

Abstract
Ischemia-reperfusion injury (IRI) due to hypotension is a common cause of human acute kidney injury (AKI). Hypoxia-inducible transcription factors (HIFs) orchestrate a protective response in renal endothelial and epithelial cells in AKI models. As human mucin 1 (MUC1) is induced by hypoxia and enhances HIF-1 activity in cultured epithelial cells, we asked whether mouse mucin 1 (Muc1) regulates HIF-1 activity in kidney tissue during IRI. Whereas Muc1 was localized on the apical surface of the thick ascending limb, distal convoluted tubule, and collecting duct in the kidneys of sham-treated mice, Muc1 appeared in the cytoplasm and nucleus of all tubular epithelia during IRI. Muc1 was induced during IRI, and Muc1 transcripts and protein were also present in recovering proximal tubule cells. Kidney damage was worse and recovery was blocked during IRI in Muc1 knockout mice compared with congenic control mice. Muc1 knockout mice had reduced levels of HIF-1α, reduced or aberrant induction of HIF-1 target genes involved in the shift of glucose metabolism to glycolysis, and prolonged activation of AMP-activated protein kinase, indicating metabolic stress. Muc1 clearly plays a significant role in enhancing the HIF protective pathway during ischemic insult and recovery in kidney epithelia, providing a new target for developing therapies to treat AKI. Moreover, our data support a role specifically for HIF-1 in epithelial protection of the kidney during IRI as Muc1 is present only in tubule epithelial cells.
Keywords: acute kidney injury, ischemia, Muc1, hypoxia-inducible factor-1 (HIF-1), AMP-activated protein kinase
sublethal and lethal damage to renal tubular epithelial cells remains centrally important to the overall pathophysiology of acute kidney injury (AKI). Ischemia-reperfusion injury (IRI) due to hypotension or sepsis is the most common cause of human AKI (28, 32). The associated reduction in tissue oxygenation alters tubular epithelial cell metabolism, which, in turn, enhances the production of reactive oxygen species (ROS) (34). ROS can react with lipids to alter membrane fluidity and permeability, modify proteins by oxidation of sulfhydryls, carbonyl formation, and deamination, and induce single-strand breaks in DNA (1, 34).
Cells can respond effectively to hypoxic conditions to adjust their metabolism and upregulate elaborate protective mechanisms, including the hypoxia-inducible factor (HIF)-1 pathway, which shifts glucose metabolism from oxidative phosphorylation to glycolysis with lactate production, thereby reducing the mitochondrial production of ROS (39), and the β-catenin pathway, which blocks apoptosis by several cascade pathways involving Akt, Bax, survivin, and p53 (48, 49). Efficient recovery from IRI is also dependent on signaling through the EGF receptor (45).
During hypoxia, the oxygen-sensitive HIF-1 α-subunit associates with the constitutively expressed HIF-1 β-subunit, forming HIF-1, which enhances transcription in the nucleus through binding hypoxia-responsive elements (HREs) within gene promoters, including those encoding 1) glucose transporters, 2) glycolytic enzymes such as lactate dehydrogenase A (LDHA), enolase, and pyruvate kinase M2 (PKM2), and 3) regulatory kinases such as pyruvate dehydrogenase kinase (PDK)1 (30, 39). The combined inhibition of pyruvate dehydrogenase by PDK1 and increased levels of LDHA effectively shifts metabolism of pyruvate to lactate instead of acetyl-CoA, which increases glucose flux through glycolysis (21, 24, 40, 47). HIF-1 also induces transcription of prolyl hydroxylase domain 3 (PHD3) and, in a positive feedback loop, PKM2 hydroxylated by PHD3 is recruited to HREs with HIF-1, and the PKM2-PHD3 complex enhances HIF-1 activity by increasing promoter occupancy (29, 30). PKM2 also directly binds β-catenin and transactivates this protective pathway (6).
Analysis of mRNA transcripts upregulated by hypoxia in a culture of human proximal tubule (HK-2) cells revealed 48 targets, including mucin 1 (MUC1), a transmembrane protein expressed on the apical surface of the distal tubule and collecting duct (CD) in adult kidneys and in the proximal tubule (PT) during development and potentially after injury (5, 17, 27). The cytoplasmic tail of MUC1 harbors numerous sites for phosphorylation and protein docking and traffics to the nucleus, where it associates with transcription factors including β-catenin, p120-catenin, estrogen receptor-α, and p53 (12, 23, 41). There are two functional HREs for HIF-1 binding in the MUC1 promoter, and MUC1 was induced in cultured human renal tumor cells during hypoxia, whereas rodent Muc1 was induced in the kidney of a rat during IRI (2, 27). MUC1 is overexpressed in most epithelial tumors, and Chaika et al. (7) recently reported that MUC1 overexpression is directly linked to aberrant glucose metabolism in pancreatic adenocarcinomas. They found that MUC1 can bind, stabilize, and enhance HIF-1α occupancy and activity on promoters of glycolytic genes in a hypoxia-dependent manner (7, 31).
MUC1 plays a unique role in kidney function, as it has been recently reported that medullary cystic kidney disease type 1 is caused by a frame-shift mutation in one allele of MUC1, resulting in an unpredictable progression of chronic kidney disease and no other clinical phenotype (4, 22). As MUC1 plays a key role in the HIF-1-dependent cellular adaptation of metabolism in response to hypoxia in cultured cells, we assessed the role of mouse mucin 1 (Muc1) in AKI using a mouse model of kidney IRI. We observed that tubular injury was worse and recovery of kidney function and morphology was hampered in the absence of Muc1. Although Muc1 was induced during IRI, an earlier response was the appearance of Muc1 in the nucleus of tubular epithelia. We also observed that levels of HIF-1α, induction of HIF-1 gene targets, and activation of AMP-activated protein kinase (AMPK) were Muc1 dependent during IRI. As genetic stabilization of HIF activity in the kidney epithelia is cytoprotective during IRI, whereas reduced levels of HIF are associated with more severe injury (8, 14, 36), our data are consistent with Muc1 enhancement of the HIF-1 protective pathway during IRI.
MATERIALS AND METHODS
Antibodies.
Armenian hamster anti-MUC1/Muc1 cytoplasmic tail monoclonal antibody CT2 was as previously described (37). Rabbit polyclonal antibodies against LDHA (NBP1-4836), PDK1 (NB100-2384), PHD3 (NB100-303), and PKM2 (NBP1-48304) were purchased from Novus Biologicals (Littleton, CO). Rabbit polyclonal antibodies against enolase (no. 3810) and phospho-Thr172 AMPK-α (no. 3531) were purchased from Cell Signaling Technology (Danvers, MA). Rabbit polyclonal antibodies against AMPK (no. 07-350) were from Millipore (Billerica, MA), and those against HIF-1α (no. 10006421) were from Cayman Chemical (Ann Arbor, MI). Mouse monoclonal antibodies against β-actin were purchased from Sigma (A1978, St. Louis, MO).
Mouse renal ischemia-reperfusion model.
All experiments were undertaken on 12-wk-old male C57BL/6J mice. Muc1 global knockout (KO) mice on a C57BL/6J background were as previously described and are unremarkable under normal physiological conditions (42). All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Indiana University School of Medicine Animal Care and Use Committee. AKI was induced as previously described using ischemia followed by reperfusion (16, 26, 43). Briefly, animals were anesthetized with isoflurane administered as a mixture with oxygen throughout the surgery and buprenorphine HCl subcutaneously at the time of induction. After induction, animals were sterilely prepped and placed on a homeothermic table to maintain core body temperature at 37°C. A midline incision was made, and both renal pedicles were clamped using nontraumatic microaneurysm clamps. Animals were monitored for the level of anesthesia. After 19 min of ischemia, the clamps were removed, and the incision was closed in two layers. Warmed saline (0.9%) was given intraperitoneally at the time of closure. Animals remained on the warming table until they moved spontaneously. Kidneys and blood were harvested at the time of death after 0–72 h of recovery.
Serum creatinine (sCr) was determined by capillary electrophoresis at The University of Texas Southwestern O'Brien Kidney Research (Physiology) Core. One kidney from each mouse was flash frozen in liquid nitrogen, and one kidney was sliced lengthwise and placed in 4% paraformaldehyde for analysis by microscopy or in situ hybridization. Kidney samples for microscopy fixed in paraformaldehyde were embedded in paraffin to prepare slides (4 μm thick), stained with hematoxylin and eosin, and scored for tubular damage (necrosis, luminal debris, and tubular dilation) using epifluorescence microscopy. Scores were based on the percentage of tubule damage as previously described (46). In situ hybridization was carried out as previously described using a murine Muc1 probe described by EURExpress (15).
Immunoblot analysis of kidney extracts.
Frozen kidneys were sliced lengthwise and then cut in half yielding a quarter kidney (or one-half of a coronal section). The one-quarter kidney (∼0.4 mg) was homogenized in 0.2 ml HEPES-buffered saline [10 mM HEPES (pH 7.4) and 150 mM NaCl] with protease inhibitor cocktail set III and phosphatase inhibitor cocktail set II from Calbiochem (EMD Millipore, Billerica, MA) and centrifuged at 8,000 g, and the supernatant was assayed for protein using the Pierce BCA protein assay kit (Rockford, IL). An equal amount of protein (60 μg) from each mouse kidney was subjected to SDS-PAGE and immunoblot analysis using horseradish peroxidase-conjugated secondary antibodies from Jackson ImmunoResearch Laboratories (West Grove, PA) and developed with Western Lightning Plus ECL as directed by the manufacturer (Perkin-Elmer, Waltham, MA). Bands were identified either with a BioRad Versadoc or by exposing Kodak BioMax MR film and quantified using BioRad Quantity One software. Samples from Muc1 KO and wild-type mouse kidneys were developed side by side for the same time period. Data were analyzed using two-way ANOVA with a Tukey post hoc test. Student's t-test was used to compare two groups of data where indicated.
RESULTS
Muc1 is induced in kidney tubule epithelial cells during IRI.
As previously published data have indicated that human MUC1 is induced in cultured cells by hypoxia and rodent Muc1 is induced in the rat kidney during IRI (2, 7, 27), we assessed Muc1 levels in control mouse kidneys in response to ischemia. Kidneys of wild-type C57BL/6 mice were subjected to IRI, and Muc1 protein and transcript levels were assessed. Both renal pedicles of mice were clamped for 19 min before a recovery period of 0–72 h, when mice were sacrificed and blood and kidneys were harvested.
Immunoblots of whole kidney homogenates revealed that Muc1 was significantly induced 4.2-fold by 72 h of recovery (Fig. 1A). Previously published data have indicated that renal Muc1/MUC1 is expressed on the apical surface of epithelial cells primarily in the distal tubule and CD in adult kidneys and in the PT during development and potentially after injury (2, 5, 17, 27). The presence of Muc1 transcripts in the S1, S2, and S3 segments of the rodent PT was recently confirmed by deep sequencing of microdissected renal tubules (25). To determine where Muc1 is expressed and induced during IRI in a mouse model, we used 1) immunohistochemistry with an anti-MUC1 cytoplasmic tail antibody to stain kidney tissue (Fig. 1) and 2) in situ hybridization with a murine Muc1-specific probe to identify Muc1 mRNA (Fig. 2). Localization was based primarily on the morphology of tubule segments.
Fig. 1.

Mucin (Muc)1 is induced during kidney ischemia-reperfusion injury (IRI) and appears in the nucleus. Kidneys of control C57BL/6 mice were subjected to 19 min of ischemia and recovery for 0–72 h (n = 3–5 mice at each time point). A: immunoblot analysis of control mouse kidney homogenates (60 μg protein/lane) with anti-Muc1 cytoplasmic tail antibodies (small subunit Mr: 25 kDa) and then anti-β-actin antibodies as a loading control (Fig. 6D) revealed a significant 4.2-fold increase of Muc1 by 72 h using one-way ANOVA (means ± SE, *P < 0.05). B–O: slices of paraformaldehyde (PFA)-fixed tissue were subjected to immunohistochemistry with the same anti-Muc1 antibody. Images for B–K are ×400 magnification. B: no Muc1 signal was observed in kidneys of Muc1 knockout (KO) mice, as expected (negative control). C: renal Muc1 staining in sham-treated C57BL/6 mice was predominantly in the thick ascending limb (TAL), distal convoluted tubule (DCT), and collecting duct (CD) (box enlarged in O). After 19 min of ischemia [time (t) = 0], Muc1 staining was also apparent in the cytoplasm of the TAL, DCT, CD, and proximal tubules (PTs) in the cortex (D) and outer stripe of the outer medulla (H and L). At 4, 24, and 72 h of recovery, Muc1 staining was also apparent in the nucleus (E–G and I–K). Boxes in H and K are enlarged in L and M, respectively, to emphasize Muc1 staining in the cytoplasm and nucleus of recovering PT (black arrows) and on the cell surface (gray arrow). Nuclear staining was clearly evident in the CD (purple arrows) and PT (blue arrows) at 4 h of recovery and absent in the CD (green arrows) and PT (yellow arrows) in sham-treated mice (compare N and O). G, glomerulus.
Fig. 2.
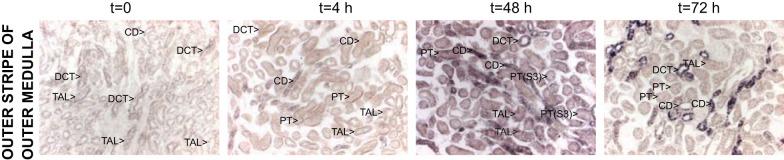
Muc1 transcripts are induced during kidney IRI. Kidneys of control C57BL/6 mice were subjected to IRI as described in Fig. 1. PFA-fixed kidneys were subjected to in situ hybridization using a Muc1-specific probe. Transcripts for Muc1 identified with in situ hybridization were evident at t = 0 but notably increased by 4 h of recovery in the TAL, DCT, and CD [outer stripe of the outer medulla (OSOM) is shown]. Transcripts were maximally increased at 48 h of recovery in all tubules, including the PT [see label for PT(S3) as shown in the third panel 48 h].
Using immunohistochemistry, we found that Muc1 was localized primarily in the thick ascending limb (TAL), distal convoluted tubule (DCT) and collecting duct (CD); Muc1 was also present in the proximal tubule (PT), albeit at lower levels. No Muc1 staining was observed in Muc1 KO kidneys (Fig. 1B). While Muc1 was found on the apical surface of tubule epithelia under sham conditions (Fig. 1, C and box enlarged in O), we also observed Muc1 in the cytoplasm immediately after 19 min of ischemia (time 0; Fig. 1, D, H, and box enlarged in L) and in the nucleus at 4, 24, and 72 h of recovery (Fig. 1, E–G, I–K, and box in K enlarged in M). The boxes in Fig. 1, E (time = 4 h cortex) and C (sham cortex), are enlarged and shown in Fig. 1, N and O, respectively, and clearly revealed nuclear staining in the CD (purple arrows) and PT (blue arrows) that was absent in sham-treated kidneys for the CD (green arrows) and PT (yellow arrows). Muc1 staining was also evident in the S3 segment of the regenerating PT in the outer stripe of the outer medulla at 72 h of recovery (compare Fig. 1, L and M) and accounted in part for the significant increase in Muc1 levels observed by immunoblot analysis at 72 h. Muc1 staining was visible in both the nuclei (black arrows) and on the cell surface (gray arrow) in flatten cells. Using in situ hybridization, we found that levels of Muc1 transcripts were notably increased in the CD, DCT, and TAL between 0 and 4 h of recovery, whereas transcripts were significantly increased in all tubule segments, including the PT, by 48 h of recovery (Fig. 2).
Our data support the hypothesis that Muc1 transcripts and protein are induced in the kidney in response to ischemia in a mouse model of IRI. Furthermore, we found that an early response of Muc1 is targeting to the nucleus within cells of many tubule segments, including the PT of the outer stripe of the outer medulla, which is the segment of the kidney tubule most sensitive to ischemia.
Muc1 is protective during IRI of the kidney.
To establish whether Muc1 plays a key role in renal adaptations to IRI, we also subjected kidneys of Muc1 KO mice to IRI and assessed kidney function, tubule damage, and changes in key adaptive proteins compared with congenic control C57BL/6 mice.
Within each group of either Muc1 KO or control mice, sCr at 24 h of recovery was significantly higher than sCr at time 0 (Fig. 3A). In control mice, sCr clearly peaked at 24 h and was significantly lower and no different than levels at time 0 by 72 h, consistent with maximal damage at 24 h and recovered kidney function by 72 h. In contrast, sCr in Muc1 KO mice was significantly higher than values for control mice at 72 h, consistent with no recovery of kidney function at 72 h. sCr profiles for Muc1 KO versus control mice were also significantly different, as judged by two-way ANOVA (P < 0.01). We concluded that Muc1 was protective for kidney function and recovery from IRI.
Fig. 3.

Muc1 protects kidney function and morphology during kidney IRI. Kidneys of Muc1 KO mice and congenic control C57BL/6 mice were subjected to IRI for 19 min and recovery for 0–72 h. A: blood was collected at the time of death and assayed for serum creatinine (sCr). Profiles for sCr between Muc1 KO mice (open circles) and congenic wild-type mice (closed circles) were significantly different by two-way ANOVA (n = 3–6 mice at each time point, P < 0.01). Values at 72 h (means ± SE) were different between Muc1 KO and wild-type mice (P < 0.001). Values at 24 h in control mouse kidneys were significantly higher than at t = 0 (*P < 0.01). Values for Muc1 KO mouse kidneys at both 24 and 72 h were significantly higher than at t = 0 (**P < 0.001). B–I: one kidney from each mouse was fixed and stained with hematoxylin and eosin and scored for tubule damage as described in the text. B–H: representative images (×200) are shown for control (B–D) and Muc1 KO (F–H) kidneys at 0, 4, and 24 h of recovery, respectively. G: karyorrhexis and karyopyknosis (arrows) and luminal casts (*) were especially evident in Muc1 KO kidneys at t = 4 h. E and I: images (×400) from 72 h of recovery are shown for control C57BL/6 (E) and Muc1 KO (I) mice. E: mitotic figures (arrows) and flattened cells in recovering tubules (arrow heads) were observed in control mouse kidneys at 72 h. I and inset: luminal casts (*) and calcium phosphate precipitates (arrowheads) were observed in Muc1 KO kidneys at 72 h.
To assess tubule morphology, one kidney from each mouse was fixed, stained with hematoxylin and eosin (Fig. 3), and scored for the percentage of damaged tubules in the outer stripe of the outer medulla using a previously described method (where 0 is none, 1 is ≤10%, 2 is 11–25%, 3 is 26–45%, 4 is 46–75%, and 5 is >76%) (46). While the morphology of control and Muc1 KO mouse kidneys was no different at time 0, there was more damage at 4 h in Muc1 KO kidneys (scores of 4, 5, and 5) than in control kidneys (scores of 2, 3, and 4) with acute tubular necrosis features, such as coagulative necrosis with luminal casts, karyorrhexis, and karyopyknosis (Fig. 3G). Whereas kidneys from both control and KO mice were maximally damaged at 24 h (all had scores of 5), tubule morphology in kidneys from control mice was notably recovered at 72 h with flattened and dividing cells apparent in the PTs (Fig. 3 E), and kidneys of Muc1 KO mice still exhibited significant PT damage as well as intracellular and extracellular precipitates of calcium phosphate (Fig. 3, I and inset). The observed lack of tubular regeneration in Muc1 KO mice at 72 h of recovery correlates with the reduced kidney function (elevated sCr) observed in this group. We concluded that Muc1 KO mice respond to IRI with more severe kidney injury as well as a reduced ability of the kidney tubules to regenerate.
Muc1 stabilizes HIF-1α and prevents metabolic stress during IRI.
A study (7) in cultured human cell lines revealed that MUC1 complexes with HIF-1α and stabilizes it by preventing its degradation. HIF-1α was previously observed in the cytoplasm of the TAL and CD in kidneys of sham-treated rats and induced in the cytoplasm and nucleus of the PT during IRI (44). As these were the same tubule segments where we localized Muc1 cytoplasmic and nuclear staining and Muc1 induction during IRI (Figs. 1 and 2), we compared HIF-1α staining in Muc1 KO and control mouse kidneys during IRI. We found that both cytoplasmic and nuclear levels of HIF-1α throughout the kidney tubules were substantially reduced in the absence of Muc1 at 4 h of recovery during IRI compared with kidneys from congenic control mice (Fig. 4, compare A and B with C and D). This was confirmed by finding a significantly lower level of HIF-1α in immunoblots of homogenates of Muc1 KO kidneys (32%) compared with control kidneys (Fig. 4E).
Fig. 4.

Muc1 stabilizes hypoxia-inducible factor (HIF)-1α levels during IRI. Kidneys of Muc1 KO mice and congenic control C57BL/6 mice were subjected to IRI for 19 min and recovery for 4 h. A–D: one kidney from each mouse was fixed and stained by immunohistochemistry with anti-HIF-1α antibody. Levels of HIF-1α were notably reduced in the Muc1 KO mouse cortex and OSOM (C and D) compared with the control mouse kidney (A and B). Examples of the glomerulus (G), PT, DCT, TAL, and CD are shown. E: immunoblot analysis of mouse kidney homogenates (60 μg protein/lane) with anti-HIF-1α antibodies revealed a significant decrease in the absence of Muc1 using Student's t-test (means ± SE, n = 3, *P < 0.05). Immunoblot analysis of β-actin was used as a loading control.
Analysis of chromatin from cultured cells using microarrays revealed that promoters of several metabolic genes were occupied by MUC1 in cultured human cell lines (7). In the same cultures, MUC1 overexpression enhanced transcription of numerous glycolytic genes under both normoxic and hypoxic conditions (7). The MUC1-mediated induction was blocked by knockdown of HIF-1α, consistent with MUC1 transactivation of HIF-1 activity (7). To determine if efficient induction of HIF-1 gene targets during IRI in our mouse model was Muc1 dependent, we immunoblotted kidney homogenates for key enzymes (LDHA, enolase, PHD3, PKM2, and PDK1) involved in the HIF-1-dependent metabolic switch in glucose metabolism in response to hypoxia/ischemia (Figs. 5 and 6). We found that levels of the glycolytic enzymes LDHA and enolase were increased two- and threefold, respectively, by 24 h of recovery during IRI in control mice (Fig. 5). Induction profiles for both LDHA and enolase in Muc1 KO mice were significantly lower compared with profiles in control mice (P < 0.01 and P < 0.05, respectively).
Fig. 5.

Muc1 transactivates gene targets of HIF-1: lactate dehydrogenase A (LDHA) and enolase (ENO). Kidneys of Muc1 KO mice and congenic control C57BL/6 mice were subjected to IRI for 19 min and recovery for 0–72 h. Products of HIF-1 gene targets were measured by immunoblot analysis of 60 μg kidney homogenates for LDHA (A and C) and ENO (B and D) and then β-actin as a loading control (E). Bands on immunoblots were quantified and presented as means ± SE relative to that of control mice at t = 0 (set as 1). Profiles of LDHA and ENO for Muc1 KO and control mouse kidneys were significantly different by two-way ANOVA (P < 0.01 and P < 0.05, respectively, as indicated). Levels of LDHA were significantly increased in control mice at 24 h (**P < 0.01) and 72 h (*P < 0.05) of recovery compared with 0 h. Levels of LDHA at 4, 24, and 72 h of recovery were significantly different between Muc1 KO and control mice (P < 0.05). Levels of ENO were significantly increased in control mice at 24 h (*P < 0.05). Levels of ENO at 24 h were significantly different between Muc1 KO and control mice (P < 0.05).
Fig. 6.

Response of gene targets of HIF-1 are Muc1 dependent: prolyl hydroxylase domain 3 (PHD3), pyruvate kinase M2 (PKM2), and pyruvate dehydrogenase kinase 1 (PDK1). Kidneys of Muc1 KO mice and congenic control C57BL/6 mice were subjected to IRI for 19 min and recovery for 0–72 h. Products of three HIF-1 gene targets were measured by immunoblot analysis 60 μg kidney homogenates for PHD3 (A and D), PKM2 (B and E), and PDK1 (C and F). Immunoblot analysis of β-actin was used as a loading control (included in D for PHD3 and shown in Fig. 5E for PKM2 and PDK1). Bands on immunoblots were quantified and presented as means ± SEM relative to that of control mice at t = 0 (set as 1). Profiles of PHD3 (P < 0.005), PKM2 (P < 0.05), and PDK1(P < 0.01) for Muc1 KO and control mouse kidneys were significantly different by two-way ANOVA. Levels of PHD3 were significantly different between control and Muc1 KO mouse kidneys at t = 0 and 4 h of recovery (P < 0.05), and levels of PHD3 in control mouse kidneys at 72 h were significantly less than at t = 0 (*P < 0.05). Levels of PKM2 were significantly different at t = 0 between control and Muc1 KO mouse kidneys (P < 0.01). Levels of PKM2 were significantly reduced in Muc1 KO mouse kidneys after 24 h (*P < 0.05) and 72 h (**P < 0.01) of recovery compared with levels at t = 0.
Levels of PHD3 were also significantly higher in control mice during IRI compared with Muc1 KO mice (P < 0.005; Fig. 6A). PHD3 levels in control mice were significantly reduced by 72 h of recovery, whereas levels were consistently lower in Muc1 KO mice at all time points. PKM2 profiles were significantly different between control and KO mice (P < 0.05), with levels in Muc1 KO mice being initially higher than control mice and significantly reduced by 24 h of recovery (Fig. 6B). In contrast, PKM2 levels appeared to increase during recovery in control mouse kidneys. Profiles of PDK1 between Muc1 KO and control mice were significantly different (P < 0.01) and remarkably higher in Muc1 KO mice (Fig. 6C). Taken together, the results indicate that regulation of LDHA, enolase, PHD3, PKM2, and PDK1 during IRI are clearly Muc1 dependent. However, the irregular profiles of PKM2 and PDK1 in Muc1 KO mice compared with control mice likely reflect a secondary response to either aberrant metabolism in response to IRI and/or changes in basal expression.
As we observed so many Muc1-dependent changes in levels of key enzymes that orchestrate the switch from oxidative to glycolytic metabolism of glucose, we asked if the increased injury found in kidneys of Muc1 KO mice during IRI could reflect a low energy state in the epithelia. To test this possibility, we first immunoblotted kidney homogenates for the energy sensor AMPK (Fig. 7). This serine/threonine kinase is rapidly activated by kidney ischemia and regulates cellular metabolism by inhibiting energy-consuming pathways and enhancing energy-producing pathways (21, 33, 35). Its activated/phosphorylated form, phospho-Thr172 AMPK-α [phospho-AMPK (pAMK)], increases in response to high [AMP]:[ATP] ratios and other cellular stresses (11, 35).
Fig. 7.

Muc1 prevents metabolic stress during IRI. Kidneys of Muc1 KO mice and congenic control C57BL/6 mice were subjected to IRI for 19 min and recovery for 0–72 h. Metabolic stress was assessed by immunoblot analysis of 60 μg kidney homogenates for activated phosphorylated AMP-activated protein kinase (pAMPK; A and D), AMPK (B and E), and β-actin (F) as a loading control. Bands on immunoblots were quantified and presented as means ± SE relative to that of control mice at t = 0 (set as 1). Data are also presented as the ratio of pAMPK to AMPK (C). Profiles of pAMPK (P < 0.001), AMPK (P < 0.01), and the ratio of pAMPK to AMPK (P < 0.001) for Muc1 KO and control mouse kidneys were significantly different by two-way ANOVA. Levels of pAMPK in Muc1 KO mice were significantly increased at 4 h (***P < 0.001) and 24 h (**P < 0.005) of recovery compared with levels at t = 0 and significantly higher at 4 and 24 h of recovery than levels for control kidney levels (P < 0.001). Levels of AMPK in Muc1 KO kidneys were significantly higher at 24 and 72 h (P < 0.05) than levels in control kidneys. The ratio of pAMPK to AMPK in Muc1 KO mice was significantly different at 24 and 72 h of recovery compared with levels at t = 0 (*P < 0.05), and the ratio was significantly higher than in control mouse kidneys at 4 and 24 h of recovery (P < 0.005).
Levels of total AMPK, as measured by blotting for the AMPK α-subunit, were significantly higher in kidneys of Muc1 KO mice during IRI compared with control mice (P < 0.01; Fig. 7B). Whereas levels of pAMPK were unchanged in kidneys of control mice during IRI, we found a significant prolonged increase in levels of pAMPK in kidney homogenates of Muc1 KO mice at 4 and 24 h of recovery compared with time 0 of both control and Muc1 KO mice (5-fold, P < 0.001; Fig. 7A). The ratio of pAMPK to AMPK was significantly higher in the profile from Muc1 KO mice than control mice after IRI, consistent with energy stress in mice lacking Muc1, likely due to aberrant adaptation of glucose metabolism (P < 0.001; Fig. 7C).
DISCUSSION
Epithelial HIF-1 mediates protection and recovery from IRI.
Several studies have shown that systemic activation of HIF in animals by inhibition of prolyl hydroxylases subsequently protects the kidneys in models of IRI (3, 14, 19). However, the mechanism of protection was not immediately obvious as HIF-1α is found in kidney tubule epithelia and only in endothelia after hypoxia, whereas HIF-2α is primarily expressed in endothelial cells, the glomerulus, and peritubular interstitial cells (9). Using endothelial-specific KO of either HIF-1α or HIF-2α, Kapitsinou et al. (20) have now shown that endothelial HIF-2 plays a critical role in protecting the kidney during IRI by regulating inflammation through suppression of VCAM-1. However, there is also evidence that genetic stabilization of HIF activity by KO of von Hippel-Lindau in all tubule epithelia, or just in the TAL alone, protects the kidney against IRI (8, 36). Our present data also support a role for epithelial HIF-1 protection of the kidney during IRI as Muc1 is present only in epithelial cells; in the absence of Muc1, renal HIF-1α levels were reduced, expression from key HIF-1 targets was blunted, tubule damage was increased, and recovery of kidney function and morphology was blocked. Our data are consistent with the phenotype expected during IRI in mice with reduced HIF activity, as Hill et al. (14) observed increased tubule damage and higher sCr levels in heterozygous HIF-1α and HIF-2α KO mice during IRI compared with control mice.
Muc1 is also a target of HIF-1 activity, and we did observe increased levels of Muc1 transcripts throughout the renal tubules and increased levels of Muc1 protein in whole kidney homogenates during IRI. However, the mechanism of Muc1 protection through HIF-1 is most evident in the appearance of Muc1 in the cytoplasm and nucleus of the epithelia early during IRI and in recovering PT epithelial cells. Taken together, our data indicate that key aspects of the epithelial HIF-1 protective pathway are Muc1 dependent in this model of ischemic AKI.
Muc1 transactivates HIF-1.
Recent studies in several human pancreatic and colon cell lines have revealed that MUC1 enhances gene transcription of numerous glycolytic enzymes under hypoxic culture conditions (hexokinase 2, enolase 1, phosphoglycerate kinase 1, phosphoglucomutase 2, and LDHA) and that MUC1 physically occupies promoters of at least two of these genes (enolase and phosphoglucomutase 2) (7). The same study showed that MUC1 overexpression increased secretion of lactate from cultured cells and enhanced cellular uptake of [3H]2-deoxyglucose in both cultured cells and MUC1-positive tumors in live mice, whereas MUC1 knockdown in cultured human cells blocked these effects. A study (7) using NMR-based differential metabolomics revealed a MUC1-dependent increase in glutamine levels and increased glucose metabolic flux through both glycolysis and the pentose phosphate pathway. MUC1 also enhanced transcription of HIF-1α and PHD3 (but not PHD1 or PHD2), coimmunoprecipitated with both PKM2 and HIF-1α, stabilized HIF-1α levels in cells independent of transcription, and enhanced HIF-1 occupation and activity at HREs within gene promoters during hypoxia (7). Taken together, the data support a role for MUC1 in facilitating metabolic changes during hypoxia by physically interacting with and stabilizing HIF-1α and enhancing transcription of HIF-1-dependent protective metabolic pathways, potentially through interactions with PKM2 and β-catenin (see model in Fig. 8).
Fig. 8.

Muc1 transactivates the HIF-1 protective pathway during IRI in the kidney. HIF-1, composed of an oxygen-regulated α-subunit and a constitutively expressed β-subunit, binds to hypoxia-responsive elements (HREs) and promotes transcription of 1) genes to shift glucose metabolism; 2) PHD3, which hydroxylates PKM2; 3) PKM2, which transactivates both HIF-1 and β-catenin; and 4) Muc1, which binds, stabilizes, and transactivates both β-catenin and HIF-1 protective pathways. See text for further discussion.
We also observed Muc1-dependent changes in profiles for PHD3, PKM2, and PDK1 in kidney tissue during IRI. There was a significant stabilization of PHD3 and induction of PKM2 during IRI in control mouse kidneys consistent with their reported HIF-1-dependent induction by hypoxia in cultured cells (29, 30). Interestingly, we observed a slow decrease in PHD3 levels while PKM2 levels increased, consistent with the reported stabilization of PKM2 by PHD3 hydroxylation in cultured cells (29). Neither PHD3 nor PKM2 were induced by IRI in kidney homogenates of the Muc1 KO mouse. The basis for the significantly increased levels of PKM2 at 0 h of recovery and the subsequent reduction is not clear. As levels of PDK1 are also significantly higher in the Muc1 KO mouse kidney but mirror the response in the control mouse kidney, it is likely that both PDK1 and PKM2 are modulated by a complex mechanism including Muc1 and/or that levels are differentially modulated in specific cell types of the kidney tubule. The S3 segment is the only part of the PT that can selectively undergo glycolysis as well as oxidative phosphorylation, as the S1 and S2 segments are not known to undergo glycolysis (for a review, see Ref. 38). Therefore, the appearance of Muc1 and its role in the upregulation of the glycolytic pathway could potentiate S3 segment recovery after ischemia.
The metabolic response to IRI reflects the HIF-1-dependent responses to hypoxia. We observed that the expression profiles of key enzymes that shift cellular glucose metabolism toward the glycolytic pathway in response to hypoxia were mostly replicated in kidneys subjected to IRI. These changes in enzyme profiles were also Muc1 dependent as there was a lack of a coordinated response after ischemia in Muc1 KO mice compared with control mice, which would likely produce aberrant handling of glucose and result in cellular metabolic stress. In support of this possibility, we observed an increased and prolonged activation of the energy sensor AMPK in the Muc1 KO mouse kidney in response to IRI, which could represent a protective response to energy stress. Levels of pAMPK remained near baseline in the control mouse kidney but were considerably elevated at 4 and 24 h in the Muc1 KO mouse, consistent with prolonged energy stress and activation of AMPK. However, AMPK can also be activated in the absence of an increased [AMP]:[ATP] ratio, as previously described for AMPK activation in cystic fibrosis airway epithelial cells (10). Kinases such as Ca2+/calmodulin-dependent protein kinase kinase-β are known to activate AMPK in response to Ca2+ stress (13, 18). Such metabolic stress-independent AMPK activation could be relevant in IRI as we have observed calcium phosphate precipitates in kidneys of Muc1 KO mice at 72 h of recovery during IRI. Future studies will address these possibilities as we found no evidence that human MUC1 is a substrate for AMPK-mediated phosphorylation using in vitro assays (data not shown).
In summary, we have established that Muc1 is a novel and significant inducer of the HIF-1 protective pathway in kidney tubule epithelia during IRI.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants DK-077124 and DK-099345 (to T. A. Sutton), DK-075048 (to K. R. Hallows), DK-084184 (to N. M. Pastor-Soler), CA-64389 (to S. J. Gendler), DK-080821 and DK-081646 (to V. H. Haase), DK-101791 (to V. H. Haase), and R01-DK-103776 (to J. Ho), the Krick-Brooks Chair in Nephrology (to V. H. Haase), NIH Grants DK-62277, DK-100287, and DK-095498 (to S. P. Monga), Department of Veterans Affairs Merit Award 1I01BX002348 (to V. H. Haase), NIH Grant DK-097889 and a Sanofi Fellowship (to M. M. Al-bataineh), a DCI Paul Teschan Research Grant (to T. A. Sutton), a Samuel and Emma Winters Foundation grant (to R. P. Hughey), NIH DK-079307 Pilot Project (to R. P. Hughey), and NIH Grant P30-DK-079307 Microscopy Core (to N. M. Pastor-Soler and S. Bastacky).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.M.P.-S., T.A.S., H.E.M., C.L.K., S.J.G., C.S.M., J.H., M.M.A.-b., K.R.H., and R.P.H. conception and design of research; N.M.P.-S., T.A.S., H.E.M., C.L.K., S.I.B., J.H., M.M.A.-b., S.S., H.K., R.P.H., and C.S.M. performed experiments; N.M.P.-S., T.A.S., H.E.M., C.L.K., S.I.B., J.H., M.M.A.-b., S.P.M., and R.P.H. analyzed data; N.M.P.-S., T.A.S., H.E.M., C.L.K., S.J.G., S.I.B., J.H., M.M.A.-b., K.R.H., S.P.M., V.H.H., and R.P.H. interpreted results of experiments; N.M.P.-S. and R.P.H. prepared figures; N.M.P.-S., T.A.S., C.L.K., S.J.G., K.R.H., V.H.H., and R.P.H. edited and revised manuscript; N.M.P.-S., T.A.S., S.J.G., S.I.B., J.H., K.R.H., S.P.M., V.H.H., and R.P.H. approved final version of manuscript; T.A.S., R.P.H., and N.M.P.-S. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Gregg L. Semenza, Dr. Youhua Liu, and Dr. Polly E. Mattila for providing antibodies for immunoblot analysis and Dr. Edwin K. Jackson, Dr. Thomas R. Kleyman, and Dr. Ora A. Weisz for helpful discussions.
REFERENCES
- 1.Adkison D, Hollwarth ME, Benoit JN, Parks DA, McCord JM, Granger DN. Role of free radicals in ischemia-reperfusion injury to the liver. Acta Physiol Scand Suppl 548: 101–107, 1986. [PubMed] [Google Scholar]
- 2.Aubert S, Fauquette V, Hemon B, Lepoivre R, Briez N, Bernard D, Van Seuningen I, Leroy X, Perrais M. MUC1, a new hypoxia inducible factor target gene, is an actor in clear renal cell carcinoma tumor progression. Cancer Res 69: 5707–5715, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Bernhardt WM, Campean V, Kany S, Jurgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Gunzler V, Amann K, Willam C, Wiesener MS, Eckardt KU. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17: 1970–1978, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoe JR, Hladik G, Klemmer P, Knohl SJ, Scheinman SJ, Vo N, Santi A, Harris A, Canaday O, Weller N, Hulick PJ, Vogel K, Rahbari-Oskoui FF, Tuazon J, Deltas C, Somers D, Megarbane A, Kimmel PL, Sperati CJ, Orr-Urtreger A, Ben-Shachar S, Waugh DA, McGinn S, Bleyer AJ Jr, Hodanova K, Vylet'al P, Zivna M, Hart TC, Hart PS. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol 9: 527–535, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braga VM, Pemberton LF, Duhig T, Gendler SJ. Spatial and temporal expression of an epithelial mucin, Muc-1, during mouse development. Development 115: 427–437, 1992. [DOI] [PubMed] [Google Scholar]
- 6.Canal F, Perret C. PKM2: a new player in the β-catenin game. Future Oncol 8: 395–398, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T, Yu F, Johnson KR, Powers R, Hollingsworth MA, Singh PK. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 α to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA 109: 13787–13792, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fahling M, Mathia S, Paliege A, Koesters R, Mrowka R, Peters H, Persson PB, Neumayer HH, Bachmann S, Rosenberger C. Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J Am Soc Nephrol 24: 1806–1819, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol 291: F271–F281, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallows KR, Fitch AC, Richardson CA, Reynolds PR, Clancy JP, Dagher PC, Witters LA, Kolls JK, Pilewski JM. Up-regulation of AMP-activated kinase by dysfunctional cystic fibrosis transmembrane conductance regulator in cystic fibrosis airway epithelial cells mitigates excessive inflammation. J Biol Chem 281: 4231–4241, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol 70: 431–457, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2: 9–19, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, Maxwell PH. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 19: 39–46, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho J, Pandey P, Schatton T, Sims-Lucas S, Khalid M, Frank MH, Hartwig S, Kreidberg JA. The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J Am Soc Nephrol 22: 1053–1063, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horbelt M, Lee SY, Mang HE, Knipe NL, Sado Y, Kribben A, Sutton TA. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol 293: F688–F695, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Howie AJ. Epithelial membrane antigen in normal and proteinuric glomeruli and in damaged proximal tubules. J Pathol 148: 55–60, 1986. [DOI] [PubMed] [Google Scholar]
- 18.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280: 29060–29066, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Kapitsinou PP, Jaffe J, Michael M, Swan CE, Duffy KJ, Erickson-Miller CL, Haase VH. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am J Physiol Renal Physiol 302: F1172–F1179, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapitsinou PP, Sano H, Michael M, Kobayashi H, Davidoff O, Bian A, Yao B, Zhang MZ, Harris RC, Duffy KJ, Erickson-Miller CL, Sutton TA, Haase VH. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest 124: 2396–2409, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Kirby A, Gnirke A, Jaffe DB, Baresova V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hulkova H, Sovova J, Vylet'al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45: 299–303, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer 9: 874–885, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107: 2037–2042, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol; doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SY, Horbelt M, Mang HE, Knipe NL, Bacallao RL, Sado Y, Sutton TA. MMP-9 gene deletion mitigates microvascular loss in a model of ischemic acute kidney injury. Am J Physiol Renal Physiol 301: F101–F109, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leonard MO, Cottell DC, Godson C, Brady HR, Taylor CT. The role of HIF-1 α in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J Biol Chem 278: 40296–40304, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Liano F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 50: 811–818, 1996. [DOI] [PubMed] [Google Scholar]
- 29.Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145: 732–744, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab 23: 560–566, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehla K, Singh PK. MUC1: a novel metabolic master regulator. Biochim Biophys Acta 1845: 126–135, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta RL, Pascual MT, Soroko S, Savage BR, Himmelfarb J, Ikizler TA, Paganini EP, Chertow GM. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 66: 1613–1621, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Mount PF, Hill RE, Fraser SA, Levidiotis V, Katsis F, Kemp BE, Power DA. Acute renal ischemia rapidly activates the energy sensor AMPK but does not increase phosphorylation of eNOS-Ser1177. Am J Physiol Renal Physiol 289: F1103–F1115, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Nath KA, Norby SM. Reactive oxygen species and acute renal failure. Am J Med 109: 665–678, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Pastor-Soler NM, Hallows KR. AMP-activated protein kinase regulation of kidney tubular transport. Curr Opin Nephrol Hypertens 21: 523–533, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Schley G, Klanke B, Schodel J, Forstreuter F, Shukla D, Kurtz A, Amann K, Wiesener MS, Rosen S, Eckardt KU, Maxwell PH, Willam C. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol 22: 2004–2015, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. J Biol Chem 276: 13057–13064, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Sekine TaE, H. Solute Transport, Energy Consumption, and Production in the Kidney. Philadelphia, PA: Elsevier, 2013. [Google Scholar]
- 39.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J 405: 1–9, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Singh PK, Hollingsworth MA. Cell surface-associated mucins in signal transduction. Trends Cell Biol 16: 467–476, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Spicer AP, Duhig T, Chilton BS, Gendler SJ. Analysis of mammalian MUC1 genes reveals potential functionally important domains. Mamm Genome 6: 885–888, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Sutton TA, Hato T, Mai E, Yoshimoto M, Kuehl S, Anderson M, Mang H, Plotkin Z, Chan RJ, Dagher PC. p53 is renoprotective after ischemic kidney injury by reducing inflammation. J Am Soc Nephrol 24: 113–124, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutton TA, Wilkinson J, Mang HE, Knipe NL, Plotkin Z, Hosein M, Zak K, Wittenborn J, Dagher PC. p53 regulates renal expression of HIF-1α and pVHL under physiological conditions and after ischemia-reperfusion injury. Am J Physiol Renal Physiol 295: F1666–F1677, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Chen JK, Wang SW, Moeckel G, Harris RC. Importance of functional EGF receptors in recovery from acute nephrotoxic injury. J Am Soc Nephrol 14: 3147–3154, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 117: 2847–2859, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang C, Jiang L, Zhang H, Shimoda LA, DeBerardinis RJ, Semenza GL. Analysis of hypoxia-induced metabolic reprogramming. Methods Enzymol 542: 425–455, 2014. [DOI] [PubMed] [Google Scholar]
- 48.Zhou D, Li Y, Lin L, Zhou L, Igarashi P, Liu Y. Tubule-specific ablation of endogenous beta-catenin aggravates acute kidney injury in mice. Kidney Int 82: 537–547, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, Hou FF, Kahn M, Liu Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol 26: 107–120, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]