

Abstract
The aim of our study was to determine the role of dystrophin hydrophobic regions in the pathogenesis of Duchenne (DMD) and Becker (BMD) muscular dystrophies, by the Kyte-Doolittle scale mean hydrophobicity profile and 3D molecular models. A total of 1038 cases diagnosed with DMD or BMD with the in-frame mutation were collected in our hospital and the Leiden DMD information database in the period 2002-2013. Correlation between clinical types and genotypes were determined on the basis of these two sources. In addition, the Kyte-Doolittle scale mean hydrophobicity of dystrophin was analyzed using BioEdit software and the models of the hydrophobic domains of dystrophin were constructed. The presence of four hydrophobic regions is confirmed. They include the calponin homology CH2 domain on the actin-binding domain (ABD), spectrin-type repeat 16, hinge III and the EF Hand domain. The severe symptoms of DMD usually develop as a result of the mutational disruption in the hydrophobic regions I, II and IV of dystrophin – those that bind associated proteins of the dystrophin-glycoprotein complex (DGC). On the other hand, when the hydrophobic region III is deleted, the connection of the ordered repeat domains of the central rod domain remains intact, resulting in the less severe clinical presentation. We conclude that mutational changes in the structure of hydrophobic regions of dystrophin play an important role in the pathogenesis of DMD.
KEYWORDS: DMD, BMD, dystrophin, Kyte-Doolittle scale mean hydrophobicity profile, 3D model, genotype–phenotype analysis
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked, recessively inherited disease, typically characterized by a progressive skeletal muscle atrophy of proximate extremities and pseudohypertrophy of the gastrocnemius muscle [1]. The onset of the disease usually occurs between the age of 3 and 5; affected individuals gradually lose their ability to walk usually at the age of 13-15 years. Most of the patients die around the age of 20 years due to respiratory failure or/and cardiac insufficiency [2-4]. DMD is caused by a genetic mutation in the X chromosome resulting in the absence of protein dystrophin on the membranes of muscle cells [5]. The clinical presentation of Becker muscular dystrophy (BMD) is similar to, but less severe than, that of DMD. Patients with BMD can maintain their ability to walk even after the age of 15 years. While DMD is considered to be a fatal genetic disorder, BMD allows for a comparatively longer life span and better quality of life [6-9].
Despite these differences in clinical severity, DMD and BMD are caused by a mutation of the same dystrophin gene (also known as the “DMD gene”), which is located in the Xp21.1 region [10]. Dystrophin is a large cytoskeletal protein, with a molecular mass of 427 kDa and is composed of an N-terminal region, a central rod domain, and a C-terminal region [11]. The N-terminal region of dystrophin binds actin, while the large central rod domain comprises 24 spectrin-like repeats, 4 hinges, and acts as another binding site for actin as well as a binding site for neuronal nitric oxide synthase (nNOS). The C-terminal region, on the other hand, binds dystrobrevins, syntrophins and β-dystroglycan, a transmembrane glycoprotein that interacts with sarcoglycans and α-dystroglycan. Furthermore, α-dystroglycan interacts with laminin-2. The membrane-spanning dystrophin glycoprotein complex (DGC) thus acts as an indirect linkage between the actin-based cytoskeleton and the extracellular matrix [12-13]. The DMD gene is the largest human gene, containing 79 exons. Many mutations in dystrophin have been found: 65% of them are deletions, 7% to 10% are duplications while 25% to 30% are point mutations [14-17]. Monaco et al. [18] proposed that if dystrophin gene mutation interrupts the reading frame, the expression of dystrophin is severely affected typically resulting in severe DMD symptoms; if the reading frame is not interrupted by the mutation, then dystrophin is only partially produced, having as an outcome the clinical presentation of BMD. According to the “reading-frame rule” out-of-frame mutations cause DMD while an in-frame mutations result in BMD [19-20]. This rule is used to explain the relationship between genotype and phenotype in the pathogenesis of DMD [21]. However, Hassan et al. found that 22.1% of patients with an in-frame mutation are presented as having DMD, which is hard to explain by the reading-frame rule only [22]. This finding suggests that the skipped structured could be important for dystrophin function.
For the large proteins, the 3D structure prediction remains a difficult and unresolved endeavor. Compared to ab initio prediction and fold recognition, a homology modeling - prediction based on the reasonable assumption that two homologous proteins will share very similar structures, is most accurate when the target and template have similar sequences. Dystrophin is a huge protein in humans, whose mapping is extremely challenging process. We used a Swiss model, a homology prediction method for remodeling parts of dystrophin. The results of the Swiss model are named batch. Each batch corresponds to one part of dystrophin structure.
The aim of our study is to identify important functional domains of dystrophin by investigating the Kyte-Doolittle mean scale hydrophobicity profile [23-24], and to model and analyze their 3D structures as well as their functions. These analyses suggested the relationship between the deletions of these regions to the mechanism of DMD/BMD pathogenesis. In this way, we have provided detailed explanations for the exceptions to the reading-frame rule.
MATERIALS AND METHODS
Case Resources
A total of 1038 in-frame deletion mutation cases diagnosed with DMD or BMD were collected in the period from 2002 to 2013. 538 cases were collected in The First Affiliated Hospital of Sun Yat-sen University, while 500 cases were obtained from an open-access internet database - Leiden Muscular Dystrophy pages (http://www.dmd.nl) [25-26]. Multiplex probe ligation-dependent amplification (MLPA) was used for genetic testing in patients. Assessment of the reading-frame was performed employing a Reading-frame Checker (DMD Exonic Deletions/Duplications Reading-Frame Checker 1.9). The diagnostic criteria for DMD and BMD used in our research were developed by the European Neuromuscular Center [27]. The main discriminating criterion between DMD and BMD was the onset of wheelchair dependency (younger than thirteen years in DMD and older than sixteen years in BMD). The onset of wheelchair dependency between twelve and sixteen years was classified as an “intermediate phenotype” and was considered as an exclusion criterion in our study.
Genotype-phenotype analysis
The MLPA test result was regarded as a genotype, while the clinical diagnosis (DMD/BMD) was considered as a phenotype. The genotypes and clinical phenotypes of 1038 in-frame deletion mutation cases were analyzed, and the numbers and percentages of cases that did not meet the reading-frame rule were calculated.
Kyte-Doolittle scale mean hydrophobicity profile analysis
The amino acid sequence of dystrophin (NCBI Reference Sequence: NP_003997.1) was obtained from GenBank (http://www.ncbi.nlm.nih.gov/genbank) and imported into BioEdit software 7.0.1 (Tom Hall Ibis Biosciences, An Abbott company, Carlsbad, CA) [28] in order to conduct a Kyte-Doolittle scale mean hydrophobicity profile analysis. After selecting the sequence→Protein→Kyte-Doolittle scale mean hydrophobicity profile analysis menus, the window size was set at 56 on the open window, the program was executed using Run Plot, and the hydrophobic peak values were read on the result figure. We compared the involvement of hydrophobic regions with their relationship with clinical types.
Modeling the 3D structure of dystrophin
The three-dimensional structure of dystrophin was modeled by importing the dystrophin amino acid sequence to Swiss-model Automatic Modeling Mode [29] (http://swissmodel.expays.org). This modeling system can turn amino acid sequences into 3D homology structures automatically. 3D models can be downloaded from My workspace on Swiss-model web server.
3D model analysis using RasMol software
The second structure information of 3D model and their corresponding functions were analyzed using RasMol software 2.7.2 (Biomolecular Structures Group, Glaxo Wellcome Research & Development, Stevenage, Hertfordshire, UK)[30]. The 3D models’ demonstration display and color allocation were set up by commands Structure and Ribbons respectively. Afterwards, we checked the secondary structural information in the Rasmol command line window and manually chose the hydrophobic regions to appear as green.
Statistical Method
SPSS V13.0 software (SPSS, Chicago, IL, USA) was employed to conduct statistical analyses. The two-sided chi-squared test was used to compare the difference between the percentages of genotypes or phenotype of DMD and BMD. The p < 0.05 was considered significantly different.
RESULTS
The genotypes and phenotypes of 1038 cases with in-frame deletion
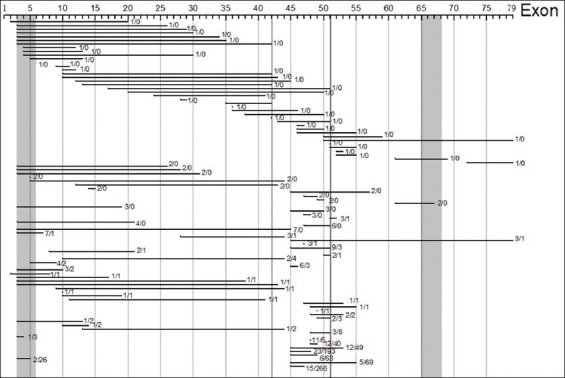
Out of the 1038 analyzed in-frame dystrophin mutation cases, 242 (or 23.3%) showed DMD pattern, not meeting thus the criteria of the reading-frame rule. On the other hand, other 796 cases (76.7%) of the in-frame dystrophin mutations presented with BMD and were hence consistent with the reading-frame rule. These 1038 cases displayed over 93 distinct types of deletions (Figure 1).
Figure 1.
The genotypes -phenotypes of 1038 cases with in-frame deletion The in-frame mutations of the 1038 analyzed patients include 93 genetically distinct mutations (horizontal lines). The numbers of cases of DMD/BMD for each of these distinct mutations are indicated on the right side of each horizontal line. 242 (23.3%) of 1038 in-frame mutation cases present symptoms of BMD, and hence meet the reading-frame rule. The number at the end of each horizontal bar is the number of cases with DMD versus BMD. The black and grey bars (from left to right) are the hydrophobic regions I-IV respectively.
Kyte-Doolittle Scale Mean Hydrophobicity Profile Analysis
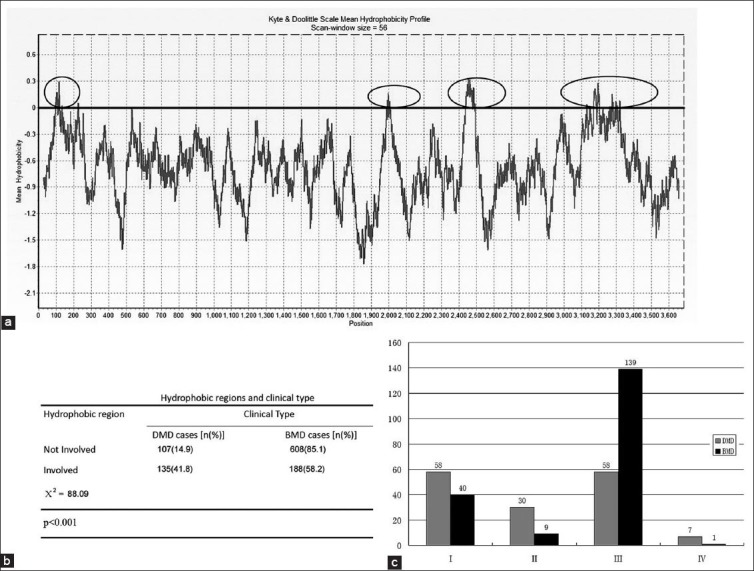
The Kyte-Doolittle plot showed that dystrophin had four hydrophobic regions. These regions encompass residues 60-132 (forming greatest part of the actin-binding domain of dystrophin), 1990-2010 (forming part of the central rod domain), 2450-2500 (forming part of the central rod domain) and 3150-3300 forming the greatest part of the cysteine-rich domain, and were coded by exons 3-6, 42, 51, and 65-68, respectively. Their corresponding peak hydrophobicity values were: 0.3, 0.15, 0.34 and 0.29. The third hydrophobic region had the highest peak value (Figure 2A).
Figure 2.
Kyte & Doolittle Scale Mean Hydrophobicity Profile Analysis (A) Kyte & Doolittle Scale Mean Hydrophobicity Profile plot. Four hydrophobic regions are located at positions 60-132, 1990-2010, 2450-2500 and 3150-3300 of the dystrophin amino acid sequence (shown in the plot as marked with gray oval circles). Their corresponding peak values are 0.3, 0.15, 0.34 and 0.29. The third hydrophobic region has the highest peak value. (B) Involved hydrophobic regions and clinical type. A total of 715 cases (68.8%) have intact hydrophobic regions and there are 107/608 cases of DMD/BMD in this group. In contrast, when a hydrophobic region is involved in the mutation, the number of DMD/BMD cases is 135/188. The proportion of DMD (cases that do not meet the criteria of the reading-frame rule) in hydrophobic region-involved group (41.8%) is higher than the group in which hydrophobic regions are left intact (14.9%) (Χ2=88.09, p < 0.001). (C) DMD/BMD proportion in 4 hydrophobic regions. BMD cases are more common when hydrophobic region III is involved in the mutation (DMD/BMD=58/139). When any of the other hydrophobic regions are involved, DMD incidences are higher than BMD (DMD/BMD 58/40, 30/9 and 7/1 in hydrophobic regions I, II, IV, respectively)
Hydrophobic regions and phenotypes
Hydrophobic regions were damaged by a gene deletion in 343 (33.0 %) of the 1038 in-frame mutations analyzed (Figure 2B). Among these 343 cases, 41.8%, presented with symptoms of DMD and the remaining 58.2% had BMD phenotype. Out of the 715 in-frame mutations not involving the hydrophobic regions, 107 mutations were associated with DMD (14.9%) and 608 cases with BMD (85.1%). The proportion of DMD in the two groups (those cases that did not meet the criteria of the reading-frame rule) was significantly different (Χ2=88.09, p < 0.001) (Figure 2B).
We have also examined contributions of each of the hydrophobic regions individually. As demonstrated in the Figure 2C, the in-frame deletions involving only hydrophobic region III correspond to the BMD phenotypes more often than they are seen in DMD cases (139 vs. 58). For the mutations involving other hydrophobic regions, DMD incidence is higher than the incidence of BMD (DMD: BMD is 58:40, 30:9 and 7:1 for hydrophobic regions I, II and IV, respectively).
3D Swiss-modeling and analysis of the hydrophobic regions of dystrophin
Homology models of the hydrophobic domains of dystrophin were built by using Swiss-model system. There are four 3D Batches including the whole or part of the hydrophobic regions. They are named as a Batch 1, a Batch 2, a Batch 3, and a Batch 4, respectively. These batches are separately included in the amino acid residues coded as 9-246, 2001-2307, 2471-2801, and 3047-3306, as demonstrated by the predicted 3D model. All of these 3D models were analyzed and displayed using RasMol software.
Batch 1 (Figure 3A) includes two dimers, where each dimer is formed by a total of 2 dystrophin actin-binding domain (ABD) monomers. Each monomer is an extended dumbbell-shaped structure (Figure 3E). The head of the dumbbell is formed by combining the two calponin homology (CH) regions. The whole hydrophobic region I (residues coded as 60-132) is included in Batch 1 (that consists of residues 9-246).
Figure 3.
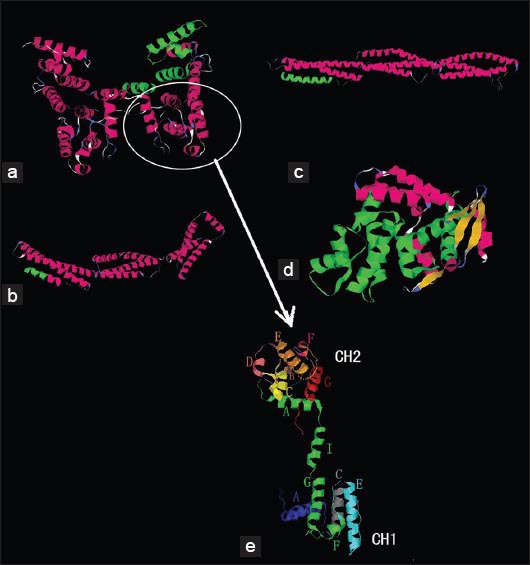
3D model of 4 hydrophobic regions-included amino acid residues (A) Batch 1 includes 2 dimers, which are formed by four dystrophin actin-binding domain (ABD) monomers. Each monomer is an extended dumbbell structure (panel E). Each dumbbell is composed of two calponin homology (CH) domains. Each monomer contains 3 actin-binding structures (ABS). Hydrophobic region I is located on the whole CH2 and the small beginning part of CH1 ABS3 (highlighted in green). (B, C) Batch 2 and 3 respectively. These batches are shown as three-stranded alpha-helical coiled coils. Hydrophobic region II contains (spectrin-type) repeat 16 of dystrophin (green on panel B). Hydrophobic region III includes 50 amino acid residues on the C-terminal part of hinge III (H III) and 30 amino acid residues on the N-terminal part of R20. H III is composed of irregular coils so that batch 3 only shows the 30 amino acids on the N-terminal part of R20 in a helical structure (highlighted in green). (D) Batch 4 is a globular structure that includes a hinge region, cysteine-rich domains on a WW domain and two helix-loop-helix EF hand-shaped structures. Hydrophobic region IV includes an EF hand-shaped region (shown in green). (E) The dystrophin actin-binding domain monomer is a dumbbell-shaped structure. Two calponin homology (CH) regions comprise the heads of the dumbbell. A triple helical bundle at the middle of monomer includes a helical linker (helix I). Seven helices form the N-terminal CH2 region (helical regions A-G). The CH1 region lacks the short B and D helices, which are replaced by a loop and an extra reverse turn on the N-terminal part of helix E. With the esception of the special highlighted parts, all the models were colored using RasMol software’s default coloring scheme for protein secondary structure: Alpha helices are colored in magenta, beta sheets are colored in yellow, turns are colored in pale blue, and all other residues are colored in white.
Batch 2 and Batch 3 (Figure 3B and Figure 3C) are presented as alpha-helical coiled-coil structures formed by folding of three helical peptides. Regions containing 52 helices with 76 turns, 11 helices with 22 turns and irregular coils are combined into these complex batches. Batches 2 and 3 represent most part of repeats (R) 16-18 and R 20-22 of the central rod domain of dystrophin, respectively. Hydrophobic region II is located on R16 (green on Figure 3B). Hydrophobic region III consists of residues 2450-2500 of dystrophin. This region includes 20 amino acid residues on the C-terminal part of the hinge III (H III) and 30 amino acid residues on the N-terminal part of R20. H III consists of irregular coils, so that Batch 3 only shows the 30 amino acid residues on the N-terminal helix of R20 (highlighted in green).
Batch 4 (Figure 3D) is a globular structure that includes one helix-loop-helix EF hand-shaped region (EF hand is a helix-loop-helix structural domain or motif, found in a large family of calcium-binding proteins, shaped like the spread thumb and forefinger of the human hand) [31-32]. Its fold includes 31 helices and 40 turns. A hinge region and the cysteine-rich domains on the WW domain (WW domain is an overlapping part of the central rod domain and the cysteine-rich domain, named after the presence of two conserved tryptophans (W)) [33-34], as well as the EF hand-shaped region, form together Batch 4. The hydrophobic region is located on the EF hand-shaped region (highlighted in green).
DISCUSSION
Dystrophin is a membrane-bound cytoskeleton protein. It has been previously shown that it has four major function regions [35]: an actin-binding domain (ABD), a central rod domain, a cysteine-rich domain and a C-terminal domain. Dystrophin connects to other glycoproteins to form the dystrophin-glycoprotein complex (DGC). In this way, this complex can stabilize the cell membrane and transport signals across the membrane [36]. The stability of protein conformation, as well as the interaction between proteins heavily, relies on its hydrophobic effects [37]. In our study, mutational impairment of the hydrophobic regions of dystrophin, even when these mutations are in-frame, has been shown to lead to DMD in most cases, suggesting their important role. We used bioinformatics techniques to model these hydrophobic regions and analyze their functions.
By using a scale mean hydrophobicity profile analysis method, we identified four hydrophobic regions in dystrophin. They are located between the residues 60 and 132, between the residues 1990 and 2010, between the residues 2450 and 2500 and between the residues 3150 and 3300 and coded by the exons 3-6, 42, 51, and 65-68, respectively.
Each monomer in the actin-binding domain is an extended dumbbell-shaped structure. The head of the dumbbell is formed by combining the two CH regions (CH1 and CH2). Hydrophobic region I includes the whole CH2 region and the small beginning part of the third actin-binding structures (ABS3) in the CH1 region. A previous investigation [28] found that actin directly combines with ABS1 and ABS3 in the actin-binding domain (ABD). ABS1 and ABS3 are located on the helix A of CH1 and the helix A of CH2, respectively. ABS2 is located on the helix G and F in the CH1 region. ABS1 and ABS2 do not stay on the same plane (Figure 3E). In order to bind actin, a conformational change occurs in the CH region, leading to the rearrangement of ABS and formation of an actin-binding surface, with the CH2 region helping to increase the affinity between the ABD and actin [38]. In our study, 59.2% of patients having an in-frame deletion in the hydrophobic region I presented with clinical symptoms of DMD and hence did not meet the reading-frame rule. These exceptions to the rule might be due to the serious alterations of the CH2 structure caused by the mutation, directly affecting the affinity between the ABD and actin. Nevertheless, not all the mutations led to the highly disruptive structural changes of the binding surface, since some patients presented with less severe BMD symptoms.
The second hydrophobic region is located in the central rod domain, which is formed by the linking of twenty-four spectrin-type repeats (STR) and four hinges [39]. Hinge I (H I) connects the N-terminal region of dystrophin and the N-terminus of the central rod domain. Hinge II (H II) connects R3 and R4; while hinge III (H III) links R19 and R20. Hinge IV (H IV) is the connection between the C-terminus of the central rod domain and the N-terminus of the cysteine-rich domain [40-41]. Batch 2 and Batch 3 of the 3D reconstruction in this study are located on the central rod domain and are made up of overlapping triple alpha-helical coiled coils. No β-strands were identified. Batch 2 contains R16 to R18, domain considered as a binding site for neuronal nitric oxide synthase (nNOS) [42-43]. R16/17 is needed to anchor nNOS onto the sarcolemma. The nNOS-sarcolemma connection requires the α2 and α3 helices of R16 and the three helices of R17. The R17 α1 helix links directly to nNOS. The two other helices can stabilize the catenating structure. The nNOS-sarcolemma connection is involved in signal transduction as well as in the post-injury recovery of muscle activity.
The second hydrophobic region is located specifically on R16. Our study demonstrated that the involvement of this hydrophobic region in in-frame mutations broke the reading-frame rule and led to DMD in a relatively large percentage (76.9%) of patients. These mutations appear to impair the stability of the nNOS-sarcolemma connection, resulting in failure of nNOS to anchor onto the sarcolemma. In the study of Cazzella et al, the damage of nNOS-sarcolemma connection might be the cause of DMD [44].
The hydrophobicity of the third hydrophobic region is the strongest one. It is made up of residues 2450-2500, including 20 amino acid residues at the C-terminus of H III and 30 residues on the N-terminal part of R20. The peptide is coded by the exons 51 and 52, and includes an STR-separating hinge area rich in proline. Formation of alpha helices and beta sheets is more difficult when its sequence contains large number of proline. Therefore, this region may be predisposed to be disordered and flexible, and it is easy to extend and distort such a structure to some degree [45-46]. Previous research [47] pointed out that the order of STR elicits significant influence on clinical symptoms. When H III is present, its irregular coil structure forms an interruption in the otherwise helical rod structure of the repeat domain. In contrast, deletion of H III yields a direct connection between two STRs (that in the native structure were not directly connected), perhaps strengthening the rod. This observation is also supported by our data, indicating that 79.6% of the patients, whose mutation involves hydrophobic region III, actually have BMD. In our study, unlike the traditional hypothesis [48] (that considers H III important in maintaining the stability of dystrophin and proposes its deletion as a cause of DMD), the deletion of H III had various influences on DGC function, applicable to BMD more than to DMD. Therefore, the exact role of H III in DGC is still to be determined in the further investigation.
Our exploration of the fourth hydrophobic region informs us about the importance of dystrophin’s ability to bind β-dystroglycan. Dystrophin’s cysteine-rich region binds β-dystroglycan, and β-dystroglycan, in turn, is connected to the extracellular matrix [49-50]. The cysteine-rich region contains a WW domain, EF hand 1, EF hand 2 and ZZ domain (include a zinc finger domain). The computerized 3D reconstruction described above shows that Batch4 has a globular structure, and contains two EF-hand structures. 31 α helices and 31 β strands form the structure of this batch, along with 40 reverse turns. Most of them are in dystrophin’s cysteine-rich WW domain and the EF-hand structure. The WW domain is composed of residues 3056-3092. When β-dystroglycan binds dystrophin, the WW domain becomes embedded into the neighboring EF-hand structure to create a bigger complex [51-52]. The proline-rich region of β-dystroglycan directly contacts the EF-hand structure, which also stabilizes the WW domain. Hydrophobic region IV is located in the EF-hand structure and is coded by exons 65-66. Mutational damage of this region thus directly influences contact between dystrophin and β-dystroglycan. As a result, the function of DGC is severely affected and, as shown in our analysis, DMD is the predominant outcome.
CONCLUSION
In a summary, there are four hydrophobic regions in dystrophin, which are found on the CH2 portion of the ABD, in R16, in H III, and on an EF hand of cysteine-rich domain. These areas are important functional areas of dystrophin. Hydrophobic regions I, II and IV, respectively, affect the binding of actin, nNOS-sarcolemma, and β-dystroglycan, associated proteins of the DGC. Mutations of these hydrophobic regions thus directly impair formation of the DGC and consequent mutational damage, even when in-frame, results in the development of DMD phenotype. Hydrophobic region III contains the H III hinge and plays an important role in the stability of the dystrophin structure. The deletion of H III, in contrast, stretches the order of STR, causing less severe clinical symptoms than in patients with the intact H III. Taken together, the discovery of these hydrophobic regions and the understanding of their functionality in pathogenesis provide supplemental information to the reading-frame rule and support the strategies for the development of exon-skipping therapy [53-54].
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
REFERENCES
- [1].Danieli GA, Mostacciuolo ML, Bonfante A, Angelini C. Duchenne muscular dystrophy. A population study. Hum Genet. 1977;35(2):225–231. doi: 10.1007/BF00393974. DOI: 10.1007/BF00393974. [DOI] [PubMed] [Google Scholar]
- [2].Pane M, Lombardo ME, Alfieri P, D’Amico A, Bianco F, Vasco G, et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. J Pediatr. 2012;161(4):705–709. doi: 10.1016/j.jpeds.2012.03.020. DOI: 10.1016/j.jpeds.2012.03.020. [DOI] [PubMed] [Google Scholar]
- [3].Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258(9):1610–1623. doi: 10.1007/s00415-011-5979-z. DOI: 10.1007/s00415-011-5979-z. [DOI] [PubMed] [Google Scholar]
- [4].Escolar DM, Zimmerman A, Bertorini T, Clemens PR, Connolly AM, Mesa L, et al. Pentoxifylline as a rescue treatment for DMD: a randomized double-blind clinical trial. Neurology. 2012;78(12):904–913. doi: 10.1212/WNL.0b013e31824c46be. DOI: 10.1212/WNL.0b013e31824c46be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonilla E, Samitt CE, Miranda AF. Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell. 1988;54(4):447–452. doi: 10.1016/0092-8674(88)90065-7. DOI: 10.1016/0092-8674(88)90065-7. [DOI] [PubMed] [Google Scholar]
- [6].López-Hernández LB, van Heusden D, Soriano-Ursúa MA, Figuera-Villanueva L, Vázquez-Cárdenas NA, Canto P, et al. Genotype-phenotype discordance in a Duchenne muscular dystrophy patient due to a novel mutation: insights into the shock absorber function of dystrophin. Rev Neurol. 2011;52(12):720–724. [PubMed] [Google Scholar]
- [7].Bushby K, Connor E. Clinical outcome measures for trials in Duchenne muscular dystrophy: report from International Working Group meetings. Clin Investig (Lond) 2011;1(9):1217–1235. doi: 10.4155/cli.11.113. DOI: 10.4155/cli.11.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Scully MA, Cwik VA, Marshall BC, Ciafaloni E, Wolff JM, Getchius TS, et al. Can outcomes in Duchenne muscular dystrophy be improved by public reporting of data? Neurology. 2013;80(6):583–589. doi: 10.1212/WNL.0b013e318282334e. DOI: 10.1212/WNL.0b013e318282334e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Soltanzadeh P, Friez MJ, Dunn D, von Niederhausern A, Gurvich OL, Swoboda KJ, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord. 2010;20(8):499–504. doi: 10.1016/j.nmd.2010.05.010. DOI: 10.1016/j.nmd.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Basumatary LJ, Das M, Goswami M, Kayal AK. Deletion pattern in the dystrophin gene in Duchenne muscular dystrophy patients in northeast India. J Neurosci Rural Pract. 2013;4(2):227–229. doi: 10.4103/0976-3147.112777. DOI: 10.4103/0976-3147.112777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. DOI: 0.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- [12].Moore CJ, Winder SJ. The inside and out of dystroglycan post-translational modification. Neuromuscul Disord. 2012;22(11):959–965. doi: 10.1016/j.nmd.2012.05.016. DOI: 10.1016/j.nmd.2012.05.016. [DOI] [PubMed] [Google Scholar]
- [13].Janke A, Upadhaya R, Snow WM, Anderson JE. A new look at cytoskeletal NOS-1 and β-dystroglycan changes in developing muscle and brain in control and mdx dystrophic mice. Dev Dyn. 2013;242(12):1369–1381. doi: 10.1002/dvdy.24031. DOI: 10.1002/dvdy.24031. [DOI] [PubMed] [Google Scholar]
- [14].Kalman L, Leonard J, Gerry N, Tarleton J, Bridges C, Gastier-Foster JM, et al. Quality assurance for Duchenne and Becker muscular dystrophy genetic testing: development of a genomic DNA reference material panel. J Mol Diagn. 2011;13(2):167–174. doi: 10.1016/j.jmoldx.2010.11.018. DOI: 10.1016/j.jmoldx.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rani AQ, Sasongko TH, Sulong S, Bunyan D, Salmi AR, Zilfalil BA, et al. Mutation spectrum of dystrophin gene in malaysian patients with Duchenne/Becker muscular dystrophy. J Neurogenet. 2013;27(1-2):11–15. doi: 10.3109/01677063.2012.762580. DOI: 10.3109/01677063.2012.762580. [DOI] [PubMed] [Google Scholar]
- [16].Lee BL, Nam SH, Lee JH, Ki CS, Lee M, Lee J. Genetic analysis of dystrophin gene for affected male and female carriers with Duchenne/Becker muscular dystrophy in Korea. J Korean Med Sci. 2012;27(3):274–280. doi: 10.3346/jkms.2012.27.3.274. DOI: 10.3346/jkms.2012.27.3.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].de Brouwer AP, Nabuurs SB, Verhaart IE, Oudakker AR, Hordijk R, Yntema HG, et al. A 3-base pair deletion, c.9711_9713del, in DMD results in intellectual disability without muscular dystrophy. Eur J Hum Genet. 2014;22(4):480–485. doi: 10.1038/ejhg.2013.169. DOI: 10.1038/ejhg.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–95. doi: 10.1016/0888-7543(88)90113-9. DOI: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- [19].Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum Mutat. 2008;29(5):728–737. doi: 10.1002/humu.20722. DOI: 10.1002/humu.20722. [DOI] [PubMed] [Google Scholar]
- [20].Zhang D, Cui S, Guo H, Jiang S. Genomic structure, characterization and expression analysis of a manganese superoxide dismutase from pearl oyster Pinctada fucata. Dev Comp Immunol. 2013;41(4):484–490. doi: 10.1016/j.dci.2013.07.010. DOI: 10.1016/j.dci.2013.07.010. [DOI] [PubMed] [Google Scholar]
- [21].Tran VK, Ta VT, Vu DC, Nguyen ST, Do HN, Ta MH, et al. Exon Deletion Patterns of the Dystrophin Gene in 82 Vietnamese Duchenne/Becker Muscular Dystrophy Patients. J Neurogenet. 2013;27(4):170–175. doi: 10.3109/01677063.2013.830616. DOI: 10.3109/01677063.2013.830616. [DOI] [PubMed] [Google Scholar]
- [22].Hassan MJ, Mahmood S, Ali G, Bibi N, Waheed I, Rafiq MA, et al. Intragenic deletions in the dystrophin gene in 211 Pakistani Duchenne muscular dystrophy patients. Pediatr Int. 2008;50(2):162–166. doi: 10.1111/j.1442-200X.2008.02538.x. DOI: 10.1111/j.1442-200X.2008.02538.x. [DOI] [PubMed] [Google Scholar]
- [23].Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Bio. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. DOI: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- [24].Ghose S, Tao Y, Conley L, Cecchini D. Purification of monoclonal antibodies by hydrophobic interaction chromatography under no-salt conditions. MAbs. 2013;5(5):795–800. doi: 10.4161/mabs.25552. DOI: 10.4161/mabs.25552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fokkema IF, den Dunnen JT, Taschner PE. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box”approach. Hum Mutat. 2005;26(2):63–68. doi: 10.1002/humu.20201. DOI: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- [26].Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557–563. doi: 10.1002/humu.21438. DOI: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- [27].Jennekens FG, ten Kate LP, de Visser M, Wintzen AR. Diagnostic criteria for Duchenne and Becker muscular dystrophy and myotonic dystrophy. Neuromuscul Disord. 1991;1(6):389–391. doi: 10.1016/0960-8966(91)90001-9. DOI: 10.1016/0960-8966(91)90001-9. [DOI] [PubMed] [Google Scholar]
- [28].Norwood FL, Sutherland-Smith AJ, Keep NH. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure. 2000;8:481–491. doi: 10.1016/s0969-2126(00)00132-5. DOI: 10.1016/S0969-2126(00)00132-5. [DOI] [PubMed] [Google Scholar]
- [29].Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–392. doi: 10.1093/nar/gkn750. Database issue. DOI: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goodsell DS. Representing structural information with RasMol. Curr Protoc Bioinformatics. 2005;(Chapter 5:Unit 5.4) doi: 10.1002/0471250953.bi0504s11. DOI: 10.1002/0471250953.bi0504s11. [DOI] [PubMed] [Google Scholar]
- [31].Kretsinger RH, Nockolds CE. Carp muscle calcium-binding protein II. Structure determination and general description. J Biol Chem. 1973;248(9):3313–3326. [PubMed] [Google Scholar]
- [32].Heidarsson PO, Otazo MR, Bellucci L, Mossa A, Imparato A, Paci E, et al. Single-Molecule Folding Mechanism of an EF-Hand Neuronal Calcium Sensor. Structure. 2013;21(10):1812–1821. doi: 10.1016/j.str.2013.07.022. DOI: 10.1016/j.str.2013.07.022. [DOI] [PubMed] [Google Scholar]
- [33].Bork P, Sudol M. The WW domain: a signaling site in dystrophin? Trends Biochem Sci. 1994;19(12):531–533. doi: 10.1016/0968-0004(94)90053-1. DOI: 10.1016/0968-0004(94)90053-1. [DOI] [PubMed] [Google Scholar]
- [34].Hiraki T, Abe F. Overexpression of Sna3 stabilizes tryptophan permease Tat2, potentially competing for the WW domain of Rsp5 ubiquitin ligase with its binding protein Bul1. FEBS Lett. 2010;584(1):55–60. doi: 10.1016/j.febslet.2009.11.076. DOI: 10.1016/j.febslet.2009.11.076. [DOI] [PubMed] [Google Scholar]
- [35].Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53(2):219–226. doi: 10.1016/0092-8674(88)90383-2. DOI: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- [36].Gumerson JD, Michele DE. The dystrophin-glycoprotein complex in the prevention of muscle damage. J Biomed Biotechnol. 2011;2011:210797. doi: 10.1155/2011/210797. DOI: 10.1155/2011/210797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mondal J, Morrone JA, Berne BJ. How hydrophobic drying forces impact the kinetics of molecular recognition. Proc Natl Acad Sci U S A. 2013;110(33):13277–13282. doi: 10.1073/pnas.1312529110. DOI: 10.1073/pnas.1312529110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Singh SM, Mallela KM. The N-terminal actin-binding tandem calponin-homology (CH) domain of dystrophin is in a closed conformation in solution and when bound to F-actin. Biophys J. 2012;103(9):1970–1978. doi: 10.1016/j.bpj.2012.08.066. DOI: 10.1016/j.bpj.2012.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Legrand B, Giudice E, Nicolas A, Delalande O, Le Rumeur E. Computational study of the human dystrophin repeats: interaction properties and molecular dynamics. PLoS One. 2011;6(8):e23819. doi: 10.1371/journal.pone.0023819. DOI: 10.1371/journal.pone.0023819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Acsadi G, Moore SA, Chéron A, Delalande O, Bennett L, Kupsky W, et al. Novel mutation in spectrin-like repeat 1 of dystrophin central domain causes protein misfolding and mild Becker muscular dystrophy. J Biol Chem. 2012;287(22):18153–18162. doi: 10.1074/jbc.M111.284521. DOI: 10.1074/jbc.M111.284521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mirza A, Menhart N. Stability of dystrophin STR fragments in relation to junction helicity. Biochim Biophys Acta. 2008;1784(9):1301–1309. doi: 10.1016/j.bbapap.2008.05.010. DOI: 10.1016/j.bbapap.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lai Y, Zhao J, Yue Y, Duan D. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A. 2013;110(2):525–530. doi: 10.1073/pnas.1211431109. DOI: 10.1073/pnas.1211431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Harper SQ. Molecular dissection of dystrophin identifies the docking site for nNOS. Proc Natl Acad Sci U S A. 2013;110(2):387–388. doi: 10.1073/pnas.1220256110. DOI: 10.1073/pnas.1220256110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cazzella V, Martone J, Pinnarò C, Santini T, Twayana SS, Sthandier O, et al. Exon 45 skipping through U1-snRNA antisense molecules recovers the Dys-nNOS pathway and muscle differentiation in human DMD myoblasts. Mol Ther. 2012;20(11):2134–2142. doi: 10.1038/mt.2012.178. DOI: 10.1038/mt.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sahni N, Mangat K, Le Rumeur E, Menhart N. Exon edited dystrophin rods in the hinge 3 region. Biochim Biophys Acta. 2012;1824(10):1080–1089. doi: 10.1016/j.bbapap.2012.06.011. DOI: 10.1016/j.bbapap.2012.06.011. [DOI] [PubMed] [Google Scholar]
- [46].Banks GB, Judge LM, Allen JM, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6(5):e1000958. doi: 10.1371/journal.pgen.1000958. DOI: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Carsana A, Frisso G, Tremolaterra MR, Lanzillo R, Vitale DF, Santoro L, et al. Analysis of dystrophin gene deletions indicates that the hinge III region of the protein correlates with disease severity. Ann Hum Genet. 2005;69(Pt 3):253–259. doi: 10.1046/J.1469-1809.2005.00160.x. DOI: 10.1046/J.1469-1809.2005.00160.x. [DOI] [PubMed] [Google Scholar]
- [48].Nicholson LV, Johnson MA, Bushby KM, Gardner-Medwin D, Curtis A, Ginjaar IB, et al. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 2. Correlations within individual patients. J Med Genet. 1993;30(9):737–744. doi: 10.1136/jmg.30.9.737. DOI: 10.1136/jmg.30.9.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chung W, Campanelli JT. WW and EF hand domains of dystrophin-family proteins mediate dystroglycan binding. Mol Cell Biol Res Commun. 1999;2(3):162–171. doi: 10.1006/mcbr.1999.0168. DOI: 10.1006/mcbr.1999.0168. [DOI] [PubMed] [Google Scholar]
- [50].Hnia K, Zouiten D, Cantel S, Chazalette D, Hugon G, Fehrentz JA, et al. ZZ domain of dystrophin and utrophin: topology and mapping of a beta-dystroglycan interaction site. Biochem J. 2007;401(3):667–677. doi: 10.1042/BJ20061051. DOI: 10.1042/BJ20061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang X, Poy F, Zhang R, Joachimiak A, Sudol M, Eck MJ. Structure of a WW domain containing fragment of dystrophin in complex with beta-dystroglycan. Nat Struct Biol. 2000;7(8):634–638. doi: 10.1038/77923. DOI: 10.1038/77923. [DOI] [PubMed] [Google Scholar]
- [52].Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta. 2014;1838(2):635–642. doi: 10.1016/j.bbamem.2013.08.023. DOI: 10.1016/j.bbamem.2013.08.023. [DOI] [PubMed] [Google Scholar]
- [53].Cirak S, Feng L, Anthony K, Arechavala-Gomeza V, Torelli S, Sewry C, et al. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. 2012;20(2):462–467. doi: 10.1038/mt.2011.248. DOI: 10.1038/mt.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mendell J, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, et al. The Eteplirsen Study Group. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–647. doi: 10.1002/ana.23982. DOI: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]