

Abstract
Programmable nucleases, which include zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and RNA-guided engineered nucleases (RGENs) repurposed from the type II clustered, regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) system are now widely used for genome editing in higher eukaryotic cells and whole organisms, revolutionising almost every discipline in biological research, medicine, and biotechnology. All of these nucleases, however, induce off-target mutations at sites homologous in sequence with on-target sites, limiting their utility in many applications including gene or cell therapy. In this review, we compare methods for detecting nuclease off-target mutations. We also review methods for profiling genome-wide off-target effects and discuss how to reduce or avoid off-target mutations.
Keywords: Cas9, CRISPR, genome editing, off-target, TALEN, ZFN
PROGRAMMABLE NUCLEASES FOR GENOME EDITING
Genome editing is a method of modifying genome sequences in cells and whole organisms via custom-designed programmable nucleases (Kim and Kim, 2014), which cleave chromosomal DNA in a targeted manner, producing site-specific DNA double-strand breaks (DSBs). These DSBs are efficiently repaired in cells by endogenous DNA repair systems known as homologous recombination (HR) and non-homologous end joining (NHEJ), often causing site-specific genome modifications. This technique is now widely used in research, medicine, and biotechnology, a phenomenon that is highlighted by the choice of genome editing as the 2011 Method of the Year and one of the 2015 Methods to Watch by Nature Methods and as a Breakthrough of the Year runner-up by Science in 2013. For example, gene knockout using engineered nucleases enables identification and validation of drug-able target genes. Gene correction in stem and somatic cells can lead to gene therapy for the treatment of various genetic and non-genetic diseases.
Three different classes of programmable nucleases have been developed: zinc finger nucleases (ZFNs) (Bibikova et al., 2003; Kim et al., 2009; 2010; Urnov et al., 2005), transcription activator-like effector nucleases (TALENs) (Kim et al., 2013a; 2013c, Miller et al., 2011), and RNA-guided endonucleases (RGENs) (Cho et al., 2013a; 2013b; Cong et al., 2013; Mali et al., 2013b) repurposed from the type II CRISPR system, an adaptive immune response in bacteria and archea.
ZFNs and TALENs consist of a common nuclease domain derived from FokI, a type IIS restriction enzyme, and distinct DNA-binding domains: ZFNs use zinc fingers (Kim et al., 1996), whereas TALENs employ TAL effectors derived from Xanthomonas, a plant pathogen (Boch et al., 2009; Moscou and Bogdanove, 2009). These DNA-binding domains can be engineered to target user-defined DNA sequences. Because the FokI nuclease domain must dimerize to cleave DNA (Bitinaite et al., 1998), these FokI-based nucleases function as pairs, contributing to their high specificities. Typically, a ZFN pair recognizes an 18-to 36-bp DNA sequence, and a TALEN pair recognizes a 30- to 40-bp DNA sequence, surpassing the complexity of the human genome (4E16 = 4.3 billion > 3.2 billion, the size of the human genome). In practice, however, these nucleases can induce off-target mutations. Furthermore, many ZFNs, especially those made using publically-available zinc-finger resources, are cytotoxic (Kim et al., 2009), which may arise from their off-target effects. Custom-made ZFNs, available from a commercial source, are more potent and specific but are expensive. ZFNs prefer guanine-rich target sequences, limiting targetable sites.
TALENs, the 2nd generation of programmable nucleases, can be designed to target almost any DNA sequence, a critical advantage over ZFNs and RGENs. Unlike zinc fingers that recognize 3-bp sub-sites, TAL effector modules recognize single bases. Four different modules, each specific to one of the four bases, are used to make TALENs. TAL effector arrays often consist of up to 20 modules, making it time-consuming and labor-intensive to prepare plasmids that encode TALENs. In general, TALENs are not cytotoxic, but can induce off-target mutations (Mussolino et al., 2011). Fortunately, TALEN off-target effects can be avoided by choosing unique target sequences that differ by at least 7 nucleotides from any other site in the genome (Kim et al., 2013a). A web-based resource (www.talenlibrary.net) provides such unique sites in each human gene.
CRISPR/Cas-derived RGENs constitute yet another class of programmable nucleases. RGENs consist of a target-specific CRISPR RNA (crRNA), a target-independent trans-activating crRNA (tracrRNA), and Cas9, the protein component originated from Streptococcus pyogenes. Essential portions of crRNA and tracrRNA can be linked to form a single-chain guide RNA (sgRNA) (Jinek et al., 2012). Both crRNAs and sgRNAs function as guide RNAs (gRNAs) to direct Cas9 to target sites. The specificity of an RGEN is determined by both the gRNA, which hybridizes with a 20-bp target DNA sequence, and Cas9, which recognizes the 5′-NGG-3′ sequence known as the protospacer-adjacent motif (PAM). New RGENs with desired specificity are prepared by replacing in vitro transcribed gRNA or gRNA-encoding plasmids, making these 3rd generation nucleases affordable and scalable. Unlike ZFNs and TALENs, however, RGENs function as monomers, initially raising concerns about their specificity. Indeed, RGENs can induce unwanted mutations at off-target sites that differ by up to several nucleotides from on-target sites (Cho et al., 2014; Cradick et al., 2013; Fu et al., 2013; Hsu et al., 2013). This means that there are thousands of potential off-target sites per RGEN in the human genome.
WHY DO NUCLEASE OFF-TARGET EFFECTS MATTER?
Programmable nucleases can cut their target sites efficiently inducing site-specific DSBs in the genome, but can also create unwanted cleavages at off-target sites with high sequence homology to on-target sites, often inducing off-target mutations. Thus, both zinc finger proteins and TAL effector arrays can bind to homologous sites, leading to off-target DNA cleavages. RGEN off-target mutations are caused by both Cas9 and gRNAs. The optimal PAM sequence recognized by Cas9 derived from S. pyogenes is 5′-NGG-3′. However, Cas9 can cleave sites with a 5′-NAG-3′ or 5′-NGA-3′ PAM albeit less efficiently (Hsu et al., 2013). A few nucleotide mismatches between a 20-nt gRNA sequence and a target DNA sequence is also tolerated by an RGEN. Mismatches in the PAM-distal sequence at the 5′ terminus are tolerated better than are those in the 10-to 12-nt PAM-proximal sequence, often termed a seed region. Furthermore, RGENs can cleave off-target sites with a few extra or missing nucleotides that can produce a DNA or RNA bulge, respectively (Lin et al., 2014).
Imprecise repair of on- and off-target DNA cleavages can give rise to gross chromosomal rearrangements such as deletions (Lee et al., 2010), inversions (Lee et al., 2012; Park et al., 2014), and translocations (Brunet et al., 2009; Cho et al., 2014), in addition to local mutations. An example is a ZFN designed to target the C-C chemokine receptor 5 (CCR5) gene that encodes an essential co-receptor of human immunodeficiency virus (HIV) infection in helper T cells. A phase I/II clinical trial with this ZNF for the treatment of acquired immune deficiency syndrome (AIDS) has been completed in the US (ClinicalTrials.gov identifier: NCT00842634) (Tebas et al., 2014). This ZFN also cleaves a highly homologous site in the CCR2 gene, leading to ∼15-kbp chromosomal deletions, duplications, and inversions of the intervening DNA segment in human cells (Lee et al., 2010; 2012). It is unknown whether the off-target mutation at the CCR2 gene and the resulting unwanted chromosomal rearrangements would cause any side effects in AIDS patients. Chromosomal rearrangements are one of the hallmarks of cancer. Because unwanted chromosomal rearrangements may activate onco- genes, they must be monitored carefully and avoided as much as possible.
METHODS FOR DETECTING ON-TARGET AND OFF-TARGET MUTATIONS
Various methods, which include Sanger sequencing, high-throughput sequencing, restriction fragment length polymorphism (RFLP) analysis, mismatch-sensitive enzymes, have been developed for detecting indels induced by erroneous NHEJ repair of DSBs. Sanger sequencing of DNA from individual clones is the gold standard for confirming nuclease-triggered mutations at on- or off-target sites, but this method is time-consuming and cost-inefficient when many samples need to be analyzed in parallel. High-throughput sequencing enables accurate measurements of indel frequencies at up to hundreds of on- and off-target sites at once. Although this method is highly sensitive, allowing detection of indels that are induced with frequencies that range from 0.01% to 1% (∼0.1% on average), care must be taken to discard false-positive sequence reads that result from PCR artifacts and to include a negative control (no nuclease expression) at each target site (Cho et al., 2014). A web-based tool (www.regenome.net) is available for obtaining indel frequencies using targeted deep sequencing data. Single-molecule real-time (SMRT) sequencing is an alternative method for measuring on- and off-target mutation frequencies (Hendel et al., 2014).
Mismatch-sensitive nucleases, which include T7 endonuclease I (T7E1) (Kim et al., 2009) and CEL-I enzyme (a.k.a. Surveyor nuclease) are widely used to measure indel frequencies in bulk populations of cells. These enzymes recognize and cleave heteroduplexes formed by hybridization of wild-type and mutant DNA sequences or of two different mutant DNA sequences. PCR amplicons treated with these enzymes are then subjected to agarose gel electrophoresis. The size and intensity of cleaved DNA bands provide accurate measurements of mutation frequencies. Although these enzymes can detect both indels and point mutations, T7E1 is more sensitive to indels than CEL-I enzyme (Vouillot et al., 2015). Because programmable nucleases rarely produce point mutations (Kim et al., 2013b), T7E1 is preferred for detecting nuclease-induced mutations. In fact, under optimal conditions, T7E1 can detect indels that are induced at frequencies below 1% (Kim et al., 2013a).
In contrast to agarose gel electrophoresis, polyacrylamide gel electrophoresis (PAGE) can be used to separate heteroduplexed DNA from homoduplexed DNA by without the use of mismatch-sensitive nucleases (Zhu et al., 2014). However, accurate quantitation of mutation frequencies using this method is difficult because multiple DNA bands are obtained.
Programmable nucleases often induce homozygous biallelic mutations in a cell or an organism, leading to a complete gene disruption or knockout. These mutations cannot be detected by T7E1 or CEL-I, because heteroduplexes are not formed. RGENs can be used for RFLP analysis to distinguish homozygous biallelic mutants from wild-type sequences or monoallelic mutations (Kim et al., 2014a). Thus, RGENs cannot cleave indel sequences induced by themselves in cells. In fact, RFLP analysis using conventional restriction enzymes was one of the first methods for detecting mutations induced by programmable nucleases in cells (Urnov et al., 2005). However, unlike RGEN-RFLP, this method is limited by the availability of appropriate restriction sites in a target DNA site. Fluorescence PCR (Kim et al., 2011) and DNA melting analysis (Parant et al., 2009) can also be used for measuring genome editing activities of programmable nucleases, but these methods require special devices.
METHODS FOR MEASURING OFF-TARGET EFFECTS IN THE GENOME
Several different methods have been used to identify nuclease off-target sites: bioinformatic prediction based on sequence homology, chromatin immunoprecipitation coupled with deep sequencing (ChIP-Seq), systematic evolution of ligands by exponential amplification (SELEX), integrase-deficient lentivirus (IDLV) capture in cells, in vitro selection using a DNA substrate library for nuclease-mediated DNA cleavage, and whole genome/exome sequencing (Figs. 1A–1C).
Fig. 1.
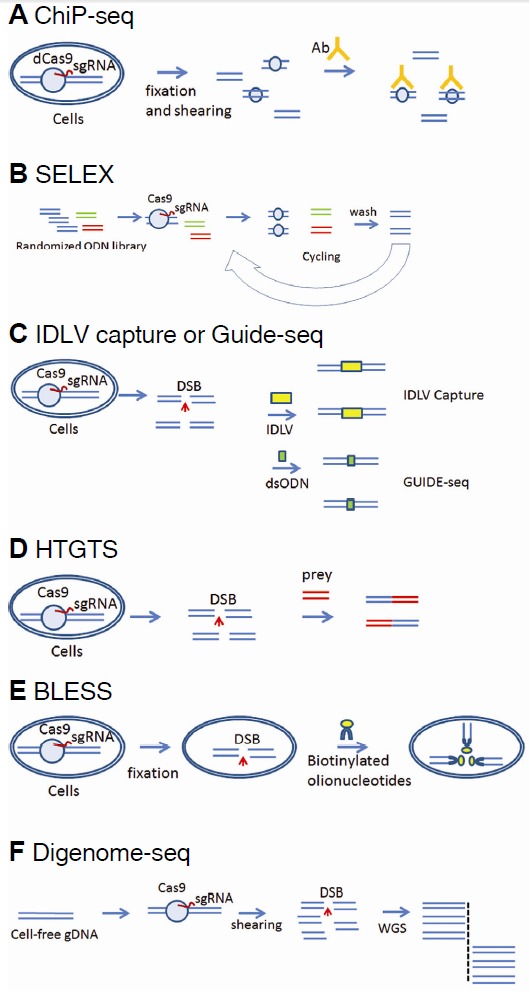
Overview of methods for measuring off-target effects in the genome. (A) Chromatin immunoprecipitation coupled with deep sequencing (ChIP-Seq). (B) Systemic evolution of ligands by exponential amplification (SELEX). (C) Integrase-deficient lentivirus (IDLV) capture or genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq). (D) High-throughput genomic translocation sequencing (HTGTS). (E) Breaks labelling, enrichments on streptavidin and next-generation sequencing (BLESS). (F) In vitro nuclease-digested genome sequencing (Digenome-seq). Ab, antibody, dCas9, catalytically-dead Cas9; DSB, double-strand break; ODN, oligodeoxynucleotide; sgRNA, small-guide RNA; WGS, whole-genome sequencing; WT, wild type.
Cas9 and other programmable nucleases can induce off-target mutations at sites that differ from their on-target sites by several nucleotides. This means that more than 10,000 potential off-target sites identified based on sequence homology must be examined. A web-based computer program, CAS-OFFinder (Bae et al., 2014), can be used to list all of these homologous sites, but measuring indel frequencies at these sites one by one is an almost impossible task. To profile genome-wide off-target effects of engineered nucleases in an unbiased manner, SELEX (Miller et al., 2011) and ChIP-Seq (Kuscu et al., 2014; Wu et al., 2014) have been used. These methods rely on DNA binding in vitro and in cells, respectively, rather than DNA cleavage. Unfortunately, DNA binding and cleavage are often uncoupled. In fact, most off-target DNA-binding sites recognized by catalytically dead Cas9 (dCas9) are not cleaved at all by Cas9 in cells.
IDLV capture and in vitro selection are two different methods that examine nuclease cleavage sites rather than binding sites. IDLV capture relies on integration of IDLV DNA at off-target sites in cells (Gabriel et al., 2011). Because the efficiency of IDLV capture at a given site is much lower than the mutation frequency at that site, many bona fide off-target sites cannot be captured by this method. In vitro selection uses nucleases to cleave a biased DNA substrate library that consists of >1012 variants of a target DNA sequence and determines cleaved library members by deep sequencing (Pattanayak et al., 2011; 2013). Although this is a powerful method of profiling nuclease specificity in vitro, most cleaved sequences do not exist in the genome. Furthermore, this approach cannot examine off-target cleavage at sequences that can hybridize with a DNA or RNA bulge.
These two methods are complementary but neither of them is comprehensive. Gabriel et al. (2011) and Pattanayak et al. (2011) have applied IDLV capture and in vitro selection, respectively, to examine off-target effects of the same CCR5-targeting ZFN that has been under clinical investigation for the treatment of HIV infection. The two groups obtained almost entirely different sets of off-target sites: only the highly homologous CCR2 off-target site was commonly identified, highlighting the limitation of the two approaches.
Whole genome/exome sequencing of clonal populations of human cells in which a gene of interest was modified using ZFNs (Yusa et al., 2011), TALENs (Smith et al., 2014; Veres et al., 2014), or RGENs (Cho et al., 2014; Kim et al., 2015) revealed remarkable specificities of these nucleases. Although these nucleases have detectable off-target effects in a bulk population of cells, off-target mutations are almost absent in the entire human exome or genome of an individual clone. This is because mutation frequencies at off-target sites are usually orders of magnitude smaller than those at on-target sites. Off-target mutations that occur at a frequency of 10% cannot be revealed by sequencing of DNA from just a few clones with a typical depth of 30X.
In the following two sections, we review recently improved methods for profiling genome-wide off-target sites of Cas9 nucleases and various approaches for reducing or avoiding their off-target effects. Some of these methods can also be applied to other programmable nucleases.
UNBIASED GENOME-WIDE PROFILING OF NUCLEASE CLEAVAGE SITES
Recently, four different methods have been reported for identifying potential off-target sites of RGENs in a bulk population of cells (Figs. 1C–1F). Genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq) represents an improvement over IDLV capture (Tsai et al., 2015). Blunt-ended, double-stranded phophothiorate oligodeoxynucleotides (dsODNs) can be captured at on-target and off-target sites, when DSBs are repaired by NHEJ in cells. These dsODN integration sites are mapped in the genome by PCR amplification and deep sequencing. High-throughput genomic translocation sequencing (HTGTS) exploits translocations that are induced by erroneous ligation of on-target and off-target sites in cells (Frock et al., 2015). HTGTS identifies off-target sites by using the on-target DSB as a ‘bait’ to catch ‘prey’ sequences that are trans-located to the on-target site. High-throughput sequencing is used to determine prey sequences that correspond to off-target sites.
Off-target DSBs can also be captured in fixed, permeable cells. Breaks labelling, enrichments on streptavidin and next-generation sequencing (BLESS) is performed by labelling DSBs present in fixed cells using biotinylated oligonucleotides, which are then enriched and subjected to deep sequencing (Crosetto et al., 2013; Ran et al., 2015). Because this method captures DSBs at a single moment, many bona fide off-target cleavage sites can be missed, resulting in poor sensitivity.
Cell-free genomic DNA can be used to profile nuclease off-target effects in vitro. Digested genome sequencing (Digenomeseq) is a method for identifying off-target sites using nuclease-digested genomic DNA (digenome) that is subjected to whole genome sequencing (Kim et al., 2015). In vitro digestion of genomic DNA with Cas9 yields sequence reads with the same 5′ ends at cleavage sites, which can be computationally identified using whole genome sequence data.
A key difference between these methods is whether genomic DNA is cleaved in cells or in vitro. Both HTGTS and GUIDE-seq rely on DNA cleavage and repair in cells. In contrast, Digenome-seq is based on digestion of cell-free genomic DNA. In BLESS, DNA is cleaved by a nuclease of interest in cells, but the resulting DSBs are captured in vitro. Note that DSBs are spontaneously generated in cells even in the absence of an engineered nuclease. These naturally-occurring nuclease-independent DSBs are captured by HTGTS, GUIDE-seq, and BLESS. Furthermore, these methods are susceptible to PCR artifacts that arise from amplification of sequences around DSB sites. These false-positive signals must be filtered out by searching for sequences around the captured sites that are homologous to the on-target site. Up to 95% of captured sites are discarded in this filtering step. Digenome-seq is not limited by Cas9-independent DSBs produced in cells or in vitro or by PCR artifacts, because these randomly-occurring DSBs are unlikely to produce the same 5′ ends and because PCR is not used to prepare Cas9-digested genomic DNA.
It is unknown to what extent programmable nucleases are limited by chromatin. Because HTGTS, GUIDE-seq, and BLESS profile nuclease cleavage sites in cells, off-target sites captured in one cell type could be different from those in other cell types, owing to differential chromatin accessibility in each cell type. Digenome-seq functions independently of the cell type, because naked, chromatin-free genomic DNA is used.
DSB repair by NHEJ in cells often result in deletions of sequences of up to hundreds of base pairs in length at cleavage sites. As a result, neither HTGTS nor GUIDE-seq can pinpoint off-target sites: One must search for potential off-target sites based on the sequence homology around captured sites. Both BLESS and Digenome-seq do not depend on NHEJ and can pinpoint cleavage sites at single-nucleotide resolution.
To determine which method is most sensitive and comprehensive, one needs to test the same nucleases using each of these methods. Only one sgRNA, specific to the VEGF-A site, has been tested by HTGTS (Frock et al., 2015), GUIDE-seq (Tsai et al., 2015), and Digenome-seq (Kim et al., 2015) thus far. Each of these methods revealed a different set of potential off-target sites, suggesting that no method is comprehensive. However, most of these candidate sites were invalidated by targeted deep sequencing. Importantly, these three methods commonly identified a total of 7 off-target sites in addition to the on-target site in the human genome. Notably, Digenome-seq identified one additional bona fide off-target site, with an indel frequency of 0.065%, which was validated using deep sequencing.
HOW TO REDUCE OFF-TARGET MUTATIONS CAUSED BY CRISPR-CAS9 NUCLEASES
First, the choice of unique target sequences, which differ from any other sites in the genome by at least 2 or 3 nucleotides in a 20-nt sequence, is important for avoiding off-target effects (Cho et al., 2014). RGENs discriminate efficiently against potential off-target sites with mismatches in the PAM sequence and the seed region upstream of the PAM sequence. A web-based computer algorithm is available (http://www.rgenome.net/casoffinder), which searches for potential off-target sites and unique target sequences in the genomes of humans and 20 other organisms of research interest (Table 1). Alternative web-based tools are also available: CRISPR Design (http://crispr.mit.edu) introduces a scoring system for choosing sgRNAs with minimum off-target ites. ChopChop (http://chopchop.re.fas.harvard.edu) provides a user-friendly sgRNA designer program. Second, the use of four different forms of modified sgRNAs can reduce off-target mutations by orders of magnitude without sacrificing mutation efficiencies at on-target sites (Fig. 2A). First, sgRNAs with two extra, target independent guanine nucleotides at the 5′ terminus are much more specific than conventional sgRNAs at least in human cells (Cho et al., 2014). The addition of two extra guanine nucleotides also allows sgRNAs that begin with nucleotides other than guanine to be made, which is required for transcription under the control of U6 promoter in cells or the T7 promoter in vitro. It is unknown how the two additional guanine nucleotides make sgRNAs more specific. Second, truncated sgRNAs (tru-sgRNAs) with 17-nt, rather than 20-nt, of complementarity enhance the specificity of RNA-guided genome editing (Fu et al., 2014). Tru-sgRNAs are more sensitive to mismatches, owing to their reduced binding energy at the sgRNA-DNA interface. It is still important to choose unique 17-nt target sites to minimize off-target effects. Unfortunately, 17-nt sites that differ from any other sites in the human genome by at least 2 or 3 nucleotides are much rarer than such 20-nt full-length sites.
Table 1.
Web-based tools available for guide-RNA design
| Name | Developer | Address |
|---|---|---|
| Cas-OFFinder | Jin-Soo Kim lab, Seoul National University | http://www.rgenome.net/ |
| CHOPCHOP | George Church lab, Harvard University | https://chopchop.rc.fas.harvard.edu/ |
| CRISPR Design | Feng Zhang lab, Massachusetts Institute of Technology | http://crispr.mit.edu/ |
| CRISPR Design tool | The Broad Institute of Harvard and MIT | http://www.broadinstitute.org/mpg/crispr_design/ |
| CRISPR/Cas9 gRNA finder | Jack Lin lab, University of Colorado | http://spot.colorado.edu/~slin/cas9.html |
| CRISPRfinder | Christine Pourcel lab, Université Paris-Sud 11 | http://crispr.u-psud.fr/Server/ |
| E-CRISP | Boutros lab, DKFZ German Cancer Research Center | http://www.e-crisp.org/E-CRISP/ |
| ZiFiT | Keith Joung lab, Harvard University | http://zifit.partners.org/ZiFiT |
Fig. 2.
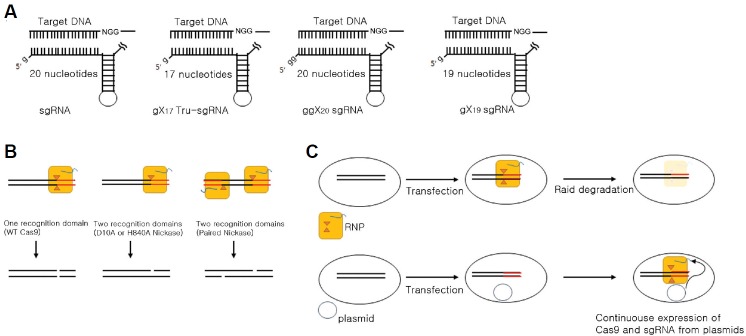
Schematic strategies for reducing off-target mutations. (A) Four different forms of sgRNAs. sgRNAs with two extra guanines (ggX20) or truncated sgRNAs (gX17) enhance the specificity of RNA-guided genome editing, compared to conventional sgRNAs (gX19 or gX20). (B) Use of paired nickases to generate two single-strand breaks or nicks on different DNA strands. (C) Use of Cas9-sgRNA ribonucleoprotein (RNP) complexes, rather than the Cas9 and sgRNA-encoding plasmids to avoid continuous expression of Cas9 and sgRNA from plasmids.
Third, paired nickases can generate two single-strand breaks or nicks on different DNA strands, producing a composite DSB and doubling the specificity of genome editing (Fig. 2B) (Cho et al., 2014; Kim et al., 2012; Mali et al., 2013a; Ran et al., 2013). Both ZFNs and Cas9 can be converted to nickases by inactivating one active site. Cas9 has two active sites, each cleaving either the Watson or Crick strand. Among the two nickase forms, D10A Cas9 appears more efficient and robust than H840A Cas9. One caveat to this approach is that two active sgRNAs are required to make a functional Cas9 nickase pair. Furthermore, target sequences must contain two PAM sequences, limiting the choice of targetable sites.
Fourth, the use of recombinant Cas9 protein [commercially available from ToolGen (www.toolgen.com)], rather than the Cas9-encoding plasmid, further reduces off-target mutations (Fig. 2C) (Kim et al., 2014b). Cas9-sgRNA ribonucleoprotein (RNP) complexes induce mutations at target sites almost immediately after delivery into cells and are degraded rapidly by endogenous proteases, reducing off-target effects without compromising on-target mutation frequencies. In fact, RGEN RNP delivery is more efficient and less stressful than plasmid transfection in human primary cells and pluripotent stem cells, in which the cyclic GMP-AMP synthase (cGAS) signalling pathway is active. In most cancer cells, in which the cGAS signalling pathway is inactive, plasmid transfection and RNP delivery produce comparable results. Furthermore, RGEN RNP delivery avoids the possibility of unwanted integration of plasmid fragments. RGEN RNPs can be delivered into cells via electroporation (Kim et al., 2014b) or lipofection (Zuris et al., 2015) or protein transdction (Ramakrishna et al., 2014).
CONCLUSION
Since we and others have reported RNA-guided genome editing in human cells in January, 2013 (Cho et al., 2013a; Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013b), the CRISPR-Cas9 system has been widely used in many labs all around the world to modify genomes in various organisms and cells. Although Cas9 nucleases have off-target effects, whole genome/exome sequencing of gene-modified clones shows that these nucleases are highly specific. Recent methods such as Digenome-seq and GUIDE-seq that profile genome-wide off-target sites in a bulk population of cells reveal a broad spectrum of sgRNA specificities. Certain sgRNAs are remarkably specific, resulting in no measurable off-target mutations, whereas others are promiscuous. To find rules that govern sgRNA specificity, one needs to profile the off-target effects of as many sgRNAs as possible at the genome-wide level. Digenome-seq is appealing in this regard, because it can be multiplexed without increasing the sequencing depth. Hundreds of sgRNAs can be tested in a single assay.
Genome-wide off-target profiling methods yield a list of potential off-target sites that are cleaved under certain conditions. Bona fide off-target sites must be validated by targeted deep sequencing. Unfortunately, validation of off-target effects is limited by intrinsic errors of sequencing platforms, which are in the range of 1% to 0.01% (0.1% on average). A more sensitive method is needed to confirm that certain sgRNAs do not induce off-target mutations with indel frequencies below 0.01% in the entire genome. Highly specific and efficient nucleases will enable applications in somatic gene and cell therapy and possibly in human germline genome editing to prevent the transmission of fatal genetic mutations.
Acknowledgments
This work was supported by a grant from IBS (IBS-R021-D1).
REFERENCES
- Bae S, Park J, Kim JS. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Brunet E, Simsek D, Tomishima M, DeKelver R, Choi VM, Gregory P, Urnov F, Weinstock DM, Jasin M. Chromosomal translocations induced at specified loci in human stem cells. Proc. Natl. Acad. Sci. 2009;106:10620–10625. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013a;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Cho SW, Lee J, Carroll D, Kim JS, Lee J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 2013b;195:1177–1180. doi: 10.1534/genetics.113.155853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/as9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods. 2013;10:361–365. doi: 10.1038/nmeth.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Hendel A, Kildebeck EJ, Fine EJ, Clark JT, Punjya N, Sebastiano V, Bao G, Porteus MH. Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep. 2014;7:293–305. doi: 10.1016/j.celrep.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Lee HJ, Carroll D. Genome editing with modularly assembled zinc-finger nucleases. Nat. Methods. 2010;7:91. doi: 10.1038/nmeth0210-91b. author reply 91–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Um E, Cho SR, Jung C, Kim JS. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat. Methods. 2011;8:941–943. doi: 10.1038/nmeth.1733. [DOI] [PubMed] [Google Scholar]
- Kim E, Kim S, Kim DH, Choi BS, Choi IY, Kim JS. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012;22:1327–1333. doi: 10.1101/gr.138792.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 2013a;31:251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim JS. TALENs and ZFNs are associated with different mutation signatures. Nat. Methods. 2013b;10:185. doi: 10.1038/nmeth.2364. [DOI] [PubMed] [Google Scholar]
- Kim YK, Wee G, Park J, Kim J, Baek D, Kim JS, Kim VN. TALEN-based knockout library for human microRNAs. Nat. Struct. Mol. Biol. 2013c;20:1458–1464. doi: 10.1038/nsmb.2701. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kim D, Kim S, Kim JS. Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat. Commun. 2014a;5:3157. doi: 10.1038/ncomms4157. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014b;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Kim E, Kim JS. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kweon J, Kim E, Kim S, Kim JS. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012;22:539–548. doi: 10.1101/gr.129635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, Wile BM, Vertino PM, Stewart FJ, Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013a;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013b;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant JM, George SA, Pryor R, Wittwer CT, Yost HJ. A rapid and efficient method of genotyping zebrafish mutants. Dev. Dyn. 2009;238:3168–3174. doi: 10.1002/dvdy.22143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Kim J, Kweon J, Son JS, Lee JS, Yoo JE, Cho SR, Kim JH, Kim JS, Kim DW. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. 2014;111:9253–9258. doi: 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014;24:1020–1027. doi: 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, Wang Y, Brodsky RA, Zhang K, Cheng L, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouillot L, Thelie A, Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 2015;5:407–415. doi: 10.1534/g3.114.015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordonez A, Hannan NR, Rouhani FJ, et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Xu Y, Yu S, Lu L, Ding M, Cheng J, Song G, Gao X, Yao L, Fan D, et al. An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci. Rep. 2014;4:6420. doi: 10.1038/srep06420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]