

Abstract
Controlled release of medications remains the most convenient way to deliver drugs. In this study, we precipitated gold nanoparticles with quercetin. We loaded gold-quercetin into poly(DL-lactide-co-glycolide) nanoparticles (NQ) and tested the biological activity of NQ on HepG2 hepatocarcinoma cells to acquire the sustained release property. We determined by circular dichroism spectroscopy that NQ effectively caused conformational changes in DNA and modulated different proteins related to epigenetic modifications and c ell cycle control. The mitochondrial membrane potential (MMP), reactive oxygen species (ROS), cell cycle, apoptosis, DNA damage, and caspase 3 activity were analyzed by flow cytometry, and the expression profiles of different anti- and pro-apoptotic as well as epigenetic signals were studied by immunoblotting. A cytotoxicity assay indicated that NQ preferentially killed cancer cells, compared to normal cells. NQ interacted with HepG2 cell DNA and reduced histone deacetylases to control cell proliferation and arrest the cell cycle at the sub-G stage. Activities of cell cycle-related proteins, such as p21WAF, cdk1, and pAkt, were modulated. NQ induced apoptosis in HepG2 cells by activating p53-ROS crosstalk and induces epigenetic modifications leading to inhibited proliferation and cell cycle arrest.
Keywords: apoptosis, gold-nanoparticles, histone deacetylation, p53-ROS crosstalk, quercetin
INTRODUCTION
Some naturally occurring active compounds interact with nucleic acids to reduce cell proliferation and induce production of reactive oxygen species (ROS) and oxidative DNA damage (Bishayee et al., 2013, Lemarie and Grimm, 2011). These events may, in turn, affect the ability of acetyl-transferases to interact with histones (Hurley, 2002) and could be helpful for developing personalized medicines for cancer and other common diseases (Liu, 2012). Naturally derived anticancer compounds have problems of preservation and bio-availability. Unfortunately, the concentrations of polyphenols that appear to be effective in vitro are often an order of magnitude higher than levels measured in vivo (Bhadoriya et al., 2011). Therefore, delivering these compounds requires product formulations to maintain the active molecular form until consumption, as well as to preserve stability, bio-activity, and bio-availability, which is the central goal of developing a nanoparticle (NP)-based system.
NPs are ubiquitous in the environment and are widely used in medical science. Moreover, their effectiveness in cancer treatment has been reported repeatedly (Maitland and Schilsky, 2011). Due to their unique physicochemical properties, NPs can cross many barriers, including the blood brain barrier, which is not possible for traditional drugs (Bhattacharyya et al., 2011). Nevertheless, exposing cells to NPs and their subsequent interactions with organelles and macromolecules can result in negative effects, as NPs can be toxic and induce cell death, particularly in cancer cells; thus, NP-encapsulated drugs are useful for cancer therapy, where the ability to induce cell death is advantageous for controlling cancer proliferation and growth (Puhl et al., 2011). Surface functionalization of gold nanoparticles (AuNPs) is essential for biomedical applications that target them to specific disease areas and allow them to selectively interact with cells or biomolecules. Surface conjugation is usually achieved by adsorption of the ligand to the surface of gold. Additionally, long-term circulating NPs are desirable in systemic applications, such as passive targeting of tumors and inflammatory sites. Poly(ethylene glycol) (PEG)/poly (lactic-co-glycolic acid (PLGA)-modified NPs have a long-term circulating property, as they can evade macrophage-mediated uptake and removal from systemic circulation (Havsteen, 2002).
Target-specific drug therapy against cancer has been effective. Suppressing cell cycle and proliferation rates depends on different parameters, such as DNA structural changes and inhibiting the activities of histone deacetylases (HDACs). These anti-growth promoting activities could make drugs more cancer specific. In earlier studies, we found aberrant expression of HDACs during cellular oncogenesis, which resulted in repression of anti-proliferative genes (Noh et al., 2011; Talluri and Dick 2012). HDAC inhibitors kill cancer cells in vitro and in vivo by altering gene expression, mutating genes, modifying proteins, and destabilizing chromosomes (Shahbazian et al., 2012). Related alterations/modifications have been reported for AuNPs, as they modulate heterochromatin connections with lamin proteins and core histones (Brannon-Peppas and Blanchette, 2004; Liu, 2012), suggesting that NPs could be considered epigenetic agents.
The objectives of this study were to determine: i) whether gold nanoparticles(AuNPs) can be precipitated using quercetin as a reducing agent, ii) if AuNPs can be successfully nanoencapsulated with PLGA to increase bio-availability, and iii) whether the quercetin-reduced AuNPs could be endowed with a sustained release property to a target site, thereby reducing unwanted gold toxicity to normal cells but not cancer cells. We investigated the effects of formulated NPs on several cancer cell lines in vitro and identified possible molecular pathways involved in inducing apoptosis in the cancer cell lines. In brief, we found that nano-quercetin (NQ) could penetrate cell membranes. We also showed that the reduction in cancer cell viability was due to the induction of apoptosis by ROS. NQ also downregulated expression of HDAC1 and 2, suggesting its role in inhibiting cancer cell proliferation.
MATERIALS AND METHODS
NQ preparation
High performance liquid chromatography-grade quercetin was purchased from HiMedia Laboratories (Mumbai, India) in an anhydrous powdered form.
Synthesis of gold-quercetin
AuNPs were synthesized by reducing 1 mM gold chloride with a freshly prepared quercetin solution in absolute alcohol. The pale-yellow solution turned to deep red as the NQ were formed.
Synthesis of PLGA-capsulated NQ
PLGA (50 mg) was added to an aqueous dispersion of AuNPs. Then, we added this mixture drop wise to 20 ml of an aqueous solution containing a stabilizer (1% polyoxyethylene-polyoxypropylene; F68). The mixture was stirred at 400 rpm and 4°C until the organic solvent had evaporated completely. The redundant stabilizer was removed by repeated washing and centrifugation (25,000 × g and 4°C for 30 min), and the pellet was resuspended in Milli-Q water. The NPs were stored at 4°C for further use.
Fluorescent dye was conjugated to the gold surface by adding FITC dye to the PLGA and NQ mixture, and the steps described above in “Synthesis of PLGA-capsulated gold-quercetin nanoparticle” were followed in the dark.
Particle size determination by dynamic light scattering (DLS)
We determined mean size and the size distribution of the polymeric micelles by DLS using a Zetasizer Nano-ZS instrument (Malvern Instruments, UK). The intensity of scattered light was detected at 90° to an incident beam. Freeze-dried powder was dispersed in aqueous solution, and measurements were taken after the aqueous micellar solution was filtered with a 0.22 μm pore size membrane extruder (Millipore, USA). We analyzed the data in the automatic mode. Size is presented as the mean value of 20 runs, with triplicate measurements for each run. We measured the zeta potential of the NPs in the same instrument using the same procedure.
Scanning electron microscopic (SEM) study
Surface morphology of the formulated NP-loaded drug was determined by SEM (S 500, Hitachi, Japan), equipped with a 15 kV SE detector with a collector bias of 300 V. Lyophilized samples were spread over double-sided conductive tape (12 mm) fixed on the metallic stud.
In vitro cell culture
HepG2 (hepatocarcinoma), HeLa (cervical cancer), A375 (skin melanoma), and WRL-68 (normal) cells were obtained from the National Centre for Cell Science (India) and maintained in DMEM medium with 10% fetal bovine serum in a humidified CO2 incubator under 5% CO2. The cells were harvested in a 0.025% trypsin-EDTA (Gibco, USA) with phosphate buffered saline solution, plated at the required cell number, and allowed to adhere before drug treatment. The cells were exposed to different doses of NQ for 24 h to check cell viability.
Determination of cytotoxity
Cytotoxicity of NQ was determined by conventional MTT assay (Mosmann, 1983). HepG2, HeLa, A375 and WRL-68 were dispensed in 96-well flat bottom microtiter plates (Tarsons, India) at a density of 1×103 cells per well and treated with various concentrations of NQ (10 to 60 μg/ml) and incubated for 24 h. Thiazolyl Blue Tetrazolium Bromide (MTT) (Sigma, USA) was then added to the well at a concentration of 10 μM and the plates were then incubated for 2–3 h at 37°C in dark. To stop the reaction and develop blue colour, acidic isopropanol was used and OD was taken at 595 nm in a microtitter plate reader (Thermo, USA).
Treatment and control
The dried drug was stored in cool place (4°C), before treatment, the desired amount of dried NQ was mixed with DMEM culture medium and the mixture was sonicated with a probe sonicator (Sonics, USA) for use in subsequent experiments.
The 24 μg/ml of NQ was used for the experiments to study time-dependent effects, if any. In the experiments set up to determine dose-dependent effects, if any, two doses- 12 (NQ1) and 24 (NQ2) μg/ml- were used; the treatment time was fixed at 24 h for all.
Equal amount of media without drug served as control. As there was insignificant difference in data between the control and drug untreated groups, for the sake of clarity and brevity, we only considered the media as control for comparing the experimental data.
Study of NQ’s cellular uptake
The cellular uptake of NQ in HepG2 cells was analysed by the fluorescence method. In brief, cells were washed twice with PBS. The washed cells were re-suspended in media and then incubated with NQ for different time periods. Cells were then examined under a fluorescence microscope (Carl Zeiss, Germany), and images were captured using an Axioscope Plus 2.
Study of NQ’s binding capability with cellular DNA
For assessment of NQ and nuclear DNA interaction, HepG2 cells were exposed to NQ for 0, 6, 12, 18 and 24 h. After incubation, the nuclear DNA of the cells were extracted and purified by using GeneiPure™ Mammalian Genomic DNA Purification Kit (Genei, India). The collected DNA was used to analyse the CD spectra obtained in the spectroscope (JASCO J720, Japan), using Origin 8 pro software.
Proliferation assay
The effect of NQ on cellular growth, proliferation assay was performed adopting the methodologies used by Bishayee et al (2013). Treated and untreated cells were harvested after 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22 and 24 h, washed twice with PBS and trypsinized. The cell suspension was then transferred for cell counting and counted with the help of a haemocytometer. This procedure was repeated for all samples at each time point, and the experiments were repeated 3 times.
Cell cycle analysis
To evaluate the cell cycle, control and NQ-treated cells were trypsinized and fixed by chilled 70% methanol. After RNase treatment, cells were incubated with PI for 20–30 min and the fluorescence intensities were measured by FL-2A filter using BD FACS machine.
Study of MMP (Δψ)
Cells were washed with PBS after the treatment and harvested using trypsin-EDTA solution. The cells were then made RNA free and incubated in Rhodamine 123 for 30 min to study depolarization of MMP. The fluorescence was measured using a BD FACS machine fitted with FL-1H filter.
p53 siRNA transfection into HepG2 cells
In the six welled tissue culture plate, 2 × 105 cells per well were seeded in 2 ml of antibiotic free normal growth medium supplemented with FBS and the cells were transfected with 300 pmol of p53-siRNA (Santacruz Biotechnology Inc., USA) and lipofectamine-2000 for 12 h by following siRNA transfection protocol supplied by Santa Cruz Biotechnology Inc. Confirmation of transfection was made through western blotting.
Study of intra-cellular ROS generation
Changes in accumulation of ROS were analysed by deploying flowcytometer. Control (wild-type p53-expressing HepG2, ascorbic acid (A.A.) treated and p53-siRNA treated) and treated cells (A.A+NQ, p53 siRNA+NQ and only NQ) were collected and fixed with 70% chilled methanol. Fixed cells were incubated with H2DCFDA for 20 min in dark. Fluorescence intensity was measured using FL-1H filter of flowcytometer. Data were analysed using Cyflogic software.
Study of apoptosis
To study apoptosis, control (wild-type p53-expressing HepG2, ascorbic acid (A.A.) treated and p53-siRNA treated) and treated cells (A.A+NQ, p53 siRNA+NQ and only NQ) were washed and harvested using trypsin-EDTA solution. Cells were then made RNA free and incubated with annexin V stain for 30 min in dark. Fluorescence intensities were measured by a BD FACS machine using FL-1H filter.
Study of NQ-induced chromosomal condensation and DNA damage
Control and treated cells were fixed with 70% ice-cold methanol and DAPI staining method was deployed to check the chromatin condensation. Analysis of DNA nick formation or strand break was done by flow cytometry using terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay, as described by Loo (2002). Cells incubated with FITC-conjugated mouse secondary antibody was analysed under FL-1H filter for fluorescence emission by flow cytometry.
Subcellular fraction
Subcellular fractionation of cells was achieved by Dounce homogenization in isotonic Buffer (250 mM sucrose, 10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, pH 7.4) and differential Centrifugation yielding nuclear (500 × g pellet), mitochondrial (15,000 × g pellet), and remaining supernatant was cytosolic fraction (Mikhailov et al., 2001).
To isolate total cellular protein, treated and control cells were washed twice with ice-cold PBS and cell lysate was prepared using lysis buffer (10 mM Tris-HCl, pH 7.4, 1 mM MgCl2, 1 mM EDTA, 0.1 mM PMSF, 5 mM β-mercaptoethanol, 0.5% CHAPS, 10% glycerol and a cocktail of protease inhibitor). Cell lysate was cleared by centrifugation at 5,000 g for 20 min at 4°C.
Study of cytochrome c activity
The activity of cytochrome c was measured in the mitochondrial and cytoplasmic fraction using conventional ELISA method as described by the manufacturer’s protocol.
Study of rac-1 activity
Time dependent study of rac1 was performed using “Active Rac1 pull down and detection kit” (catalogue no 16118, Thermo scientific, USA).
Study of caspase 3 activity
Caspase 3 activity was measured by (i) spectrophotometry assay; and (ii) flowcytometry assay.
To determine caspase 3 activity, the spectrophotometric detection of chromophore p-nitroaniline (p-NA) was adopted. The experiment was performed using Caspase 3 Assay Kit (colorimetric) (ab39401) from Abcam. pNA after cleavage from the substrate DVED-pNA was quantified by microtiter plate reader at 405 nm. Comparison of the absorbance of p-NA from NQ treated sample with control allows determination of the fold increase in caspase 3 activity.
The caspase 3 activity assay was performed using indirect staining protocol after the incubation of 24 h with the drugs and a control set in HeLa cells. The 5 × 106 cells were resuspended in ice cold phosphate buffer saline with 10% FBS and 1% sodium azide. The 10 μg/ml of anti-caspase primary antibody was added and then cells were incubated in room temperature for 30 min. After this, cells were further incubated in FITC tagged secondary antibody for 20 min in dark. The fluorescence was determined by flowcytometer using FL-IH filters. Data were analysed by using Cyflogic software.
Study of histone de-acetylase activity
Histone de-acetylase activity was studied using histone de-acetylase assay kit (colorimetric) from Upstate Millipore. The working protocol supplied with the material was adopted for this study.
Western blot analysis
For direct western blot analysis, a total of 50 μg of protein was resolved using SDS-PAGE and transferred to PVDF membranes for western blotting using the relevant antibodies e.g., anti-cytochrome c, anti-Bax, anti-p53, anti-Apaf1, anti-PARP, anti-HDAC1, anti-HDAC2, anti-p-Akt, anti-Akt, anti-cdk1, anti-cyclinD1, anti-p21WAF, anti-Mcl-1, anti-Bcl-2, anti-Bcl-xL and anti-caspase 9 The blots were developed with NBT/BCIP (1:1) (Ghosh et al., 2014).
Statistical analysis
Experiments were performed in triplicate and analyzed using SPSS.20 software (SPSS, Inc., USA). Statistical analyses were performed by one-way analysis of variance with least significant difference post-hoc tests to identify differences between mean values of different groups. Results are expressed as mean ± standard error. A p < 0.05 was considered significant.
RESULTS
NQ characterization
The surface morphology of the NQ was studied under SEM. The images show spherically shaped NPs with a smooth surface with no pinholes or cracks. The DLS data show that mean NQ diameter was 113.7 nm, the polydispersity index was 0.21, and the zeta potential was −17.4 mV (Figs. 1A–1C).
Fig. 1.
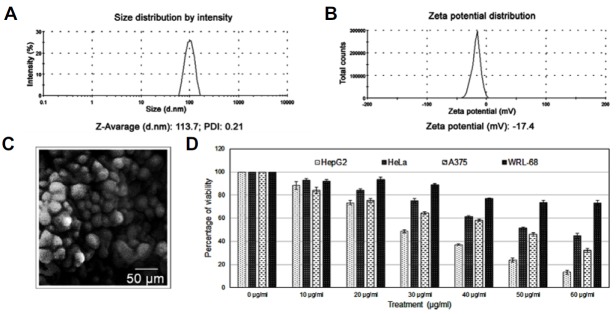
Particle size and charge: (A) Average particle size obtained from the nanoparticle DLS data. (B) Zeta potential of PLGA encapsulated NQ. (C) NQ structure by SEM (D) Percentage cell viability: 10–60 μg/ml NQ was added to different cancer cell line cultures for 24 h. Cell viability was determined by the MTT assay, and the graph shows a gradual decrease in cancer cell viability. The LD50 values of HepG2, HeLa, and A375 cells were 24.1 ± 1.6, 53.1 ± 1.8, and 44.2 ± 0.7 μg/ml, respectively. The same drug concentrations were added to normal cells, such as WRL-68 and PMBCs. The viability curves indicate that cells death was less side than that in the cancer cells. The LD50 value of WRL68 cells was 75.1 ± 1.4 μg/ml.
NQ was more toxic to cancer cells than to normal cells
Selecting a specific cancer model for therapeutic use is necessary to study new drugs. In this experiment, three cancer cell lines of different origins were chosen to determine whether NQ was specific. Thus, a 24-h dose-dependent (0–60 μg/ml) study using HepG2, HeLa, and A375 cells was conducted. As results, HepG2 cell viability decreased from 100% to 13.3 ± 1.8%, that for HeLa cells decreased from 100% to 44.6 ± 2.1%, and that for A375 cells decreased from 100% to 32.0 ± 1.7% (Fig. 1D). As NQ was more toxic to HepG2 cells, all subsequent experiments were conducted using HepG2 cells.
WRL-68 cells were treated with NQ to evaluate toxicity to normal hepatocytes. As a result, cell viability decreased but to a much lesser degree than that in the cancer cell lines (Fig. 1D).
NQ interacts with DNA
Entry of a drug into cells and its binding capacity with DNA are two major ways to generate cytotoxicity. Fluorescence microscopy images indicated that NQ reached a maximum level inside the cells after a 3 h incubation (Fig. 2A). The drug also interacted with DNA. The circular dichroism (CD) spectroscopic results revealed that NQ disturbed the native B-confirmation of DNA in HepG2 cells, with a gradually decreasing negative signal and positive hypo-chromism signals with a peak shift. Such changes were likely the result of structural alterations in the double helical DNA structure induced by NQ (Fig. 2B).
Fig. 2.
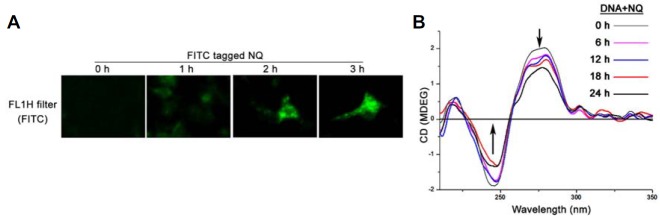
(A) Bio-availability of NQ in HepG2 cells: The cells were incubated with 24 μg/ml NQ for 1 to 3 h. The cells were visualized under fluorescence microscope at the indicated time intervals. Greenish cell fluorescence indicates the presence of NQ inside the cell. (B) Drug-DNA interaction: 24 μg/ml NQ was added to the HepG2 cell cultures. DNA from the cells was isolated at different time intervals, and the interaction between the drug and DNA was studied by CD spectroscopy. The interaction was more intense after a 12 h incubation with NQ.
NQ reduces cancer cell proliferation
A proliferation assay was performed to determine the anti-proliferative action of NQ. The results revealed that the growth rates of cancer cells treated with NQ decreased compared to that of the control (Fig. 3A). We confirmed Akt and pAkt expression by Western blotting. The results showed that expression of the activated form of Akt- pAktSer473 was downregulated in a time-dependent manner after NQ treatment (Fig. 3B). This result indicates that NQ impacted the Akt regulatory domain; thus, reducing survivability and proliferative potential of HepG2 cells.
Fig. 3.
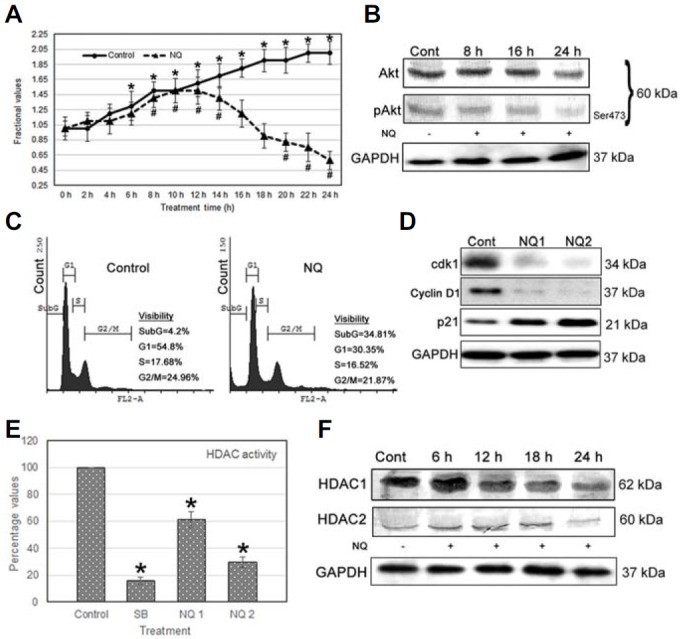
(A) Proliferation assay: A comparative population growth curve was prepared between untreated control and NQ-treated cells. NQ (24 μg/ml) was administered and growth of HepG2 cells decreased after 12 h. (B) Expression of proteins in relation with growth: pAkt expression decreased in HepG2 cells incubated with 24 μg/ml NQ but not that of total Akt. (C) Cell cycle analysis: 24 μg/ml NQ was added to HepG2 cells for 24 h, and the cell cycle was assessed. An increase in the sub-G cell population and a reduction in the S-phase population were observed, indicating reduced DNA synthesis after NQ treatment. (D) Cdk1, cyclin D1 and p21WAF activity: cdk1 expression decreased and p21WAF expression increased after a 24 h incubation with NQ. (E) Histone deacetylase (HDAC) assay: HDAC enzyme activity decreased following NQ treatment. Sodium butyrate, which is a known HDAC suppressor, was used as a positive control. (F) Expression of proteins related to growth and acetylation: HDAC 1 and 2 as well as Akt/pAkt expression decreased gradually in HepG2 cells after incubation with 24 μg/ml NQ. [NQ1 = 12 μg/ml and NQ2 = 24 μg/ml].
NQ arrests the cell cycle
Decreased histone deacetylation activity and cell cycle arrest are two simultaneous processes. NQ reduced the S-phase cell population and increased the sub G-phase population, indicating induction of apoptosis (Fig. 3C). In addition, expression of p21WAF1, a cell-cycle protein, increased, whereas that of cdk1 and cyclin D1 decreased, suggesting that these proteins were associated with inducing cell cycle arrest (Fig. 3D) in the NQ-treated cells.
NQ acts as an HDAC inhibitor
We found that NQ regulated expression of p21waf1 and other cell cycle regulators; thus, we were interested in determining whether NQ could reduce HDAC activities. We measured total HDAC activity and the expression levels of HDAC 1 and 2, which are regulators of deacetylase activity. The results show that total HDAC activity was suppressed after a 24 h NQ treatment (8 mM sodium butyrate was used as an HDAC inhibitor) (Fig. 3E), and that HDAC1 and HDAC2 expression decreased after NQ treatment. HDAC1 expression decreased significantly after 12 h, whereas that of HDAC2 decreased at 24 h of NQ treatment (Fig. 3F). These results indicate that NQ suppressed HDAC action, demonstrating that NQ is a repressor of transcription and proliferation.
NQ initiates rac-1-related ROS activity to induce cancer cell apoptosis
To understand the molecular mechanism behind cell death, we determined ROS activity in NQ-treated cells using flow cytometry. The results showed that ROS were generated in HepG2 cells after NQ treatment (Fig. 4A). p53 silencing suppressed ROS generation (Fig. 4B). ROS scavenger treatment with NQ increased cell viability (Fig. 4C). This result confirms that ROS generation following NQ treatment was the major event controlling the cell death pathway and generation of ROS was not controlled by p53. So to understand the mechanism behind the generation of ROS, we determined rac-1 activity and found that rac-1 activity increased early in the treatment and then returned to the baseline level (Fig. 4D). The accumulation of ROS increased after rac-1 was activated and increased further later in the treatment (Fig. 4E). These results suggest that rac-1 initiates the increase of ROS in cancer cells, which returned to the basal level later.
Fig. 4.
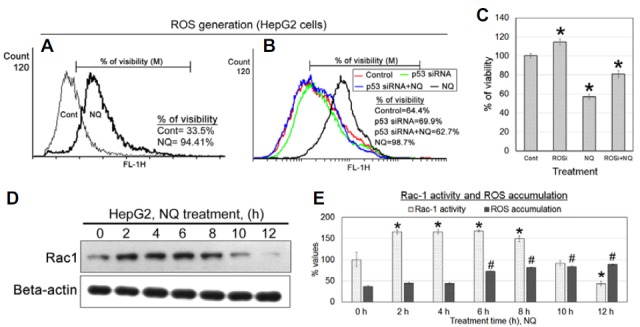
ROS generation: (A) 24 μg/ml NQ was added to HepG2 cells, and ROS accumulation was evaluated by flow cytometry. The results indicated increased ROS accumulation in cancer cells after NQ treatment. (B) NQ (24 μg/ml) was added to HepG2 cells, and ROS generation was negligible when NQ was added to p53-silenced HepG2 cells, but ROS were generated in native p53 expressing HepG2 cells. (C) Treatment with ROS scavenger with NQ increases the viability of the cells. (D) Rac1 activity was measured using active pull down assay. Rac1 activity was increased at earlier hr, but decreased further at late intervals. (E) ROS accumulation and rac1 activity were compered during different intervals of treatment.
Effect of ROS on p53 activity and apoptosis
Wild-type p53 expressing HepG2 cells, ascorbic acid (AA)-treated (50 μM)-HepG2 cells, and p53-siRNA-transfected HepG2 cells were treated with NQ to determine the dependency of ROS on p53 activity. The results showed that the AA-treated cells had a low or basal level of p53 expression compared to that of NQ-treated cells (Fig. 5A). In parallel, silencing p53 also suppressed ROS activity but greatly increased cancer cell viability and reduced apoptosis. Treatment with ZDVD-FMK (caspase 3 inhibitor) with NQ significantly nullified the effect of NQ (Figs. 5B–5E) in respect of cellular viability. Taken together, we propose that NQ accelerates rec-1 activity in HepG2 cells, leading to increased ROS production, which further controls cell death.
Fig. 5.
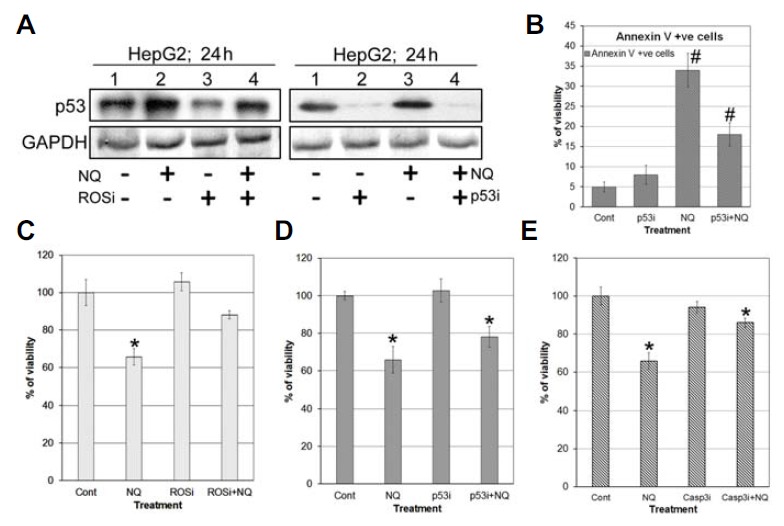
p53 and apoptosis: (A) Western blot of p53 using ROS scavenger. (B) Annaxin V assay to determine the effect of silencing of p53 on apoptosis. We found that apoptosis percentage was gone down in p53 silenced cells devoid of NQ treatment. (C–E) Viability assay using ROSi (ascorbic acid), p53i (p53 siRNA), Casp3i (ZDEVD-FMK) with NQ.
NQ depolarizes the mitochondrial outer membrane
Increased ROS generation and mitochondrial membrane depolarization are two integrative processes involved in inducing apoptosis via the intrinsic pathway. We treated HepG2 cells with NQ with or without AA and then stained the cells with rhodamine 123. The results showed that the mitochondrial membrane was depolarized following NQ treatment (Figs. 6A–6B) and that the AA pre-treatment inhibited this process (Fig. 6C). We also found that Bax was translocated to the mitochondrial outer membrane after NQ treatment, which initiated the apoptosis stimulus and contributed to the formation of mitochondrial outer membrane pores, loss of membrane potential, and depolarization. Cytochrome c was also released into the cytosol (Figs. 6D–6E). The NQ-mediated damage in the mitochondrial network and release of cytochrome c into the cytosol acted as a catalyst to induce apoptosis in HepG2 cells.
Fig. 6.
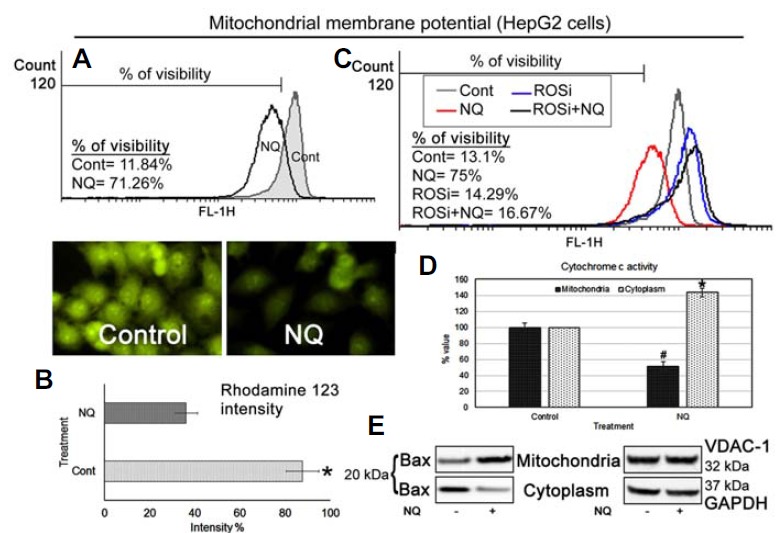
Mitochondrial membrane potential: (A–B) Flow cytometric and fluorescence images indicate depolarization of the MMP after HepG2 cells were treated with 24 μg/ml NQ. (C) Flow cytometric analysis of MMP using ROS scavenger (ROSi) (D) Cytochrome c activity: ELISA for cytochrome c in the HepG2 cell mitochondrial and cytosolic fractions after treatment with 24 μg/ml NQ indicated decreased cytochrome c activity in the mitochondrial fraction but increased activity in the cytosolic fraction. (E) Bax activity: Bax expression decreased in the cytoplasmic fraction but increased in the mitochondrial fraction when HepG2 cells were treated with 24 μg/ml NQ.
NQ induces apoptosis by activating caspase 3
Activating apoptosis requires suppressing anti-apoptotic proteins and activating pro-apoptotic proteins, which activate caspase 3 to induce apoptosis. We investigated the cellular morphological changes and DNA damage induced by NQ. NQ induced the formation of blebs and rounding off of cellular structures. DAPI staining indicated characteristic nuclear swelling and chromatin condensation, and the terminal deoxynucleotidyl transferase dUTP nick end labeling assay confirmed DNA damage after NQ treatment (Fig. 7A). We used immunoblotting to determine the role of different proteins related to apoptosis. The overall results showed decreased expression of Mcl-1, Bcl-2, and Bcl-xL and increased expression of apaf1, caspase 9, and caspase 3, as well as formation of cleaved poly(ADP-ribose) polymerase (PARP) when HepG2 cells were incubated with NQ for 24 h (Fig. 7D). Taken together, these results suggest that NQ generates DNA damage and induces caspase 3-mediated apoptosis in HepG2 cells.
Fig. 7.
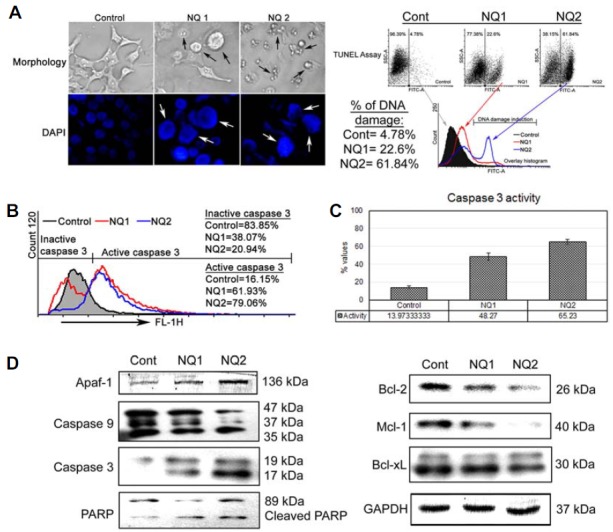
Apoptosis analysis: (A) morphology, DAPI staining, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay: NQ (12 [NQ1] and 24 [NQ2] μg/ml) was added to HepG2 cells for 24 h and morphological changes were photographed. DAPI staining indicated swelling and nick formation in nuclei after NQ treatment. The TUNEL assay indicated DNA damage in NQ-treated cells. The damage was more extensive and prominent at higher NQ doses. (B) Caspase 3 assay by flow cytometry: The same drug doses were used for 24 h, and caspase 3 activity was assayed in HepG2 cells. The results showed increased caspase 3 activity in a dose-dependent manner. (C) Spectophotometric caspase 3 assay: Caspase 3 activity increased in a dose-dependent manner in NQ-treated cells compared with those without NQ. (D) Western blotting: The same drug doses were used to study on different pro- and anti-apoptotic proteins in HepG2 cells.
DISCUSSION
Uses for NPs as carrier systems for chemotherapeutic drugs are gaining attention due to the specificity of NPs for cancer cells, enhanced efficiency, and reduced systemic toxicity (Lee and Odom, 2015). AuNPs have several advantages, including a bio-compatible core, which makes them an ideal starting point for a nano-carrier system (Jain et al., 2012). Moreover, AuNPs undergo functionalization of multiple surfaces, which renders them highly versatile for targeting. However, some researchers have pointed out disadvantages of AuNPs, such as high toxicity (Arvizo et al., 2010); thus, reducing undesirable toxicity and improving sustained release ability and bio-availability of drugs are important considerations. Here, we encapsulated reduced AuNPs with quercetin and PLGA for the first time. PLGA encapsulation or PEG tagging protects the compound inside from attack by immune cells, such as macrophages or other phagocytes. This study was designed to evaluate the molecular mechanism of the cancer-killing effect of this new drug. In brief, we found that NQ showed remarkable anticancer effects and toxicity towards cancer cells, and more effectively towards HepG2 cells than other cancer cell types, such as HeLa and A375 cells (Fig. 1).
Ligand/DNA targeted therapy was developed recently (Ajazuddin Saraf, 2010; Kumari et al., 2010), in which DNA acts as a large receptor for vast numbers of small molecules and becomes a major target for designing anti-cancer agents (Shahabuddin et al., 2009). CD spectroscopic data support that NQ has the potential to interact with DNA. Thus, NQ cytotoxicity could be explained as inhibition of the normal DNA synthetic process at some level. The static blocking capacity of NQ made it possible to induce apoptosis in HepG2 cells, and intercalating NQ to nuclear DNA also induced nuclear damage. The CD-spectroscopic data revealed that the interaction between NQ and cellular DNA was more pronounced 12 h after NQ application (Fig. 2). The interaction with DNA increased the capability of the drug to reduce cell proliferation and induce cell cycle arrest (Shobha Devi et al., 2013). In contrast, increased p21WAF1 levels control cell differentiation, and the cell cycle is associated with increased p21WAF1 expression (Huang et al., 2005). In addition, p21WAF1, as a cyclin-dependent kinase (CDK1) inhibitor, influences G1 progression in both positive and negative ways (Kim et al., 2012). Thus, the cellular level of p21WAF may be an important factor determining whether the cell differentiates or undergoes apoptosis. For example, lower levels of p21WAF can cause cells to differentiate and higher levels may direct the cell into an apoptotic pathway (Harper et al., 1993).
Histone deacetylase inhibitors (HDIs) have been developed as a cancer treatment or adjunct (Falkenberg and Johnstone, 2014). The exact mechanisms by which these compounds work are unclear, but epigenetic pathways have been proposed. HDIs induce p21WAF1 expression, which is a p53 tumor suppressor activity regulator. As HDIs modulate p21 and p53 protein expression, they induce growth arrest, differentiation, and/or apoptotic death in a wide variety of cultured transformed cells, including neuroblastoma, melanoma, and leukemia cells, as well as in cells from breast, prostate, lung, ovary, liver, and colon cancers. For example, suberoylanilide hydroxamic acid induces terminal cell differentiation in several cell lines, as evaluated by morphological and cell cycle arrest experiments (Marks et al., 2000). HDIs induce apoptotic cell death in human cell lines, and apoptosis has been assayed by DNA fragmentation analysis, and deletion of the sub-G1 (< 2N ploidy) population has been detected by flow cytometry (Duan et al., 2005). Overexpression of HDAC1 and HDAC2, which are key histone deacetylation regulators, is implicated in cancer, partly through aberrant recruitment and consequent silencing of the tumor suppressor genes p21WAF1 and p53, which are associated with histone acetylation at the promoter region, and can be reversed by HDI treatment (Hagelkruys et al., 2011).
NQ possesses a similar ability to induce ROS as quercetin. Quercetin has efficacy for inducing apoptosis in p53-expressing HeLa and p53-null prostate cancer PC3 cells (Bishayee et al., 2013; Vijayababu et al., 2006) by increasing ROS. The mechanism behind the increase in ROS is increased rec-1 activity (Hanahan and Weinberg, 2011). rac-1 and ROS are correlated, but higher rac-1 activity has a negative effect on tumor suppressor genes, such as p53, and a positive effect on proliferative proteins, such as cyclin D1, which is why elevated rec-1 activity occurs in different cancer cells. Earlier study shows that the effect of rec-1 could be nullified by the Hace-1 enzyme under stress and p53 activated conditions (Daugaard et al., 2013). We also found that rec-1 activity decreased after a certain treatment period, which may have occurred due to activation of p53 and increased ROS beyond a specific threshold level.
Elevated ROS and p53 activation are simultaneous processes. ROS-dependent apoptosis involving p53 generally depends on damage to the mitochondrial membrane. Two studies (Karbowski and Youle, 2003; Yu et al., 2006) have described dramatic alterations in mitochondrial morphology during the early stages of apoptotic cell death, including network fragmentation and remodeling of cristae. Surprisingly, proteins that control mitochondrial morphology also appear to participate in apoptosis, and proteins associated with regulating apoptosis affect mitochondrial ultrastructure. Translocation of Bax to the mitochondrial membrane creates pores, which depolarize the mitochondrial membrane potential. Then, cytochrome c moves into the cytosol and initiates formation of apoptosomes along with the adopter molecule Apaf-1 and other pro-caspase molecules, such as caspase-9 (Fulda and Debatin, 2006).
We hypothesize that NQ downregulates HDACs and p21, which may be correlated with cell cycle arrest. The decrease in Cdk1 and pAkt may have resulted in arrest of HepG2 cell proliferation. The flow cytometry analysis demonstrated that NQ arrested the HepG2 cell cycle in the sub-G phase and decreased cell populations in the synthetic and mitotic phases. As most anti-neoplastic drugs in clinical use block the cell cycle in the S or G2/M phases, whereas NQ blocked the cell cycle in the sub-G phase, a combination of NQ with currently used drugs could improve the efficacy of current hepatocarcinoma therapies (Fig. 8). In general, NQ treatment induced ROS and mitochondrial membrane depolarization, and the release of cytochrome c induced apoptosis in HepG2 cells by activating caspase 3. Thus, ROS and p53 shared a combined role.
Fig. 8.
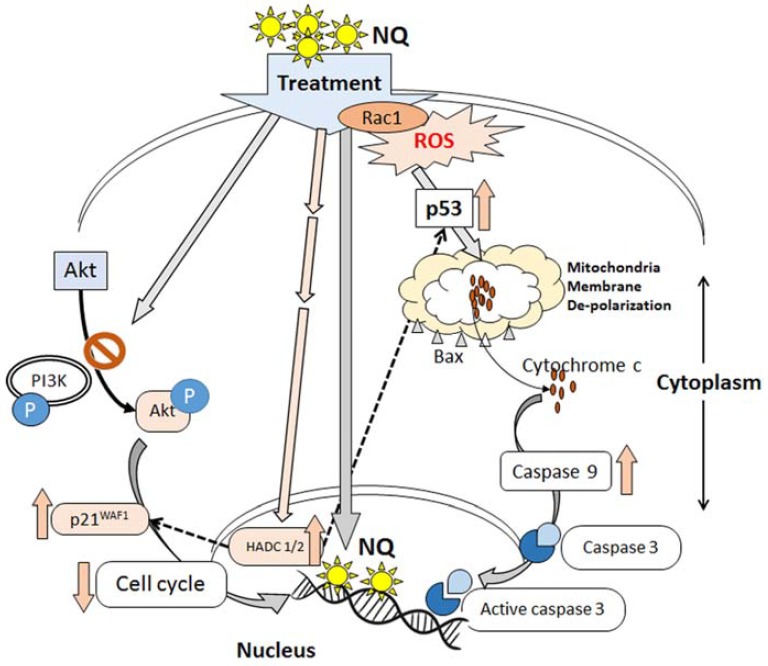
Schematic representation of the action of the nano-particles on HepG2 cells
Acknowledgments
This study was financially supported, in part, by the UGC, New Delhi, Govt. of India though the Emeritus Fellowship to ARK-B and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2009-0094071). Sincere thanks are due to Professor Tarak Das Basu of the Department of Biochemistry and Biophysics, University of Kalyani for allowing us to use the FACS and DLS machines.
REFERENCES
- Ajazuddin, Saraf S. Applications of novel drug delivery system for herbal formulations. Fitoterapia. 2010;81:680–689. doi: 10.1016/j.fitote.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Arvizo R, Bhattacharya R, Mukherjee P. Gold nanoparticles: opportunities and challenges in nanomedicine. Expert Opin. Drug Deliv. 2010;7:753–763. doi: 10.1517/17425241003777010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadoriya SS, Mangal A, Madoriya N, Dixit P. Bioavailability and bioactivity enhancement of herbal drugs by “Nanotechnology”: a review. J. Curr. Pharmaceutical. Res. 2011;8:1–7. [Google Scholar]
- Bhattacharyya SS, Paul S, De A, Das D, Samadder A, Boujedaini N, Khuda-Bukhsh AR. Poly (lactide-co-glycolide) acid nanoencapsulation of a synthetic coumarin: Cytotoxicity and bio-distribution in mice, in cancer cell line and interaction with calf thymus DNA as target. Toxicol. Appl. Pharmacol. 2011;253:270–282. doi: 10.1016/j.taap.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Bishayee K, Ghosh S, Mukherjee A, Sadhukhan R, Mondal J, Khuda-Bukhsh AR. Quercetin induces cytochrome c release and ROS accumulation to promote apoptosis and arrest the cell cycle in G2/M, in cervical carcinoma: signal cascade and drug–DNA interaction. Cell Prolif. 2013a;46:153–163. doi: 10.1111/cpr.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishayee K, Paul A, Ghosh S, Sikdar S, Mukherjee A, Biswas R, Boujedaini N, Khuda-Bukhsh AR. Condurango-glycoside-A fraction of Gonolobus condurango induces DNA damage associated senescence and apoptosis via ROS-dependent p53 signalling pathway in HeLa cells. Mol. Cell. Biochem. 2013b;382:173–183. doi: 10.1007/s11010-013-1732-5. [DOI] [PubMed] [Google Scholar]
- Brannon-Peppas L, Blanchette OJ. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S, Castillo-Lluva S, Rotblat B, Li L, Malliri A, et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat. Commun. 2013;4:2180. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Bishayee K, Khuda-Bukhsh AR. graveoline isolated from ethanolic extract of ruta graveolens triggers apoptosis and autophagy in skin melanoma cells: a novel apoptosis-independent autophagic signaling pathway. Phytother. Res. 2014;28:1153–1162. doi: 10.1002/ptr.5107. [DOI] [PubMed] [Google Scholar]
- Hagelkruys A, Sawicka A, Rennmayr M, Sei C. The biology of HDAC in cancer: the nuclear and epigenetic components. Handb. Exp. Pharmacol. 2011;206:13–37. doi: 10.1007/978-3-642-21631-2_2. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Havsteen B. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002;6:67–202. doi: 10.1016/s0163-7258(02)00298-x. [DOI] [PubMed] [Google Scholar]
- Huang BH, Laban M, Leung CH, Lee L, Lee CK, Salto-Tellez M. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404. doi: 10.1038/sj.cdd.4401567. [DOI] [PubMed] [Google Scholar]
- Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- Jain S, Hirst DG, O’Sullivan JM. Gold nanoparticles as novel agents for cancer therapy. Br. J. Radiol. 2012;85:101–113. doi: 10.1259/bjr/59448833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Sun Y, Yang H, Kim NH, Jeon SH, Huh SO. Involvement of the cAMP response element binding protein, CREB, and cyclin D1 in LPA-induced proliferation of P19 embryonic carcinoma cells. Mol. Cells. 2012;34:323–328. doi: 10.1007/s10059-012-0163-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloid. Surfaces. B. 2010;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Lee H, Odom TW. Controlling ligand density on nanoparticles as a means to enhance biological activity. Nanomedicine. 2015;10:177–180. doi: 10.2217/nnm.14.204. [DOI] [PubMed] [Google Scholar]
- Lemarie A, Grimm S. Mitochondrial respiratory chain complexes: apoptosis sensors mutated in cancer. Oncogene. 2011;300:3985–4003. doi: 10.1038/onc.2011.167. [DOI] [PubMed] [Google Scholar]
- Liu S. Epigenetics advancing personalied nanomedicine in cancer therapy. Adv. Drug Deliv. Rev. 2012;64:1532–1543. doi: 10.1016/j.addr.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo DT. TUNEL Assay: An Overview of Techniques. In: Didenko VV, editor. In in situ detection of DNA damage: methods and protocol. Spinger; 2002. pp. 313–323. [Google Scholar]
- Maitland ML, Schilsky RL. Clinical trials in the era of personalized oncology. CA Cancer J. Clin. 2011;61:365–381. doi: 10.3322/caac.20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Pulkrabek DJ, Dong Z, Venkatachalam MA, Saikumar P. Bcl-2 prevents Bax oligomerization in the mitochondrial outer membrane. J. Biol. Chem. 2001;276:18361–18374. doi: 10.1074/jbc.M100655200. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ, Xie HJ. Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcinoma cells by deregulating expression of G1/S cell cycle proteins. PLoS One. 2011;6:e28103. doi: 10.1371/journal.pone.0028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhl AC, Fagundes M, dos Santos KC, Polikarpov I, das Graças MF, da Silva F, Fernandes JB, Vieira PC, Forim MR. Preparation and characterization of polymeric nanoparticles loaded with the flavonoid luteolin, by using factorial design. Int. J. Drug Deliv. 2011;3:683–698. [Google Scholar]
- Shahabuddin MS, Gopal M, Raghavan SC. Intercalating cytotoxic, anti-tumour activity of 8-chloro and 4-morp96holinopyrimido [4′,5′:4.5]thieno(2,3-b) quinolone. J. Photochem. Photobiol. B. 2009;94:13–19. doi: 10.1016/j.jphotobiol.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Shahbazian D, Sznol J, Kluger HM. Vertical pathway targeting in cancer therapy. Adv. Pharmacol. 2012;65:1–26. doi: 10.1016/B978-0-12-397927-8.00001-4. [DOI] [PubMed] [Google Scholar]
- Shobha Devi C, Anil Kumar D, Singh SS, Gabra N, Deepika N, Kumar YP, Satyanarayana S. Synthesis, interaction with DNA, cytotoxicity, cell cycle arrest and apoptotic inducing properties of ruthenium(II) molecular “light switch” complexes. Eur. J. Med. Chem. 2013;64:410–421. doi: 10.1016/j.ejmech.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Talluri S, Dick FA. Regulation of transcription and chromatin structure by pRB. Cell Cycle. 2012;11:3189–3198. doi: 10.4161/cc.21263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayababu MR, Kanagaraj P, Arunkumar A, Ilangovan R, Dharmarajan A, Arunakaran J. Quercetin induces p53-independent apoptosis in human prostate cancer cells by modulating Bcl-2-related proteins: a possible mediation by IGFBP-3. Oncol. Res. 2006;16:67–74. doi: 10.3727/000000006783981224. [DOI] [PubMed] [Google Scholar]
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]