

Abstract
The role of Toll-like receptor 4 (TLR4) in the regulation of inflammation and fibrosis in sterile wounds was investigated in TLR4 signal-deficient (C3H/HeJ or TLR4−/−) and control mice using the subcutaneously implanted polyvinyl alcohol sponge wound model. Total and differential wound cell counts 1, 3, and 7 days after injury did not differ between C3H/HeJ and C3H/HeOuJ animals. Blood monocytes from both strains expressed CCR2 equally. Day one wounds in C3H/HeJ mice contained fewer Gr-1high wound macrophages, CCL3, and CCL5, and more CCL17 than those in controls. The accumulation of CCL2, CX3CL1, tumor necrosis factor-α, interleukin (IL)-6, IL-10, IL-12, and interferon-γ in wound fluids was not TLR4 dependent. Wound macrophages from C3H/HeJ and C3H/HeOuJ mice expressed CCR4 and CCR5, but not CCR1 or CCR3. Wound macrophage recruitment was not altered in CCR5−/− mice or in C3H/HeOuJ animals injected with neutralizing anti-CCL3 and anti-CCL5 antibodies. Neutralization of the CCR4 ligand CCL17 in C3H/HeJ mice did not alter wound macrophage populations. There was a twofold increase in collagen content and number of neovessels in 21-day-old wounds in C3H/HeJ vs. C3H/HeOuJ mice. There were no differences between strains in the number of myofibroblasts in the wounds 7 or 21 days post-wounding. The increased fibrosis and angiogenesis in wounds from /HeJ mice correlated with higher concentrations of transforming growth factor-β and fibroblast growth factor 2 in wound fluids from these animals. Wound fluids did not contain detectable lipopolysaccharide and did not induce IκBα degradation in J774.A1 macrophages. Results support a role for endogenous ligands of TLR4 in the regulation of inflammation and repair in sterile wounds.
The healing of wounds requires the orderly participation of a variety of cell types derived from the circulation and from tissues local to the wound. Many of the cell types involved in repair markedly alter their naïve phenotype upon arrival at the wound. Examples for phenotypic maturation, differentiation, and polarization in wound-healing cells include the acquisition of a “repair” phenotype by wound macrophages derived from circulating monocytes,1 the transdifferentiation of fibroblasts into myofibroblasts, the activation of vascular endothelia during neovascularization, and the migration and proliferation of keratinocytes. The combined and coordinated activities of these cells are required for the normal healing of wounds and the replacement of injured tissue by scar.
A number of mediators, including growth factors, chemokines, cytokines, lipids, and others, have been shown to modulate the phenotype of cells participating in sterile wound healing. Additional signals conveyed by microorganisms through pathogen-associated molecular patterns (PAMPs) contribute to the phenotypic modulation of wound-healing cells in infected wounds. As a counterpart to the PAMPs present in infected wounds, a variety of molecules arising from injured or necrotic cells have been identified and are grouped under the denomination of endogenous danger-associated molecular patterns (DAMPs) or alarmins.2–6 These endogenous mediators, which in many cases signal through receptors canonically associated with the sensing of microorganisms such as the Toll-like receptors (TLR), have been reported to modulate and modify inflammatory cell function and may, thus, impact wound healing.2,5–9
The role for DAMPs and TLRs in sterile wound healing has, to the best of the authors’ knowledge, only been reported in a publication by Bettinger et al., who found accelerated collagen deposition in wounds inflicted onto mice expressing a mutated, nonsignaling form of TLR4 (strain C3H/HeJ).10 Bettinger et al. concluded that the improvement of repair in these animals stemmed from a marked suppression of the inflammatory response to tissue injury that was manifested by a reduction in inflammatory cell influx and a 60% decrease in the tumor necrosis factor-α (TNF-α) content of early wounds.10
Results reported here failed to confirm Bettinger et al.'s hypothesis that the accelerated repair in TLR4-deficient animals resulted from a reduced inflammatory response to wounding in C3H/HeJ or TLR4 KO mice. Findings, however, showed a more robust fibrotic and angiogenic response in wounds from C3H/HeJ mice that correlated with the increased accumulation of transforming growth factor-β (TGF-β) and fibroblast growth factor 2 (FGF2) in wound fluids from these animals.
MATERIALS AND METHODS
Animals
C3H/HeJ (henceforth /HeJ, containing a point mutation in the Tlr4 gene that interferes with TLR4 signaling11), C3H/HeOuJ (henceforth /HeOuJ, with intact TLR4 signaling), C57BL/ 10ScNJ (TLR4−/−, containing a deletion in the Tlr4 gene11), and C57BL/6J male mice were obtained from Jackson Labs (Bar Harbor, ME). B6D2F1, B6.129P2-Ccr5tm1KuzN10, and C57BL/6 male mice were purchased from Taconic Farms (Germantown, NY). Animals were housed at the Central Research Facilities at Rhode Island Hospital, fed mouse chow and water ad libitum, and were studied at 8–12 weeks of age. Mice were certified free of common pathogens by the suppliers and were monitored by Brown University/Rhode Island Hospital veterinary personnel. The Institutional Animal Care and Use Committee at Rhode Island Hospital approved animal protocols.
Polyvinyl alcohol sponge wound model
Mice were anesthetized with pentobarbital (50 mg/kg i.p.). Two to six polyvinyl alcohol (PVA) sponges (PVA Unlimited, Warsaw, IN) were inserted into individual subcutaneous pockets through a midline dorsal incision under aseptic conditions, and the skin closed with clips. Mice were euthanized by CO2 asphyxiation, sponges removed, and wound cells and cell-free wound fluids obtained at the indicated times.12 Wound cell counts were determined with a hemocytometer. Differential wound cell counts were determined by microscopic examination of stained cytocentrifuge preparations. Blood leukocytes were obtained from cardiac blood as published.13
Flow cytometry
Cells were stained using fluorochrome conjugated antibodies specific for C-C motif chemokine receptor 1 (CCR1) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), CCR2, CCR3 (R&D Systems, Minneapolis, MN), CCR4 (Biolegend, San Diego, CA), CCR5, TLR4 (eBioscience, Inc., San Diego, CA), CD68, F4/80 (AbD Serotec, Raleigh, NC), Gr-1 (BD Biosciences, San Diego, CA), or α-smooth muscle actin (Abcam, Cambridge, MA). Cells were interrogated using a Becton-Dickinson FACSort or FACSAria and analyzed using FlowJo software (TreeStar, Ashland, OR). Macrophages in wound cell suspensions were identified by forward and side scatter (FSC/SSC) properties and either surface F4/80 or intracellular CD68 staining. The gating strategies used to identify wound macrophages were as follows. Forward and side scatter were used to detect viable cells (generally >85% of all cells) and exclude red blood cells, cell doublets, and cellular debris. Neutrophils (SSChighF4/80-) and eosinophils (SSChighF4/80+) were excluded from analysis. Macrophages were identified as SSClowF4/80+ cells. When using intracellular CD68 to identify wound macrophages (SSClowCD68high), neutrophils (SSChighCD68low) and eosinophils (CD68-) were excluded from analysis. The purity of the isolated macrophage preparations was verified by morphology using Cytospin (Thermo, Astmoor, United Kingdom) preparations. Both staining protocols (SSC-F4/80 vs. SSC-intracellular CD68) identify overlapping populations of wound macrophages. Blood monocytes were identified in buffy coat suspensions by FSC/SSC properties and F4/80 staining.
Neutralization of wound fluid chemokines
Systemic neutralization of C-C motif chemokine ligand 3 (CCL3) and CCL5 in /HeOuJ mice was accomplished by i.p. injection of 30 μg of neutralizing antibodies (R&D Systems) 1 hour before and 12 hours after wounding. CCL17 was neutralized in /HeJ mice in a similar fashion using a neutralizing polyclonal antibody (R&D Systems). Control animals received injections of isotype control antibodies or goat IgG. The complete neutralization of chemokines was confirmed by enzyme-linked immunosorbent assay (ELISA) analysis of the individual chemokines in wound fluids of antibody-treated mice.
Cells and cell culture
The murine macrophage cell line J774A.1 was obtained from ATCC (Rockville, MD). Cells were plated in Dulbecco's modified Eagle medium (Invitrogen, Grand Island, NY) supplemented with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Sigma-Aldrich, St. Louis, MO), 100 U/mL penicillin-streptomycin, and 1% fetal bovine serum (Hyclone, Logan, UT). Cells were treated with lipopolysaccharide (LPS) (Escherichia coli serotype 055:B5, Sigma-Aldrich, 100 ng/mL) with or without polymyxin B (Sigma-Aldrich, 50 ng/mL), day 1 or day 7 wound fluids, or normal mouse serum.
Western blot
Cell lysates were size fractionated by SDS-polyacrylamide gel electrophoresis, and immunoblotted with antibodies specific for IκBα (Santa Cruz Biotechnology) or inducible nitric oxide synthase (BD Biosciences).
Assays
Cytokines were measured by ELISA (CCL2, CCL3, CCL5, CCL17, CCL22, C-X3-C motif chemokine ligand 1 [CX3CL1], TGF-β, FGF2 [basic fibroblast growth factor], and vascular endothelial growth factor [VEGF] [the assay detects VEGF120 and VEGF164] [R&D Systems]), or by Cyto-metric Bead Array (TNF-α, interleukin 6 [IL-6], IL-10, IL-12, and interferon-γ [IFN-γ] [BD Biosciences]). LPS was assayed by the chromogenic limulus amebocyte lysate assay (Lonza, Hopkington, MA).
Computerized histomorphometry
Two sponges were inserted as just described and excised 21 days later, along with overlying skin and surrounding tissue. The sponges were fixed in formalin and embedded in paraffin. Sections from the center of each sponge were stained with Gomori's Trichrome, anti-CD31 antibody (Abcam), or anti-α-smooth muscle actin antibody (Abcam). Irrelevant antibodies were used for negative control stains. Images were scanned using an automated Aperio ScanScope (Aperio Technologies, Vista, CA) at 200× magnification. Images were analyzed using Image-Pro Plus7 (Media-Cybernetics, Bethesda, MD). The area of fibrosis for each sponge was determined by dividing the total area of mature collagen (trichrome positive tissue) that penetrated each sponge by the total area of the sponge. The depth of collagen penetration was determined by measuring the distance from the edge of the sponge to the deepest point of collagen penetration, divided by the maximal thickness of the sponge. The four most vascular and myofibroblast-rich fields from each sponge were used for the quantitation of microvessel density and myofibroblast enumeration. The threshold of hue, saturation and intensity of the immunohistochemically stained objects of interest (endothelium, myofibroblasts) was established, followed by a semiautomatic segmentation to measure the relative percent area of the microvessels and to count the number of myofibroblasts per microscopic field. Data from each of the two sponges were averaged to produce a single data point for each animal.
Statistical analysis
The number of animals used in each experiment is indicated in the text or in the legends to figures or tables. Results shown are from a representative of two to six independent experimental repeats, except for the examination of the accumulation of TGF-β, FGF2, and VEGF in wound fluids over time that was performed once. Data shown are means ± standard deviations (SD). Statistical analysis was performed using Mann–Whitney's U-test in the case of comparisons between two strains at one time point, or ANOVA/Newman–Keuls for comparisons between two strains across multiple time points. The value of alpha was established at p < 0.05.
RESULTS
TLR4 is expressed on wound macrophages
The expression of TLR4 on SSClow F4/80+ wound macrophages from wild type mice was examined by flow cytometry. TLR4 was expressed on wound macrophages at 1, 3, and 7 days after injury (Figure 1).
Figure 1.

Wound macrophages express Toll-like receptor 4 (TLR4). Wound cells were isolated from B6D2F1 mice at 1, 3, and 7 days after sponge insertion. Open histograms show staining for TLR4 on macrophages (SSClow F4/80+); filled histograms show staining with isotype control antibody.
Effects of TLR4 on the recruitment of blood monocytes to sterile wounds
It has been proposed that the two major circulating murine monocyte populations distinguished by the relative expression of the surface marker Gr-1 are sequentially recruited into sites of inflammation, with cells expressing high levels of Gr-1 arriving earlier than those with lesser levels of marker expression. Gr-1high macrophages exhibit phenotypic traits associated with classical activation, while Gr-1low cells present features of alternative activation and are thought to be responsible for wound repair.1,14 This pattern of cellular recruitment in the subcutaneous sponge wound model has been previously reported by this laboratory.13
The level of Gr-1 expression by macrophages present in day 1 wounds in /HeJ mice differed from those of /HeOuJ mice. Figure 2A and B show that fewer macrophages in day 1 wounds from /HeJ mice expressed the Gr-1high phenotype than in /HeOuJ mice. The wound macrophage expression of Gr-1 did not differ between /HeJ vs. /HeOuJ mice at later times. The lower number of Gr-1high wound macrophages in /HeJ mice did not result from a reduced frequency of Gr-1high blood monocytes in these animals, because Gr-1 expression in blood monocytes did not differ between /HeOuJ and /HeJ naïve mice (Figure 2C).
Figure 2.

Gr-1 staining in wound macrophages and blood monocytes from /HeOuJ and /HeJ mice. A. Wound cells were isolated from /HeOuJ and /HeJ mice at 1, 3, and 7 days after sponge insertion. Open histograms show Gr-1 staining in macrophages (SSClow intracellular CD68high); filled histograms show staining with isotype control antibody. B. Gr-1 staining in day 1 wound macrophages, mean of three experiments, including at least six mice per strain, *p < 0.05 vs. /HeOuJ, Mann–Whitney's U-test. C. Gr-1 staining in blood monocytes from nonwounded mice. Graphs show the values of the means and SD n = 5 each group.
Despite the difference in Gr-1 expression in macrophages from /HeJ and /HeOuJ mice, no differences in total and differential wound cell counts between strains were found at 1, 3, or 7 days after injury (Table 1). Experiments using TLR4−/− mice confirmed these observations. Day 1 wounds from C57BL/6 control animals contained 1.4 ± 0.8 × 106 inflammatory cells and those from TLR4−/− contained 1.4 ± 0.6 × 106 inflammatory cells (p > 0.05).
Table 1.
Wound cell counts from /HeOuJ and /HeJ mice
| Postwound day | Strain | Wound cells (×106/mouse) | Neutrophils (%) | Macrophages (%) | Lymphocytes (%) | Eosinophils (%) |
|---|---|---|---|---|---|---|
| 1 | /HeOuJ | 1.5 ± 0.5 | 87 ± 3 | 12 ± 3 | 0.1 ± 0.1 | 0.4 ± 0.5 |
| /HeJ | 1.2 ± 0.4 | 88 ± 2 | 12 ± 2 | 0.1 ± 0.1 | 0.7 ± 0.7 | |
| 3 | /HeOuJ | 5.9 ± 2 | 33 ± 7 | 61 ± 6 | 2.1 ± 1.4 | 2.7 ± 1.3 |
| /HeJ | 5.9 ± 2 | 42 ± 18 | 52 ± 15 | 3.1 ± 1.4 | 2.1 ± 2.2 | |
| 7 | /HeOuJ | 8.3 ± 2.5 | 23 ± 7 | 76 ± 7 | 0.9 ± 0.5 | 0.1 ± 0.1 |
| /HeJ | 10.5 ± 3.0 | 28 ± 6 | 71 ± 6 | 0.7 ± 0.2 | 0.1 ± 0.1 |
Wound cells were recovered from C3H/HeOuJ and C3H/HeJ mice 1, 3, or 7 days after wounding and counted as described in Materials and Methods. There were no differences between strains in total or differential cell counts at any time point. n ≥ 4 animals per strain and postwound day.
TLR4 and the regulation of the wound microenvironment
The recruitment of Gr-1high and Gr-1low monocytes into inflammatory sites is thought to be dependent on the chemokine receptors CCR2 and C-X3-C motif chemokine receptor 1 (CX3CR1), respectively.14 Day 1 wound fluids were assayed for the chemokines that bind to these receptors. CCL2 concentrations in these fluids did not differ between /HeOuJ and /HeJ mice (/HeOuJ 41 ± 10 ng/mL vs. /HeJ 33 ± 10 ng/mL, p > 0.05, n = 6 animals per strain). Moreover, blood monocytes from the two mouse strains equally expressed CCR2 (Figure 3A and B), the receptor thought to be essential for recruitment of Gr-1high monocytes into inflammatory sites.14 There were no differences in wound fluid CX3CL1 content between the strains (/HeOuJ = 0.5 ± 0.1 vs. /HeJ = 0.5 ± 0.1 ng/mL, p > 0.05, n = 6 animals per strain).
Figure 3.

CCR2 staining on blood monocytes from /HeOuJ and /HeJ mice. A. Blood leukocytes were isolated from /HeOuJ and /HeJ mice 1 day after sponge insertion. The figure shows Gr-1 and CCR2 staining on gated blood monocytes (SSClow F4/80+). B. Median channel fluorescence of CCR2 stained blood monocytes, according to Gr-1 expression. Graphs show the values of the means and SD n > 5 animals per group.
The concentrations of additional chemokines potentially related to the recruitment of monocytes into the wound were also determined. Those chemokines chosen for analysis were selected because they reflect the polarization of macrophages toward the classical activated state (CCL3 and CCL5) or the alternative activation phenotype (CCL17).15 Substantial differences in the wound fluid concentrations of all three chemokines were observed between /HeOuJ vs. /HeJ mice at 1 day after wounding (Figure 4). Wound fluids from /HeJ mice contained three times less CCL3 and nine times less CCL5 than those in /HeOuJ mice (CCL3: /HeOuJ 507 ± 131 vs. /HeJ 176 ± 21 pg/mL, p < 0.05; CCL5: HeOuJ 632 ± 140 vs. /HeJ 68 ± 9 pg/mL, p < 0.05). In contrast, the concentration of CCL17 in wound fluids from /HeJ mice was three times greater than that in fluids from /HeOuJ animals (/HeOuJ 477 ± 132 vs. /HeJ 1311 ± 264 pg/mL, p < 0.05). Confirming this pattern, wound fluids from mice with a TLR4 deletion (TLR4−/−) also showed lower concentrations of CCL3 and CCL5 and greater concentrations of CCL17, as compared with C57BL/6 controls (CCL3: C57BL/6 794 ± 233 vs. TLR4−/− 203 ± 86 pg/mL, p < 0.05; CCL5: C57BL/6 512 ± 157 vs. TLR4−/− 81 ± 10 pg/mL, p < 0.05; CCL17: C57BL/6 2.1 ± 0.6 vs. TLR4−/− 3.5 ± 0.4 ng/mL, p < 0.05, n = 6 per strain). Supporting the concept that wound inflammatory cells are the source of these chemokines, at least for CCL5, day 1 wound cell lysates from /HeOuJ mice contained more CCL5 than those from /HeJ animals (/HeOuJ 348 ± 111 vs. /HeJ 125 ± 63 pg/106 cells, p < 0.05, n = 4 per strain).
Figure 4.
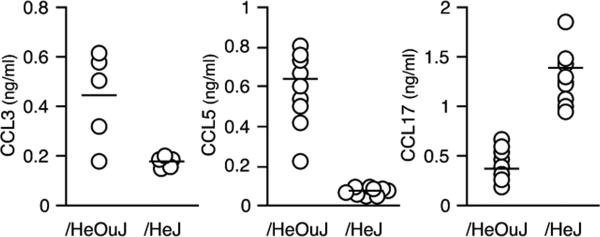
Chemokines in wound fluids from /HeOuJ and /HeJ mice. Wound fluids were obtained 1 day after sponge insertion and assayed for CCL3, CCL5, and CCL17 by ELISA. p < 0.05 between strains for each chemokine, Mann–Whitney's U-test. Open circles show values from individual animals, and the group means are represented with a horizontal bar, n > 5 per strain.
The differences in wound fluid concentrations of CCL3, CCL5, and CCL17 between /HeOuJ and /HeJ mice were restricted to the first postwounding day. Fluids obtained 3 and 7 days after injury exhibited no differences in the concentrations of these chemokines between the two animal strains (not shown).
CCL3, CCL5, and CCL17 were also measured in the plasma of /HeOuJ and /HeJ animals 1 day after wounding. No differences were found between strains in the plasma concentrations of CCL3 (not detected) or CCL5 (/HeOuJ 226 ± 30 pg/mL vs. /HeJ 206 ± 11 pg/mL, p > 0.05, n = 5 per strain). Plasma from /HeJ mice contained more CCL17 than those from /HeOuJ animals (/HeOuJ 70 ± 12 pg/mL vs. /HeJ 98 ± 10 pg/mL, p < 0.05, n = 5 per strain).
Contrasting with the substantial differences in chemokine contents of wound fluids from /HeJ and /HeOuJ mice, the concentrations of TNF-α, IL-6, IL-10, IL-12, and IFN-γ in postinjury day 1 wound fluids were the same in both strains (Table 2).
Table 2.
Wound fluid cytokines from /HeOuJ and /HeJ mice
| /HeOuJ | /HeJ | |
|---|---|---|
| TNF-α (pg/mL) | 598 ± 105 | 478 ± 94 |
| IL-6 (ng/mL) | 15 ± 6 | 15 ± 5 |
| IL-10 (pg/mL) | 429 ± 73 | 401 ± 134 |
| IL-12 (pg/mL) | 198 ± 34 | 207 ± 9 |
| IFN-γ (pg/mL) | 24 ± 7 | 16 ± 2 |
Wound fluids were collected from 1-day-old wounds in /HeOuJ and /HeJ mice, as described in Materials and Methods and cytokine concentrations determined by ELISA. n ≥ 6 animals per strain. Data are means ± SD. There were no differences between groups for any of the measured cytokines (Mann–Whitney U).
ELISA, enzyme-linked immunosorbent assay; IFN-γ; interferon-γ; IL, interleukin; TNF-α; tumor necrosis factor-α
Chemokine receptor expression in wound macrophages from /HeOuJ and /HeJ mice
Chemokines CCL3 and CCL5 signal mainly through CCR1, 3, and 5; whereas CCL17 selectively binds to CCR4.16–18 (CCL22, a CCR4 ligand, was found in similar concentrations in wound fluids from /HeOuJ [1.2 ± 0.2 ng/mL] and /HeJ mice [1.0 ± 0.2 ng/mL, p > 0.05, n = 6 per strain].) Given the difference in chemokine concentrations in wound fluids from /HeOuJ and /HeJ mice, the expression of the relevant chemokine receptors in blood monocytes and wound macrophages was examined by flow cytometry (Figure 5). CCR1 was not found in blood monocytes (not shown) or wound macrophages at 1, 3, and 7 days after wounding (Figure 5). Wound macrophages (Figure 5), but not blood monocytes (not shown), expressed CCR4, suggesting that this receptor is upregulated on entry into the wound. CCR5 was abundantly expressed on wound macrophages (Figure 5) and blood monocytes (not shown). For all receptors, no differences were detected between /HeOuJ and/HeJ mice. CCR3 was expressed exclusively in wound eosinophils (not shown).
Figure 5.

Chemokine receptors in wound macrophages from /HeOuJ and /HeJ mice. Wound cells were isolated from /HeOuJ and /HeJ mice at 1, 3, and 7 days after sponge insertion. Open histograms show chemokine receptor staining on wound macrophages (SSClow intracellular CD68high); filled histograms show staining with isotype control antibody.
The neutralization of CCL3, CCL5, or CCL17 does not alter the phenotype of early wound macrophages
The relevance of the altered chemokine composition of wound fluids in /HeJ mice to the recruitment of Gr-1low and Gr-1high monocytes into wounds was examined through the in vivo administration of neutralizing antibodies specific for these chemokines. Simultaneous neutralization of CCL3 and CCL5 using specific antibodies administered 1 hour prior to and 12 hours after wounding in /HeOuJ mice did not alter the frequency of Gr-1high vs. Gr-1low wound macrophages at 1 day after wounding (not shown). Similarly, the administration of anti-CCL17 antibody prior to and after wounding did not affect Gr-1 expression in day 1 wounds of /HeJ mice (not shown).
Given that the differences in chemokine concentrations between /HeOuJ and /HeJ mice was most marked for CCL5, additional experiments examined postwound day 1 macrophages from wild-type and CCR5 knockout mice. No differences were found in either the number or phenotype of macrophages isolated from day 1 wounds in either mouse strain (not shown).
Increased wound fibrosis and neovascularization in /HeJ mice
Sponges removed 21 days after wounding were prepared for computerized histomorphometry. Morphometric analysis of trichrome stained sections showed an increase in collagen deposition in /HeJ mice, as compared with /HeOuJ mice (Figure 6). Both the total area of collagen deposition and the maximum depth of collagen penetration into the sponge were greater in /HeJ mice than in /HeOuJ mice (p < 0.05 for either measurement). Wounds in /HeJ mice also contained a higher number of neovessels (p < 0.05) (Figure 6). No difference in the number of myofibroblasts in the wounds between strains were found by histomorphometry at day 21 postwounding (Figure 6), or by flow cytometric analysis of wound cells after α-smooth muscle actin staining on days 7 or 21 after wounding (not shown).
Figure 6.

Increased fibrosis and neovascularization in wounds in TLR4 deficient mice. Histomorphometric analysis of sponges removed 21 days after insertion in /HeOuJ or /HeJ mice. Graphs show area of fibrosis, maximum depth of fibrosis, area of the wound invaded by neovessels, and the number of myofibroblasts in the wound for each animal. Open circles show values from individual animals, and the group means are represented with a horizontal bar. p < 0.05 between strains, except for the number of myofibroblasts (P > 0.05), Mann–Whitney's U-test, n = 5 mice per group.
Fibrogenic/angiogenic growth factors in wound fluids from /HeOuJ and /HeJ mice
Given the differences in fibrosis and neovascularization between wounds from /HeOuJ and /HeJ mice, the potential correlation between these differences and the accumulation of fibrogenic/angiogenic growth factors in wound fluids from animals of both strains was examined in wound fluids harvested 1, 7, and 21 days following wounding. Results shown in Table 3 show that, congruent with the histomorphometric data, fluids from 21-day-old wounds in /Hej mice contained more TGF-β and FGF2 than those from /HeOuJ. The concentration of VEGF in wound fluids did not differ been strains at any time point.
Table 3.
Fibrogenic/angiogenic growth factors in wound fluids from /HeOuJ and /HeJ mice
| Postwound day | Strain | TGF-β (ng/mL) | FGF2 (pg/mL) | VEGF (pg/mL) |
|---|---|---|---|---|
| 1 | /HeOuJ | 1.2 ± 0.2 | 129 ± 10 | 231 ± 16 |
| /HeJ | 1.2 ± 0.2 | 114 ± 20 | 234 ± 31 | |
| 7 | /HeOuJ | 2.9 ± 0.9b | 48 ± 6 | 473 ± 133b |
| /HeJ | 2.6 ± 0.3b | 49 ± 7 | 610 ± 153b | |
| 21 | /HeOuJ | 5.1 ± 0.7b,c | 140 ± 48 | 600 ± 176b,c |
| /HeJ | 6.5 ± 1.2a,b,c | 411 ± 251a,b,c | 499 ± 179b,c |
Wound fluids collected from /HeOuJ or /HeJ mice (six animals per strain and time point) were analyzed for TGF-β, FGF2, and VEGF by ELISA. Data are means ± SD. Results were analyzed by two-way ANOVA/Newman–Keuls. For each assay
indicates p < 0.05 /HeOuJ vs. /HeJ for the indicated postwound day. Within each strain and assay
indicates p < 0.05 vs. postwound day 1
indicates p < 0.05 vs. postwound day 7.
ANOVA, analysis of variance; ELISA, enzyme-linked immunosorbent assay; FGF2, fibroblast growth factor 2; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor.
Wound fluids do not contain soluble TLR4 agonists
The presence of bacterial LPS in wound fluids was examined using the chromogenic limulus amebocyte lysate assay. Results in wound fluid samples obtained 1 or 7 days after wounding were below the limit of detection of the assay. Recovery of LPS added to wound fluid samples did not detect inhibition or enhancement of test results by the wound fluids (not shown).
To examine further the potential accumulation of soluble TLR4 agonists in wound fluids, J774.A1 murine macrophages were exposed to wound fluids from /HeOuJ and /HeJ wounds and examined for degradation of IκBα. As shown in Figure 7, wound fluids did not by themselves induce IκBα degradation (Figure 7A), nor did they interfere with or potentiate LPS-dependent NFκB activation (Figure 7B). Lastly, inducible nitric oxide synthase (iNOS) protein was not detected in wound macrophages from either /HeOuJ or /HeJ mice (not shown).
Figure 7.

Wound fluids do not induce IκBα degradation or interfere with NFκB activation by lipopolysaccharide (LPS). A. Murine J774.A macrophages were untreated (Control: C) or exposed to LPS (100 ng/mL) ± polymyxin B (PM, 50 ng/mL), or day 1 (WF1) or day 7 (WF7) wound fluids (50%, v : v) for 30 minutes in culture medium containing 1% FBS. Cell lysates were immunoblotted for IκBα. B. Cells were cultured with day 1 wound fluids (WF1) or normal mouse serum (MS) at 50% (v : v), with or without LPS (100 ng/mL), as described under (A) and assayed for IκBα content. Intervening noncontributory lanes were removed from the image for clarity.
DISCUSSION
The Toll-like receptors were originally characterized as sensors for PAMPs. TLR4, in particular, has been identified as the receptor for bacterial LPS. Beyond their role in detecting exogenous agonists, the TLRs have also been implicated in the recognition of a variety of endogenous danger signal molecules that are produced or released in areas of tissue damage.2,5,6,9,19 The only published report addressing wound healing in mice deficient in TLR4 signaling found a substantial reduction in inflammatory response to the wound and accelerated collagen deposition.10 Given present understanding of the regulation of cellular infiltration into wounds, primarily of monocytes/macrophages, present studies initially tested the hypothesis that intact signaling through TLR4 is necessary for orderly recruitment of monocytes into the wound and the establishment of a normal inflammatory response to sterile tissue injury.
In this connection, work from this and other laboratories has begun to elucidate the mechanisms for the recruitment of different blood monocyte populations into wounds and the polarization of these cells into distinct wound macrophage types. Two distinct populations of monocytes have been identified in mice, and are distinguished by the differential expression of surface markers and distinct kinetics of recruitment into sites of sterile inflammation. Monocytes expressing high levels of antigen Gr-1 (Ly6C/G) also express CCR2, migrate into wounds early after injury, and present an inflammatory phenotype defined by the preferential production of TNF-α and other pro-inflammatory cytokines. A second population of monocytes is characterized by low level expression of Gr-1. These cells are also characterized by expressing chemokine receptor CX3CR1, and migrate into inflammatory sites later than their Gr-1high counterparts. It has been postulated that Gr-1low wound macrophages contribute to repair through the production of fibrogenic and angiogenic growth factors.1,14
Examination of the inflammatory cellular infiltrate in wounds from TLR4 signal-deficient mice (/HeJ) and controls (/HeOuJ) showed decreased numbers of Gr-1high macrophages in day one wounds from /HeJ mice, without alterations in the total number of inflammatory cells or their differential counts. Findings were confirmed using TLR4−/− mice. The decreased number of Gr-1high macrophages in the wounds of TLR4 signal-deficient mice could not be explained by changes in the number of Gr-1high blood monocytes or by differences in CCR2 expression in circulating monocytes. Furthermore, changes in wound macrophage subpopulations in TLR4 signal-deficient mice did not result from alterations in chemokines thought to regulate the recruitment of Gr-1high or Gr-1low monocytes into the wound (CCL2 and CX3CL1, respectively).
Further evaluation of the chemokine profile of wound fluids from /HeOuJ and /HeJ mice revealed substantial differences in the expression of CCL3, CCL5, and CCL17. Indeed the expression of CCL5 was found to be almost completely dependent on TLR4 function.
The potential roles of those chemokines found to differ in /HeOuJ and /HeJ mice in determining the infiltration of Gr-1low or Gr-1high monocytes into the wound were examined. Neither the genetic abrogation (CCR5) nor the neutralization of these chemokines (CCL3, CCL5, CCL17) altered the recruitment of monocytes to the wound. Moreover, the expression of the canonical receptors for these chemokines did not differ between the strains that were studied. These results suggest that a redundant signaling system is required for the attraction of circulating monocytes to a site of tissue injury so that single, or even multiple, chemokine ablations, or substantial variations in specific chemokine concentrations in wounds, are unable to significantly impact on this phenomenon. It is possible, however, that additional factors critical to the recruitment of inflammatory cells into wounds were not examined or identified in the present experiments.
Present observations do not confirm that either the recruitment of inflammatory cells (Table 1) or the accumulation of TNF-α (Table 2) in wounds is TLR4 dependent, as was originally proposed by Bettinger et al.10 Further illustrating the specificity of TLR4-dependent events in acute sterile inflammation, no differences were found in the concentrations of IL-6, IL-10, IL-12, or IFN-γ in wound fluids from /HeOuJ and /HeJ mice.
A previous report on the development of fibrosis in sterile wounds showed that the deposition of collagen was accelerated in wounds in /HeJ mice.10 Experiments reported here examined the deposition of collagen and neovascularization in wound sponges 21 days after injury. At that time, the skin incision used for sponge insertion is healed, and the inflammatory response to the foreign body represented by the sponge is transitioning toward its granulomatous incorporation. Findings were of marked differences in fibrotic response to the implanted sponge, with /HeJ mice showing a twofold increase in the area of the sponge invaded by collagen, and the maximal penetration of fibrosis into the sponge matrix. Moreover, the higher collagen content of wounds of /HeJ coincided with a doubling of the number of neovessels invading the sponge. There were no differences in the number of myofibroblasts detected by histomorphometry 21 days after wounding, or by flow cytometry 7 and 21 days after injury, in the wounds of animals of either strain. These results indicate that signals conveyed through TLR4 down-regulate the fibrotic response to sterile injury, without altering the number of myofibroblasts present in the wound.
The higher collagen content and neovascularization of sponges in /HeJ mice correlated with increased accumulation of the fibrogenic and angiogenic growth factors TGF-β and FGF2 in the wounds, and are consistent with findings in other models of sterile tissue injury and fibrosis in animals with deficient or defective TLR4. Lack of TLR4 signaling has been shown to be associated with increased fibrosis in a model of bleomycin-induced pulmonary injury20 and in experimental myocardial infarctions.21 Moreover, the development of tensile strength in skin wounds was reported to be accelerated in /HeJ mice.10 As to a mechanistic correlation between TLR4 deficiency and increased TGF-β expression, it was most recently reported in a murine model of sterile arthritis that inflamed joints in TLR4−/− mice contain more TGF-β transcripts than those of control.22 The authors proposed that IL-12 and IFN-γ released under the influence of TLR4 agonists suppressed TGF-γ transcription. The concentrations of IL-12 or IFN-γ in early wound fluids from /HeOuJ or /HeJ mice did not differ in the present experiments. Further investigation is needed to determine whether the kinetics of IL-12 and IFN-γ production in wounds differ from those in arthritis, or whether another mechanism is responsible for the higher TGF-β content of wounds in /HeJ mice. No information regarding the regulation of FGF2 by TLR4 agonists was found in the literature.
Present findings of an intact inflammatory response and increased collagen deposition in sterile wounds in TLR4 signal-deficient mice add to literature reporting alterations in wound healing mediated by other TLRs. In this regard, it was shown that TLR2 activation is associated with a prolonged inflammatory phase in diabetic wounds, and that open skin wounds in TLR2 KO diabetic, but not in TLR2 KO nondiabetic mice, heal faster than those in wild-type mice.23 It was also reported that TLR9 activation by CpG oligodeoxynucleotides accelerated wound healing in normal mice, and TLR9 KO animals exhibit a delay in skin repair.24 Lastly, a substantial delay in the healing of open cutaneous wounds was found in TLR3 KO mice.25
The aforementioned studies of wound healing in TLR2, TLR9, and TLR3 KO mice utilized models of open cutaneous wounds rather than closed, sterile wounds such as those used in the present studies. Work from this laboratory conclusively showed not only that open skin wounds in mice are colonized by bacteria, but also that the presence of bacteria in wounds markedly alters the phenotype of wound repair cells.26 Thus, studies of open wounds in various TLR-deficient animals that were just discussed implicate PAMPs, but not necessarily DAMPs, in the regulation of wound healing.
Given that LPS contamination of the wounds would critically bias results through the activation of TLR4 in LPS-responsive animals, the presence of bacteria or LPS in the wounds was exhaustively investigated and excluded in these experiments. Previous work from this laboratory showed that the expression of iNOS in murine wound cells is a sensitive indicator of bacterial colonization of wounds.26 iNOS was not detected in the wounds of /HeOuJ or /HeJ mice in the present study (data not shown). Further evidence for the sterility and lack of bacteria-derived TLR4 agonists in the wounds under study is given by negative bacteriologic culture of sponges upon retrieval from the animals, and the lack of detectable LPS in wound fluids. In addition, wound fluids did not induce IκBα degradation or modulate LPS-induced activation of NFκB in J774.A1 macrophages. In toto, then, evidence argues against the possibility that LPS contamination of the wounds or the presence of bacteria is responsible for the observed differences between wounds in control and TLR4 defective/deficient mice.
The absence of soluble TLR4 agonists in wound fluids raises the question as to the nature of the TLR4 stimulus or stimuli responsible for the differences found between wounds in /HeOuJ and /HeJ mice. Results are compatible with the hypothesis that TLR4 effects on wound macrophages are derived from signals presented in the context of extracellular matrix. This proposal is supported by reports that DAMPs, including matrix components (such as hyaluronan fragments, biglycan, fibronectin, etc.)6,27–29 and compounds bound to matrix (such as high-mobility group protein B1),30 can provide proinflammatory signals through TLR4 and other receptors.
In conclusion, results show that TLR4 is required for the orderly recruitment of a “normal” complement of monocytes into early sterile wounds, and that TLR4 regulates chemokine expression in early wounds. Findings also give evidence for the remarkable redundancy and robustness of the mechanisms regulating the selective migration of circulating cells into wounds. This is so because neither substantial differences in the concentrations of key chemokines in wounds from TLR4 competent and incompetent mice, nor the immune neutralization of selective chemokines, altered the number of inflammatory cells in early wounds. Additionally, data show that signals conveyed through TLR4 suppress the accumulation of collagen and neovascularization of sterile wounds, a response that may be mechanistically related to the lower concentrations of TGF-β and FGF2 in wounds in /HeOuJ vs. /HeJ mice.
Present work focused exclusively on the role of TLR4 signaling in sterile wound repair. It is apparent that DAMPs can convey signals through other receptors. Some of these receptors are expressed in all wound cells in mice (e.g., TLR2, unpublished observation), while others (e.g., receptors for advanced glycosylation products) are not detectable in any murine wound cell (unpublished observation). The identity and potential role of additional DAMP receptors in the healing of sterile wounds remains to be investigated.
ACKNOWLEDGMENTS
The authors thank William Henry and Sandra Augusto for technical support, and Patty Young for editorial assistance.
Source of Funding: Present work was supported by T32-GM-065085 (SKB), the Armand Versaci Research Scholar in Surgical Sciences Award (AAT), NIH grant RO1-GM 042859 (JEA), NIH grant RO1-GM066194 (JSR), KO8-GM079227 (JMD), and by funds allocated to the Department of Surgery by Rhode Island Hospital/Lifespan.
Footnotes
Conflict of Interest: The authors have no conflicts of interest to reveal.
REFERENCES
- 1.Brancato SK, Albina JE. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am J Pathol. 2011;178:19–25. doi: 10.1016/j.ajpath.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Grote K, Schutt H, Schieffer B. Toll-like receptors in angiogenesis. Scientific World Journal. 2011;11:981–91. doi: 10.1100/tsw.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11:91–9. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 5.Sabroe I, Parker LC, Dower SK, Whyte MK. The role of TLR activation in inflammation. J Pathol. 2008;214:126–35. doi: 10.1002/path.2264. [DOI] [PubMed] [Google Scholar]
- 6.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010:1–21. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–7. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- 8.Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta. 2012;1832:1049–60. doi: 10.1016/j.bbadis.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Q, Li M, Fang D, Fang J, Su SB. The essential roles of Toll-like receptor signaling pathways in sterile inflammatory diseases. Int Immunopharmacol. 2011;11:1422–32. doi: 10.1016/j.intimp.2011.04.026. [DOI] [PubMed] [Google Scholar]
- 10.Bettinger DA, Pellicane JV, Tarry WC, Yager DR, Diegelmann RF, Lee R, et al. The role of inflammatory cytokines in wound healing: accelerated healing in endotoxin-resistant mice. J Trauma. 1994;36:810–3. doi: 10.1097/00005373-199406000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 12.Daley JM, Reichner JS, Mahoney EJ, Manfield L, Henry WL, Jr, Mastrofrancesco B, et al. Modulation of macrophage phenotype by soluble product(s) released from neutrophils. J Immunol. 2005;174:2265–72. doi: 10.4049/jimmunol.174.4.2265. [DOI] [PubMed] [Google Scholar]
- 13.Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 16.Mueller A, Strange PG. The chemokine receptor, CCR5. Int J Biochem Cell Biol. 2004;36:35–8. doi: 10.1016/s1357-2725(03)00172-9. [DOI] [PubMed] [Google Scholar]
- 17.Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem. 1997;272:15036–42. doi: 10.1074/jbc.272.23.15036. [DOI] [PubMed] [Google Scholar]
- 18.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Lin Q, Yang XP, Fang D, Ren X, Zhou H, Fang J, et al. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1024–32. doi: 10.1161/ATVBAHA.111.224048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180:275–92. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–64. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 22.Kim HS, Chung DH. TLR4-mediated IL-12 production enhances IFN-gamma and IL-1beta production, which inhibits TGF-beta production and promotes antibody-induced joint inflammation. Arthritis Res Ther. 2012;14:R210. doi: 10.1186/ar4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dasu MR, Thangappan RK, Bourgette A, DiPietro LA, Isseroff R, Jialal I. TLR2 expression and signaling-dependent inflammation impair wound healing in diabetic mice. Lab Invest. 2010;90:1628–36. doi: 10.1038/labinvest.2010.158. [DOI] [PubMed] [Google Scholar]
- 24.Sato T, Yamamoto M, Shimosato T, Klinman DM. Accelerated wound healing mediated by activation of Toll-like receptor 9. Wound Repair Regen. 2010;18:586–93. doi: 10.1111/j.1524-475X.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Q, Fang D, Fang J, Ren X, Yang X, Wen F, et al. Impaired wound healing with defective expression of chemokines and recruitment of myeloid cells in TLR3-deficient mice. J Immunol. 2011;186:3710–7. doi: 10.4049/jimmunol.1003007. [DOI] [PubMed] [Google Scholar]
- 26.Mahoney E, Reichner J, Bostom LR, Mastrofrancesco B, Henry W, Albina J. Bacterial colonization and the expression of inducible nitric oxide synthase in murine wounds. Am J Pathol. 2002;161:2143–52. doi: 10.1016/s0002-9440(10)64492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–81. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 28.Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–33. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 30.Castiglioni A, Canti V, Rovere-Querini P, Manfredi AA. High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. 2011;343:189–99. doi: 10.1007/s00441-010-1033-1. [DOI] [PubMed] [Google Scholar]