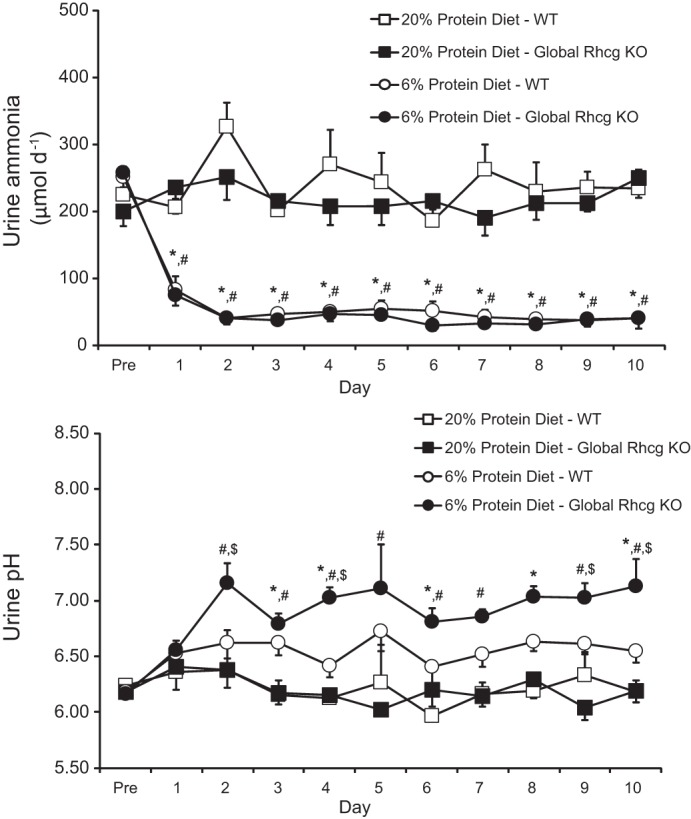
Effect of Rhcg deletion on the response to dietary protein restriction. Top: urinary ammonia excretion. Dietary protein restriction induced a rapid decrease in urinary ammonia in both control and global Rhcg deletion mice. There was no significant difference in urinary ammonia excretion between mice with global Rhcg deletion and those with intact Rhcg expression, either while receiving basal, 20%, protein diet or after initiation of 6% protein diet. Bottom: urinary pH. There was no significant difference in urinary pH between mice with global Rhcg deletion and those with intact Rhcg expression when fed 20% protein diet; however. in mice 6% protein diet, Rhcg deletion mice excreted more alkaline urine than Rhcg intact mice. *P < 0.05, 20 vs. 6% protein diet in wild-type (WT) mice; #P < 0.05, 20 vs. 6% protein diet in global Rhcg KO mice; $P < 0.05, WT vs. global Rhcg KO mice in 6% protein diet; n = 11/group on 20% protein and 6% protein diet on WT mice and n = 5/group on 20% protein and 6% protein diet on global Rhcg KO mice.
As discussed previously, adaptive changes to the genetic deletion of a protein can sometimes mask its role in a physiologic response. Thus we examined whether there were adaptive responses to Rhcg deletion. In mice with intact Rhcg expression, dietary protein restriction had only a very minimal, ∼6% decrease, effect on PDG expression. In contrast, in Rhcg KO mice, dietary protein restriction decreased PDG expression by ∼30% to a level not different than observed in mice with intact Rhcg expression (Fig. 8). Thus, adaptive changes in PDG expression compensate for the absence of Rhcg during normal dietary protein intake, but are not necessary during dietary protein restriction.
Fig. 8.

Effect of Rhcg deletion on ammoniagenic enzyme response to low-protein diet. Top: immunoblot analysis of PDG expression in renal cortical proteins in mice with Rhcg deletion in response to dietary protein restriction. While on control, 20% protein diet, PDG expression was significantly greater in Rhcg KO mice than in WT mice. In response to 6% protein diet, PDG expression decreases in both genotypes and does not differ between mice with intact Rhcg expression or with Rhcg deletion. Middle: immunoblot analysis of effect of Rhcg deletion on PEPCK expression. Similar to PDG, Rhcg deletion induced an increase in PEPCK expression on control 20% protein diet. Dietary protein restriction with 6% protein diet decreased PEPCK expression in both genotypes, and expression did not differ in response to Rhcg deletion. Bottom: effect of Rhcg deletion on glutamine synthetase expression. Rhcg deletion induced decreased glutamine synthetase expression while on 20% protein diet. Dietary protein restriction increased glutamine synthetase expression in both genotypes, and expression did not differ in the 2 genotypes. Values are means ± SE; n = 5/group on WT mice and global Rhcg KO mice.
Rhcg deletion increased PEPCK expression and decreased glutamine synthetase expression in mice while on normal protein diet. Following induction of dietary protein restriction, PEPCK expression decreased and glutamine synthetase expression increased in both WT and Rhcg deletion mice (P < 0.05 for each), and the relative changes did not differ between the two genotypes (P > 0.05; Fig. 8). Thus PEPCK and glutamine synthetase, similar to PDG, exhibit adaptive changes in expression in response to Rhcg deletion while on 20% protein diet but not during dietary protein restriction with a 6% protein diet.
DISCUSSION
The current studies provide a detailed evaluation of the effects of dietary protein restriction on renal ammonia metabolism and excretion. Dietary protein restriction results in a substantial decrease in ammonia excretion, with maximal response present on the second day. The decrease in ammonia excretion involves coordinated changes in multiple aspects of renal ammonia metabolism. Multiple components of ammoniagenesis, including PDG, PEPCK, and glutamine synthetase, exhibited altered expression consistent with decreased net ammoniagenesis. Urine pH increases, decreasing protonation of NH3, the ammonia molecular species secreted by the collecting duct, and thereby decreasing the gradient for collecting duct NH3 secretion. Rhbg expression increases paradoxically and is not necessary for the difference in ammonia excretion between mice fed 6 and 20% protein as determined from studies examining the effect of collecting duct-specific Rhbg deletion. Rhcg expression appears to be important for the response to variations in dietary protein as evidenced from adaptive changes in PDG in response to Rhcg deletion. Finally, the altered ammonia metabolism does not appear to involve changes in expression of key proteins involved in proximal tubule and thick ascending limb ammonia transport, NHE3 and NKCC2.
Dietary protein restriction is commonly used in the management of patients with chronic kidney disease to try to slow the progressive loss of renal function and to minimize the symptoms of azotemia (1, 17, 20, 49). Dietary protein restriction decreases glomerular hydrodynamic pressures, and this mechanism is believed to account in large part for the beneficial effects on progressive chronic kidney disease (26, 44, 45). In addition, in view of evidence that metabolic acidosis can worsen progressive renal injury (10, 19), dietary protein restriction may also have beneficial effects related to reductions in endogenous acid production and subsequent decrease in ammonia metabolism, as demonstrated in the current study. Conditions associated with increased ammonia metabolism, such as metabolic acidosis and hypokalemia, are also associated with progressive chronic kidney disease (10, 19, 47, 52), and ammonia may contribute to this progression via ammonia-mediated complement activation (11, 43). The current study adds to this information by showing that dietary protein restriction results in rapid and substantial decreases in renal ammonia metabolism and excretion and thus we speculate that decreased renal ammonia metabolism may contribute to the beneficial effects of a low-protein diet in patients with chronic kidney disease.
The time course of changes in renal ammonia excretion in response to dietary protein restriction appears to be more rapid than the response to either metabolic acidosis or hypokalemia. In these other conditions, which are associated with increased ammonia excretion, maximal changes in ammonia metabolism are not identified until 4–5 days after initiation of the stimulus (4, 34–37). In contrast, dietary protein restriction induced the maximal change in ammonia excretion after only 2 days. This difference may result because increased ammonia excretion requires synthesis of multiple proteins involved in ammonia production and transport, whereas decreased ammonia excretion response to protein restriction primarily requires decreased expression or activity, and protein “removal” or inactivation may occur more rapidly than new protein synthesis. In addition, because the increased ammoniagenesis in metabolic acidosis and hypokalemia requires increased skeletal muscle glutamine release, delays in activation of skeletal muscle breakdown may contribute to the delayed increase in ammonia excretion.
Decreased renal ammonia excretion in response to dietary protein restriction involves multiple components of renal ammonia generation. PDG, a mitochondrial protein that catalyzes the initial enzymatic step in ammoniagenesis, exhibits decreased expression with protein restriction, as does PEPCK, a cytoplasmic enzyme. Quantitatively, the decrease in PEPCK expression is greater than the decrease in PDG expression. However, it is important to recognize that decreased PEPCK-mediated glutamate metabolism is known to cause feedback inhibition of PDG (18, 33). Thus dietary protein restriction appears to inhibit multiple aspects of ammonia generation.
The renal response to dietary protein restriction may also involve increased intrarenal glutamine regeneration. The protein glutamine synthetase catalyzes the reaction of ammonium (NH4+) with glutamate to regenerate glutamine (54). Dietary protein restriction increases glutamine synthetase expression, which likely increases glutamine regeneration, increases ammonium utilization, and decreases net ammoniagenesis. Physiologically, the increased glutamine regeneration via glutamine synthetase serves as a nitrogen-sparing benefit. Moreover, regulated glutamine synthetase expression may be a common component of the regulation of renal ammonia metabolism, with changes occurring in response to protein restriction (current study), metabolic acidosis, and hypokalemia (5, 12, 34, 37, 54).
Somewhere between 60 and 80% of total urinary ammonia is secreted into the luminal fluid in the collecting duct (21, 55), and the Rhesus glycoprotein Rhcg has a critical role in this process. In the current study, adaptive changes in PDG, PEPCK, and glutamine synthetase expression were required to maintain normal ammonia excretion in mice with global Rhcg deletion while on 20% protein diet. However, during dietary protein restriction with a 6% protein diet, ammonia excretion was not altered by Rhcg deletion and these adaptive changes were not present. Thus Rhcg expression appears to contribute to ammonia excretion while on normal protein diet (current study) and a high-protein diet (7), but neither its expression nor changes in its expression appear to be necessary for the adaptive changes in ammonia excretion during dietary protein restriction. Thus Rhcg expression appears necessary for normal ammonia excretion during basal conditions (current study, 6, 34), metabolic acidosis (6, 34, 35), hypokalemia (36), and high-protein intake (7), conditions of increased ammonia excretion, but not during dietary protein restriction (current study) where ammonia excretion is decreased. Changes in urine pH in response to dietary protein restriction were greater in mice with Rhcg deletion than in those with intact Rhcg expression; the mechanism of this difference is not clear at this time.
Rhbg is another Rhesus glycoprotein involved in collecting duct ammonia transport. Although an initial report found no effect of Rhbg deletion on either basal or acidosis-stimulated ammonia excretion (9), other studies clearly demonstrate a role for Rhbg in ammonia excretion. Under baseline conditions, intercalated cell-specific Rhbg deletion resulted in adaptive changes in glutamine synthetase expression that appeared to have compensated for the lack of Rhbg, indicating a role for Rhbg in baseline ammonia excretion (5). Studies examining metabolic acidosis, using a different model that resulted in a greater stimulation of ammonia excretion than in the earlier study, found that Rhbg deletion impaired acidosis-stimulated ammonia excretion (5). Rhbg deletion also impaired the hypokalemia-induced stimulation of ammonia excretion, and the effect of Rhbg deletion was greater than the effect of Rhcg deletion (4, 36). Thus Rhbg appears to have an important role not only in basal ammonia metabolism but also in response to both metabolic acidosis and hypokalemia.
Unexpectedly, dietary protein restriction resulted in increased Rhbg expression in the outer medulla in type A intercalated cells despite the decrease in urinary ammonia excretion. This increased expression is not necessary for normal ammonia metabolism, as shown by the absence of an effect of collecting duct-specific Rhbg deletion. These observations suggest that increased Rhbg expression is not likely to be contributing to the decrease in ammonia excretion. Rhbg has also been suggested to function as a CO2 transporter (16, 42), but because intercalated cell CO2 transport is necessary for intracellular H+ generation used for urinary acidification, and dietary protein restriction causes urine alkalinization, this function is unlikely to underlie its increased expression during dietary protein restriction. Importantly, collecting duct Rhbg expression does not appear to be necessary for the changes in renal ammonia excretion in response to dietary protein restriction.
Changes in urine ammonia excretion in response to dietary protein restriction, ∼85% decrease, substantially exceeded the change in expression of any the proteins discussed above. This suggests that changes in expression of a single one of these proteins are not solely responsible for the overall change in ammonia metabolism and instead that an integrated interaction of multiple components of renal ammonia metabolism explains this dramatic difference in ammonia excretion. Moreover, the finding of decreased expression of proteins involved in ammonia production, i.e., PDG and PEPCK, and increased expression of a protein involved in ammonia utilization for glutamine regeneration, glutamine synthetase, suggests that there exists synergistic regulation of these separate components of net ammoniagenesis.
Titratable acid excretion decreased in response to dietary protein restriction. However, the magnitude of change in titratable acid excretion was less than the change in ammonia excretion, and the time course was slower. This quantitatively lesser role for changes in titratable excretion than changes in ammonia excretion is consistent with previous findings in both metabolic acidosis and hypokalemia (4, 34). Titratable acid excretion decreased slightly more in mice with global Rhcg deletion than in those with intact Rhcg expression. This finding is consistent with the slightly higher urine pH in these mice.
In summary, the current studies demonstrate significant new findings with regards to ammonia metabolism in response to dietary protein restriction. There is a substantial and rapid decrease in urinary ammonia excretion, and the time course of this decrease is faster than that observed with conditions that increase ammonia metabolism. Dietary protein restriction induces adaptive responses in multiple proteins involved both in renal ammoniagenesis and in renal ammonia utilization to regenerate glutamine. Rhcg appears to play a role in the response to normal dietary protein intake but not during dietary protein restriction. Rhbg expression increases in type A intercalated cells in the inner stripe of the OMCD, but Rhbg expression is not necessary for the adaptive response to dietary protein restriction, suggesting that the increased Rhbg expression may have physiologic roles in addition to its role in ammonia transport.
GRANTS
The studies were supported by funding from National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-045788 and from the Department of Veterans Affairs (1I01BX000818).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.-W.L., J.W.V., and I.D.W. conception and design of research; H.-W.L., G.O., M.E.H., H.G., and J.W.V. performed experiments; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. analyzed data; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. interpreted results of experiments; H.-W.L. and I.D.W. prepared figures; H.-W.L. and I.D.W. drafted manuscript; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. edited and revised manuscript; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Gina Cowsert for secretarial assistance and Sharon W. Matthew, Tanisha S. Thomas, and Diana Chu of the University of Florida College of Medicine Electron Microscopy Core Laboratory for excellent tissue processing for immunohistochemical studies.
Footnotes
Ammonia exists in two molecular forms, NH3 and NH4+. When referring specifically to the molecular form NH3, we state specifically “NH3.” When referring specifically to NH4+, we either state specifically either “ NH4+” or we use the term “ammonium.” In this article, the term “ammonia” refers to the combination of both molecular forms.
REFERENCES
- 1.Aparicio M, Bellizzi V, Chauveau P, Cupisti A, Ecder T, Fouque D, Garneata L, Lin S, Mitch WE, Teplan V, Zakar G, Yu X. Protein-restricted diets plus keto/amino acids–a valid therapeutic approach for chronic kidney disease patients. J Ren Nutr 22: S1–21, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Attmane-Elakeb A, Amlal H, Bichara M. Ammonium carriers in medullary thick ascending limb. Am J Physiol Renal Physiol 280: F1–F9, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Bergeron MJ, Gagnon E, Wallendorff B, Lapointe JY, Isenring P. Ammonium transport and pH regulation by K+-Cl− cotransporters. Am J Physiol Renal Physiol 285: F68–F78, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Bishop JM, Lee HW, Handlogten ME, Han KH, Verlander JW, Weiner ID. Intercalated cell-specific Rh B glycoprotein deletion diminishes renal ammonia excretion response to hypokalemia. Am J Physiol Renal Physiol 304: F422–F431, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop JM, Verlander JW, Lee HW, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the Rhesus glycoprotein, Rh B glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol 299: F1065–F1077, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biver S, Belge H, Bourgeois S, Van Vooren P, Nowik M, Scohy S, Houillier P, Szpirer J, Szpirer C, Wagner CA, Devuyst O, Marini AM. A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature 456: 339–343, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Bounoure L, Ruffoni D, Muller R, Kuhn GA, Bourgeois S, Devuyst O, Wagner CA. The role of the renal ammonia transporter rhcg in metabolic responses to dietary protein. J Am Soc Nephrol 25: 2040–2052, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourgeois S, Bounoure L, Christensen EI, Ramakrishnan SK, Houillier P, Devuyst O, Wagner CA. Haploinsufficiency of the ammonia transporter Rhcg predisposes to chronic acidosis. Rhcg is critical for apical and basolateral ammonia transport in the mouse collecting duct. J Biol Chem 288: 5518–5529, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambrey R, Goossens D, Bourgeois S, Picard N, Bloch-Faure M, Leviel F, Geoffroy V, Cambillau M, Colin Y, Paillard M, Houillier P, Cartron JP, Eladari D. Genetic ablation of Rhbg in mouse does not impair renal ammonium excretion. Am J Physiol Renal Physiol 289: F1281–F1290, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Abramowitz MK. Treatment of metabolic acidosis in patients with CKD. Am J Kidney Dis 63: 311–317, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark EC, Nath KA, Hostetter MK, Hostetter TH. Role of ammonia in tubulointerstitial injury. Miner Electrolyte Metab 16: 315–321, 1990. [PubMed] [Google Scholar]
- 12.Conjard A, Komaty O, Delage H, Boghossian M, Martin M, Ferrier B, Baverel G. Inhibition of glutamine synthetase in the mouse kidney: a novel mechanism of adaptation to metabolic acidosis. J Biol Chem 278: 38159–38166, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Franch HA, Mitch WE. Navigating between the scylla and charybdis of prescribing dietary protein for chronic kidney diseases. Annu Rev Nutr 29: 341–364, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Frassetto LA, Todd KM, Morris RC Jr, Sebastian A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am J Clin Nutr 68: 576–583, 1998. [DOI] [PubMed] [Google Scholar]
- 15.Garvin JL, Burg MB, Knepper MA. Active NH4+ absorption by the thick ascending limb. Am J Physiol Renal Fluid Electrolyte Physiol 255: F57–F65, 1988. [DOI] [PubMed] [Google Scholar]
- 16.Geyer RR, Parker MD, Toye AM, Boron WF, Musa-Aziz R. Relative CO2/NH3 permeabilities of human RhAG, RhBG and RhCG. J Membr Biol 246: 915–926, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giordano M, Ciarambino T, Castellino P, Paolisso G. Light and shadows of dietary protein restriction in elderly with chronic kidney disease. Nutrition 29: 1090–1093, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Goldstein L. Relation of glutamate to ammonia production in the rat kidney. Am J Physiol 210: 661–666, 1966. [DOI] [PubMed] [Google Scholar]
- 19.Goraya N, Wesson DE. Acid-base status and progression of chronic kidney disease. Curr Opin Nephrol Hypertens 21: 552–556, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Goraya N, Wesson DE. Dietary management of chronic kidney disease: protein restriction and beyond. Curr Opin Nephrol Hypertens 21: 635–640, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol Renal Fluid Electrolyte Physiol 253: F595–F605, 1987. [DOI] [PubMed] [Google Scholar]
- 22.Han KH, Lee HW, Handlogten ME, Whitehill FM, Croker BP, Clapp W, Verlander JW, Weiner ID. Expression of the ammonia transporter family member, Rh B glycoprotein, in the human kidney. Am J Physiol Renal Physiol 304: F972–F981, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han KH, Kim HY, Croker BP, Reungjui S, Lee SY, Kim J, Handlogten ME, Adin CA, Weiner ID. Effects of ischemia-reperfusion injury on renal ammonia metabolism and the collecting duct. Am J Physiol Renal Physiol 293: F1342–F1354, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Han KH, Lee HW, Handlogten ME, Bishop JM, Levi M, Kim J, Verlander JW, Weiner ID. Effect of hypokalemia on renal expression of the ammonia transporter family members, Rh B glycoprotein and Rh C glycoprotein, in the rat kidney. Am J Physiol Renal Physiol 301: F823–F832, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haser WG, Shapiro RA, Curthoys NP. Comparison of the phosphate-dependent glutaminase obtained from rat brain and kidney. Biochem J 229: 399–408, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol Renal Fluid Electrolyte Physiol 241: F85–F93, 1981. [DOI] [PubMed] [Google Scholar]
- 27.Karim Z, Szutkowska M, Vernimmen C, Bichara M. Renal handling of NH3/NH4+: recent concepts. Nephron Physiol 101: 77–81, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Karim Z, Szutkowska M, Vernimmen C, Bichara M. Recent concepts concerning the renal handling of NH3/NH4+. J Nephrol 19, Suppl 9: S27–S32, 2006. [PubMed] [Google Scholar]
- 29.Kikeri D, Sun A, Zeidel ML, Hebert SC. Cell membranes impermeable to NH3. Nature 339: 478–480, 1989. [DOI] [PubMed] [Google Scholar]
- 30.Kim HY, Verlander JW, Bishop JM, Cain BD, Han KH, Igarashi P, Lee HW, Handlogten ME, Weiner ID. Basolateral expression of the ammonia transporter family member Rh C glycoprotein in the mouse kidney. Am J Physiol Renal Physiol 296: F545–F555, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293: F1238–F1247, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Kinsella JL, Aronson PS. Interaction of NH4+ and Li+ with the renal microvillus membrane Na+-H+ exchanger. Am J Physiol Cell Physiol 241: C220–C226, 1981. [DOI] [PubMed] [Google Scholar]
- 33.Krebs HA. Metabolism of amino-acids: the synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem J 29: 1951–1969, 1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HW, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID. Effect of intercalated cell-specific Rh C glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 299: F369–F379, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HW, Verlander JW, Bishop JM, Handlogten ME, Han KH, Weiner ID. Renal ammonia excretion in response to hypokalemia: effects of collecting duct-specific Rh C glycoprotein deletion. Am J Physiol Renal Physiol 304: F410–F421, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HW, Verlander JW, Handlogten ME, Han KH, Weiner ID. Effect of collecting duct-specific deletion of both Rh B glycoprotein (Rhbg) and Rh C glycoprotein (Rhcg) on renal response to metabolic acidosis. Am J Physiol Renal Physiol 306: F389–F400, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levey AS, Greene T, Beck GJ, Caggiula AW, Kusek JW, Hunsicker LG, Klahr S. Dietary protein restriction and the progression of chronic renal disease: what have all of the results of the MDRD study shown? Modification of Diet in Renal Disease Study group. J Am Soc Nephrol 10: 2426–2439, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Lim SW, Ahn KO, Kim WY, Han DH, Li C, Ghee JY, Han KH, Kim HY, Handlogten ME, Kim J, Yang CW, Weiner ID. Expression of ammonia transporters, Rhbg and Rhcg, in chronic cyclosporine nephropathy in rats. Nephron Exp Nephrol 110: e49–e58, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mak DO, Dang B, Weiner ID, Foskett JK, Westhoff CM. Characterization of transport by the kidney Rh glycoproteins, RhBG and RhCG. Am J Physiol Renal Physiol 290: F297–F305, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitch WW. Dietary protein restriction and progressive renal insufficiency. Am J Kidney Dis 30: 297–298, 1997. [DOI] [PubMed] [Google Scholar]
- 42.Nakhoul NL, Hamm LL. Characteristics of mammalian Rh glycoproteins (SLC42 transporters) and their role in acid-base transport. Mol Aspects Med 34: 629–637, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nath KA, Hostetter MK, Hostetter TH. Increased ammoniagenesis as a determinant of progressive renal injury. Am J Kidney Dis 17: 654–657, 1991. [DOI] [PubMed] [Google Scholar]
- 44.Nath KA, Kren SM, Hostetter TH. Dietary protein restriction in established renal injury in the rat. Selective role of glomerular capillary pressure in progressive glomerular dysfunction. J Clin Invest 78: 1199–1205, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neuringer JR, Brenner BM. Hemodynamic theory of progressive renal disease: a 10-year update in brief review. Am J Kidney Dis 22: 98–104, 1993. [DOI] [PubMed] [Google Scholar]
- 46.Quentin F, Eladari D, Cheval L, Lopez C, Goossens D, Colin Y, Cartron JP, Paillard M, Chambrey R. RhBG and RhCG, the putative ammonia transporters, are expressed in the same cells in the distal nephron. J Am Soc Nephrol 14: 545–554, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Ray PE, Suga S, Liu XH, Huang X, Johnson RJ. Chronic potassium depletion induces renal injury, salt sensitivity, and hypertension in young rats. Kidney Int 59: 1850–1858, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Remer T, Manz F. Estimation of the renal net acid excretion by adults consuming diets containing variable amounts of protein. Am J Clin Nutr 59: 1356–1361, 1994. [DOI] [PubMed] [Google Scholar]
- 49.Rosman JB, ter Wee PM, Meijer S, Piers-Becht TP, Sluiter WJ, Donker AJ. Prospective randomised trial of early dietary protein restriction in chronic renal failure. Lancet 2: 1291–1296, 1984. [DOI] [PubMed] [Google Scholar]
- 50.Seshadri RM, Klein JD, Kozlowski S, Sands JM, Kim YH, Handlogten ME, Verlander JW, Weiner ID. Renal expression of the ammonia transporters, Rhbg and Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F397–F408, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Seshadri RM, Klein JD, Smith T, Sands JM, Handlogten ME, Verlander JW, Weiner ID. Changes in the subcellular distribution of the ammonia transporter Rhcg in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F1443–F1452, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Suga SI, Phillips MI, Ray PE, Raleigh JA, Vio CP, Kim YG, Mazzali M, Gordon KL, Hughes J, Johnson RJ. Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am J Physiol Renal Physiol 281: F620–F629, 2001. [DOI] [PubMed] [Google Scholar]
- 53.Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID. Localization of the ammonium transporter proteins Rh B glycoprotein and Rh C glycoprotein in the mouse kidney. Am J Physiol Renal Physiol 284: F323–F337, 2003. [DOI] [PubMed] [Google Scholar]
- 54.Verlander JW, Chu D, Lee HW, Handlogten ME, Weiner ID. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal Physiol 305: F701–F713, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiner ID, Hamm LL. Molecular mechanisms of renal ammonia transport. Annu Rev Physiol 69: 317–340, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weiner ID, Miller RT, Verlander JW. Localization of the ammonium transporters, Rh B glycoprotein and Rh C glycoprotein, in the mouse liver. Gastroenterology 124: 1432–1440, 2003. [DOI] [PubMed] [Google Scholar]
- 57.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol 2014. July 30 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiner ID, Verlander JW. Role of NH3 and NH4+ transporters in renal acid-base transport. Am J Physiol Renal Physiol 300: F11–F23, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weiner ID, Verlander JW. Renal acidification mechanisms. In: Brenner and Rector's The Kidney, edited by Taal M, Chertow GM, Marsden PA, Skorecki K, Yu AS, Brenner BM. Philadelphia, PA: Elsevier Saunders, 2012, p. 293–325. [Google Scholar]
- 60.Weiner ID. Renal acid-base regulation via ammonia transport in mammals. In: Epithelial Transport Physiology, edited by Gerencser GA. New York: Humana, 2010, p. 299–322. [Google Scholar]
- 61.Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Compr Physiol 3: 201–220, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiner ID, Verlander JW. Ammonia transport in the kidney by Rhesus glycoproteins. Am J Physiol Renal Physiol 306: F1107–F1120, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]