

Abstract
Visualizing tissue structures in three-dimensions (3D) is crucial to understanding normal and pathological phenomena. However, staining and imaging of thick sections and whole mount samples can be challenging. For decades, researchers have serially sectioned large tissues and painstakingly reconstructed the 3D volume. Advances in automation, from sectioning to alignment, now greatly accelerate the process. In addition, immunofluorescent staining methods allow multiple antigens to be simultaneously detected and analyzed volumetrically. The objective was to incorporate multi-channel immunofluorescent staining and automation in 3D reconstruction of serial sections for volumetric analysis. Paraffin-embedded samples were sectioned manually but were processed, stained, imaged and aligned in an automated fashion. Reconstructed stacks were quantitatively analyzed in 3D. By combining automated immunofluorescent staining and tried-and-true methods of reconstructing adjacent sections, we were able to visualize, in detail, not only the geometric structures of the sample but also the presence and interactions of multiple proteins and molecules of interest within their 3D environment. Advances in technology and software algorithms have significantly expedited the 3D reconstruction of serial sections. Automated, multi-antigen immunofluorescent staining will significantly broaden the range and complexity of scientific questions that can be answered with this methodology.
Keywords: Automation, serial section, three-dimension reconstruction, volumetric analysis
BACKGROUND
Visualization of the three-dimensional (3D) cellular and tissue environment has become more and more crucial in understanding normal and pathological biological phenomena.[1] However, visualizing specific proteins and molecules in high-resolution can be extremely challenging in thick tissue blocks or whole-mount samples. Creation of animals expressing endogenous fluorophores attached to specific protein targets is costly, not to mention impossible in human samples. In addition, immunostaining of large samples has many issues such as improper fixation and incomplete penetration of the antibodies. Our laboratory has struggled to stain 60+ μm sections of various mouse and human tissues with varying success. Only in certain regions of certain samples do we observe good staining with a small subset of antibodies [Figure 1a]. This inconsistency in staining results is a big problem. Even when the staining is successful, penetration of excitation lasers and detection of emission light from thick samples often require special sample processing, clearing and/or microscopy.
Figure 1.
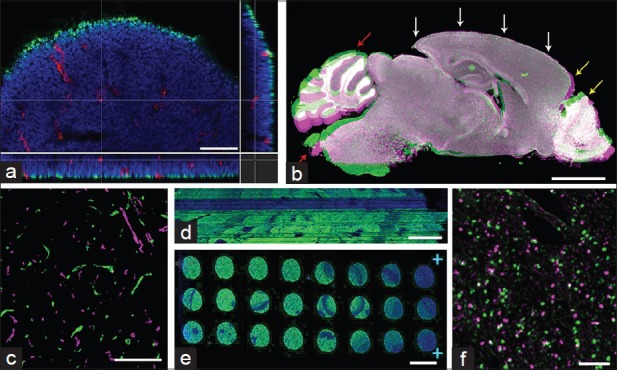
Examples of difficulties visualizing three-dimension structures. (a) 60 μm-thick 13.5d mouse embryo section was stained for Ki67 (green) and CD31 (red). The brain area was imaged using a confocal microscope with × 40 objective. XYZ section view shows that while penetration of anti-CD31 antibody is complete, anti-Ki67 antibody only detected cells in the very superficial layer of cells. The stack had to be imaged with laser compensation in order to view the signal in deeper layers. (b) Sagittal serial sections of an adult mouse brain stained with 4’,6-diamidino-2-phenylindole only, were manually aligned using AutoAligner. The snapshot from the software displays one section in green and its adjacent section in magenta. The two sections were manually aligned along the cortical outline (white arrows). When this region is well-aligned, one sees that the cortex has expanded (yellow arrow). Olfactory bulb, cerebellum, and brain stem regions have rotated around relative to the cortex and cannot be aligned well (red arrows). (c) Snapshot from AutoAligner showing adjacent breast tumor xenograft sections stained for CD31 clearly see that these two adjacent sections cannot be aligned in any meaningful way, suggesting that there was more than a 5 μm gap between the two adjacent sections. (d) XZ view showing the aligned sections of seminiferous tubules stained for DAZL (green) shows the inconsistency in the DAZL staining across the sections. (e) Whole-slide view of serial sections that were mounted and stained on one slide with the anti-DAZL antibody (green). While discovery XT automated stainer gives much more reproducible and reliable results, this particular slides exhibits uneven staining across the slide. (f) Zoomed view of E9.5 mouse embryo stained for nuclear pHH3. Snapshot from AutoAligner overlaying the pHH3 staining from two adjacent sections (green and magenta) shows the difficulty in aligning small signals such as nuclear staining. Scale bars: A =50 μm, B = 2 mm, C = 200 μm, D = 50 μm, E = 5 mm, F = 100 μm
For decades, scientists have circumvented these problems by serially sectioning large samples, staining individual sections, and painstakingly reconstructing the sections back into a 3D volume. While this method does cause some destruction of the 3D environment, the advantage of antigen detection being performed on typical thin sections (~5 μm) has made it the only option to visualize tissues in 3D in many cases. The downside of reconstructing serial sections is two-fold. One is the fact that it is very time and labor intensive, especially if one would like to visualize over a 100 μm–thick tissue sample. Another drawback is that in most studies, serial sections are visualized using chromogen-based staining protocols, which limits the number of proteins that can be visualized at the same time.[2,3,4]
The current study aims to combat these two disadvantages through the use of automation and immunofluorescent staining protocols. In recent years, there has been great technological development in automation of many of the routine procedures in immunostaining and imaging. From sectioning to staining and slide digitizers, high throughput machines have replaced repetitive manual labor. Automation significantly improves productivity because it can run with minimal human intervention, and it minimizes many of the human errors and biases that occur during manual procedures. It has been repeatedly demonstrated that automation can result in much more consistent and reliable results.[5,6] In the reconstruction of serial sections, the consistency in morphology as well as staining from one section to the next is very important. Incorporation of automated machines will be not only time-saving but necessary in the future.
The advantage of immunofluorescent staining has been demonstrated in many studies.[7] The intensity of the fluorescent signal can be accurately quantified and linearly correlated to the relative amount of targeted proteins/molecules.[8] Also with the advent of various fluorophores with distinct spectral characteristics, multiple antigens can be stained and visualized with clear separation. For some reason, however, immunofluorescent staining has rarely been used in the 3D reconstruction of serial sections. One reason may be consideration of reagent cost; another may be concerns that immunofluorescent staining is not permanent and may be less sensitive than, for example, DAB staining. Whole-slide digitization allows permanent storage and ensures that the data are captured permanently. And with appropriate amplification steps and use of automated staining machines, immunofluorescent staining can be easily and reliably incorporated into the serial section reconstruction. One then gains the ability to observe the localization and interaction of multiple proteins and molecules.
Here, we present multiple examples of successful 3D reconstruction of serial sections stained fluorescently and processed with as much automation as possible. We also discuss the issues associated with each step of reconstruction that are ameliorated by or persist despite automation.
METHODS
Sample Preparation
Mouse samples were obtained from the Memorial Sloan-Kettering Cancer Center Mouse Genetics Core Facility or by the laboratory of Dr. Joan Massague of Cancer Biology and Genetics Program. Tumor samples were kindly donated by MSKCC Pathology Department. All samples were fixed in freshly prepared 4% paraformaldehyde in phosphate buffer saline (PBS) (Electron Microscopy Sciences) for 16–24 h at 4°C. All tissues were washed three times in PBS for a total of 90 min and kept in 70% ethanol for several hours up to several days at 4°C. Tissue was processed for paraffin embedding using Leica ASP6025 tissue processor and embedded in paraffin (ParaPlast Plus paraffin; Leica Microsystems, Wetzlar, Germany).
Sectioning
Paraffin blocks were trimmed at 15 μm until the desired tissue depth was reached. The trimmed paraffin blocks were soaked in the ice-water mixture for 30 min, then automated paraffin microtome (RM2265, Leica MicroSystems) was used to obtain ribbons of 15–25 serial, 5 μm thick sections. The ribbon was stretched over the water surface in a floatation water bath kept at 42°C. The ribbon was broken down to smaller pieces that were arranged in order onto SuperfrostPlus microscope slides (Fisher Scientific). As a control, 60 μm thick sections were also obtained and stained in a similar fashion. The slides were air-dried overnight at room temperature and baked for 60 min in a 42°C oven. The slides were stored at 4°C until ready for staining.
Staining
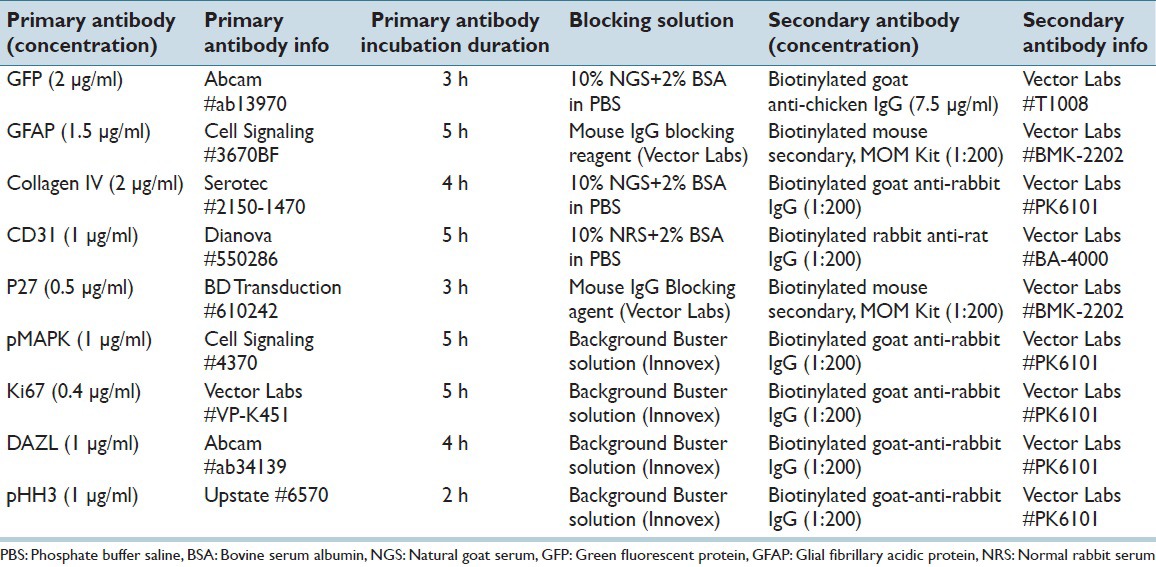
Slides were loaded onto the autostainer Discovery XT processor (Ventana Medical Systems) and de-waxed by being heated up to 98°C with EZPrep solution (Ventana Medical Systems). Antigen retrieval was performed with CC1 buffer (Ventana Medical Systems) before subsequent steps.Table 1 shows the detailed information of the antibodies and blocking solutions used. In brief, the tissue sections were incubated in blocking solution for 30 min prior to primary antibody incubation which lasted for 2–5 h. The biotinylated secondary antibody was applied for 60 min. Fluorescent detection of the antibodies was performed with Blocker D, Streptavidin-HRP D (Ventana Medical Systems), followed by incubation with Tyramid-Alexa Fluor of various wavelengths (Invitrogen): Alexa-488 (#T20922), 546 (#T20933), 594 (#T20935) or 647 (#T20936). Chromogen-based detection was performed with DAB MAP kit (Ventana Medical Systems) according to the manufacturer instructions.
Table 1.
Detailed protocol of automated staining
4’,6-diamidino-2-phenylindole Staining and Mounting
After staining was completed in the Ventana stainer machines, the slides were washed and incubated in 5 μg/ml 4’,6-diamidino-2-phenylindole (DAPI) (Sigma Aldrich) in PBS (Sigma Aldrich) for 10 min and rinsed in PBS twice. Finally, few drops of mounting media (Mowiol 4–88, Calbiochem) were applied to the slide surface and #1.5 cover glass was mounted manually avoiding any air bubbles. The slides were kept for at least several hours up to several days at – 20°C before imaging.
Slide Scanning
The slides were equilibrated to room temperature and digitally scanned using Pannoramic Flash (3DHistech, Hungary) with 20x/0.8NA objective (0.325 μm/pixel). The filters used were customized to optimize fluorescence detection and separation. The following is the detailed information on excitation, dichroic, and emission filters for each Alexa Fluor: Alexa488 (482/18 – 488 – 525/39), Alexa546 (530/23 – 550 – 572/28), Alexa594 (586/15 – 594 – 631/36), Alexa647 (650/13 – 669 – 700/50). Autofocus was performed on the DAPI channel (using the stock filter set), and all channels were imaged sequentially at each field of view. The same scanner was used to image DAB-stained slides as well (0.22 μm/pixel).
Alignment of Serial Sections
The initial step of alignment was accomplished using Voloom software (version 2.2.0, MicroDimensions, München, Germany). This software directly reads the raw 3DHistech scanner data, automatically detects tissues on each slide, orders them according to your specifications, and performs low magnification alignment. The reconstructed volume was reviewed and zoomed into areas of interest. Automatic re-alignment of zoomed area was performed by the software. The final stack was exported as series of tiff files or as an Imaris file. If further adjustment to the alignment is required, the images were imported into AutoAligner (Bitplane) for manual rotation/translation of individual slices.
Confocal Imaging
A 60 μm mouse embryo section was imaged with Leica TCS SP5-II (Leica Microsystems) using 40x/1.25NA oil objective, zoom of 1.0 (0.378 μm/pixel). 60 μm-thick z-stacks were taken from the top of the tissue section with laser compensation to obtain similar signal intensity throughout the thickness of the section. The step size was kept constant at 0.4 μm.
Three-dimensional Analysis
Aligned fluorescent stacks and confocal data were visualized and analyzed by Imaris (Bitplane, Zurich, Switzerland). Histogram normalization was performed for stacks in which the signal intensity varied greatly from slice to slice. Segmentation or spots analysis of the data was performed using the software's built-in wizard. Distance analysis was performed using MatLab XT tools through Imaris. Statistical analysis and graphing were conducted using Prism 6 (GraphPad Software, La Jolla, CA, USA).
RESULTS
Sectioning
The most crucial factor in the 3D alignment of serial sections is the consistency in the morphology of tissue sections. Prior to sectioning, any inadvertent alteration that may occur to the sample is global and does not induce difficulties in subsequent reconstruction. However, during the sectioning step, where the 3D sample is cut and separated into individual sections, independent morphological changes can occur and are best to be minimized.
While most steps in our protocol call for automated, machine-based processing, sectioning and mounting of paraffin-embedded samples were performed manually using Leica paraffin microtome. And not surprisingly, biggest obstacles in accurate alignment of serial sections came from slight differences in the morphology from one section to another. While the machine may be set to section at 5 μm interval, the actual thickness of the tissue may vary depending on (consistency of) speed of sectioning, the size of the paraffin block, and humidity, among other factors. The variation in the thickness not only changes the intensity of immunostained signals, but it can also cause differences in the amount of tissue shrinkage/expansion that may occur during subsequent processing and staining. For example, Figure 1b showing two adjacent 5 μm sections (pseudo-colored in green and magenta) of a DAPI-stained sagittal mouse brain, demonstrates the distortion that can occur from one section to another. The two serial sections were manually aligned along the cortical outline (white arrows), but the expansion of the magenta section causes the frontal cortex and olfactory bulb to not be aligned properly (yellow arrows).
More obviously, folds and other destruction of the tissue can occur during sectioning and mounting onto slides. A relatively small and intact tumor sample may be significantly easier to section consistently without any artifacts while larger convoluted sample will more likely have distortions. In a sagittal section of an adult mouse brain, cerebellum, and brain stem regions can bend relative to the rest of the brain [Figure 1b, red arrows].
If a large sample cannot be entirely sectioned within one session, remounting of the block the next day can cause a large gap from the previous section.Figure 1c shows CD31 blood vessel staining from the last section of the ribbon (green) and the first section from the next ribbon (magenta). Even though these two sections are supposed to be adjacent to one another, we could not automatically or manually align these two sections. All of these aberrations in the tissue morphology will pose difficulties in the alignment, 3D reconstruction, and subsequent analysis steps.
A solution may be to utilize fully automated sectioning machine such as Kurabo AS-200S from Kurabo Industries in Japan.[9] Automation will greatly improve consistency in thickness, minimize folds and tears, and eliminate the need to pause in the middle of an extensive sectioning process. As Onozato et al. describe in their paper,[9] the advantages of using automated sectioning machine would be most apparent and appreciated in 3D reconstruction of serial sections.
Staining
Staining of mounted sections was performed using Discovery XT processor from Ventana Medical Systems.[10,11] Once again, automation reduces the human error and variability of staining from one slide to the next. Inconsistency in staining levels becomes quite apparent when 3D reconstructed [Figure 1d]. Because volumetric analysis of immunofluorescence involves thresholding by signal intensity, variability in brightness from slice to slice can greatly alter quantitative results. One can perform normalization for visual effects but then forfeit any comparison of intensity between groups. It is best to perform automated staining to obtain the most consistent staining results.
On a cautionary note, automated machines are not artifact or error-proof. We have encountered instances where one out of many slides would come out unstained or with staining artifacts [Figure 1e]. Sections from such slides must be removed from the further analysis. It is crucial that such variability be minimized by maintaining the machines in optimal condition and by double-, triple-checking the protocol.
Scanning
The immunostained slides were then scanned with the whole-slide digitizer. There are many different slide scanners out in the market today, automating the imaging of histological slides,[12] some specifically for serial section reconstruction. At our facility, we routinely use 3D Histech's Pannoramic Flash scanner. In our experience, tile-scanning compared to line-scanning technology results in better quality scans albeit longer scan time. We were also able to easily install customized excitation and emission filters in order to cleanly separate the secondary antibodies. We often perform triple and even quadruple staining, and it is impossible to image Alexa 488, 546, 594, and 633 with minimal bleed-through on stock filters.
Using scanners to digitize slides with serial sections is not only time-saving but crucial for the quality of 3D reconstruction. Digitized slides have the ability to view the entire tissue at a lower magnification and to zoom into a specific area at higher magnification and resolution. Manually capturing images of areas of interest can be extremely time-consuming depending on the number of sections to be aligned. Human error can also be introduced in exposure times, naming of image files, deciding which areas to image, among others. Reconstructing a different area of interest could require re-capturing of images from every serial section.
As with the autostainers, it is important to maintain the scanners, cameras, light sources, and filters in their best condition. While slides can be re-scanned if the results are not optimal, one does not want to risk bleaching the fluorescent signals. The slides should be well-cleaned and free of dust. Cover slips should completely cover the sections with uniformly spread out mounting media. Coverslip mounting machines can be useful here as with all other automations.
Alignment
There are multiple 3D reconstruction and image alignment/registration software out in the market as well as freely available open-source plugins. In our case, we specifically used Voloom (MicroDimensions, Germany) for its ability to directly read 3DHistech scan files. Once the data are imported and ordered, the software automatically aligns the series at a low-magnification. We then zoomed in on specific areas of interest, and re-alignment at that higher magnification and resolution was performed automatically. When automated alignment result is not optimal, slice-by-slice manual adjustment was performed using AutoAligner (Bitplane).
At low magnification, the outline of the tissue sample as well as clear strong signals helps optimize the alignment. At this step, the imperfections during the sectioning and staining steps can become obvious. Voloom software as well as some others does not perform any stretching of the images: Only rotational and translational adjustments. Therefore, shrinkage or expansion of sections will result in misaligned stack.
Alignment of images at higher magnification will also be affected by tissue quality, but the morphology of the stained antigen will influence the alignment more heavily. Filamentous and continuous staining such as vasculature markers or extracellular matrix proteins will ease the alignment process, both automated and manual.
There is a limitation in the size of immunosignal that can be aligned well. Because the aggregate of antigens must appear in minimum two consecutive adjacent slices to be aligned, the smallest immunosignal that can be accurately reconstructed is twice the thickness of our sections. In our experience, alignment of nuclear markers such as pHH3 can be extremely difficult in some samples.Figure 1f shows the nuclear pHH3 signal from two adjacent sections (green and magenta), and one can see that some signals can be aligned well while others appear only in one or the other sections. Even with vascular protein like CD31, small capillary-like vessels that may run horizontally across a single section cannot be aligned well across multiple serial sections. For visualizing such small signals in the 3D environment, optimizing thick section staining and capturing confocal z-stacks may be a better solution.
Another current limitation is that the Voloom software only allows reconstruction of fluorescent images with 3 or less channels. At our facility, we routinely perform triple and quadruple immunostaining, resulting in 4+ numbers of separate channels. Future development of the software should allow alignment of tissues stained for a greater number of antigens.
Analysis
For quantitative analysis of 3D reconstructed, immunofluorescent volumes, we use Imaris software (Bitplane). Following segmentation of an individual channel, we can analyze the signal volume, cell counts, spot distribution in 3D and other analyses.Figure 2 represents a small sample of volumetric analyses that can be performed on reconstructed volumes.Figure 2a shows vasculature in a breast tumor stack, and one can measure its total volume, number of branching points and length of each segment, etc.
Figure 2.
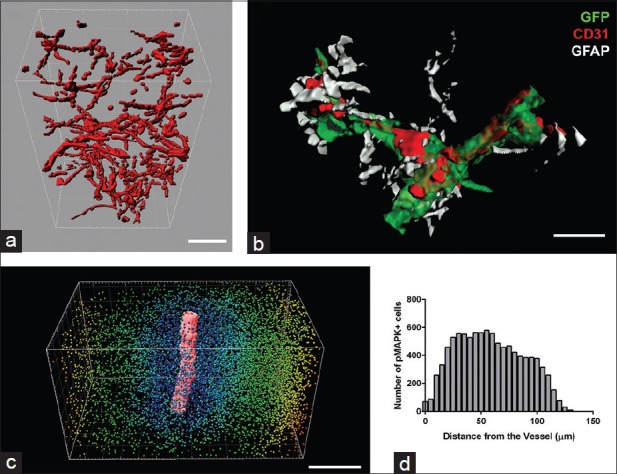
Volumetric analysis of three dimension reconstructed serial sections. (a) CD31 blood vessel staining in reconstructed mixfibrosarcoma tissue was surface rendered using Imaris. The total volume of the vasculature as well as branching points and segment lengths can then be easily calculated. (b) Reconstructed brain region was highly zoomed in and rendered to show the intricate interaction between blood vessels (red), cancer cells (green) traveling along the blood vessels, and the astrocytes (white) supporting and interacting with cancer cells as well as blood vessels. Such detailed visualization of the 3 antigens can only be conducted with reliable and specific immunofluorescent staining. (c) The Imaris snapshot shows the surface rendering of a large CD31-positive blood vessel in the center (red) and spot detection of pMAPK-positive cells in the surrounding area. The color of each spot depicts the different distances from the blood vessels. (d) The graphic representation shows the distribution of the pMAPK-positive cells relative to the blood vessel. Scale bars: A =50 μm, B = 20 μm, C = 150 μm
Figure 2b shows rendered close-up view of blood vessels (red) in the brain with green fluorescent protein-positive cancer cells (green) wrapped around the vessels and GFAP-positive glial cells (white) supporting the both cancer cells as well as blood vessels. Such zoomed-in view is possible after alignment of sagittal sections of the entire adult mouse brain.
In Figure 2c, we spot detected pMAPK-positive cells within a mixofibrosarcoma tissue and measured the shortest distance from each cell to the CD31-labeled major blood vessel segment. Distribution of these distances is graphed in Figure 2d and shows where the pMAPK-positive cells are located in relation to a large blood vessel coursing through the tumor tissue. These figures are just a few examples of what can be quantitatively analyzed from reconstructed volumes.
The biggest concern may be how accurate it is to analyze 3D reconstructed serial sections compared to analyzing in 2D or whole mount samples. In one instance, we reconstructed a portion of mouse ovary stained against p27 protein using DAB staining (total of 30 sections 5 μm thick). On one hand, the images were aligned, and the number of p27-positive follicles was counted via Imaris software [Figure 3a]. Alternatively, the follicles were counted manually by reviewing the images in sequence and tallying the number of incidences where the p27 signal was first detected. The counts were 55 follicles by 3D and 47 by hand. The difference of roughly 15% stems from the fact that Imaris software detected the folding of the tissue and resultant shading as DAB signal. If the p27 antigen was detected fluorescently, these artifacts would influence the data less, and the analysis results between automated and manual would be more similar.
Figure 3.
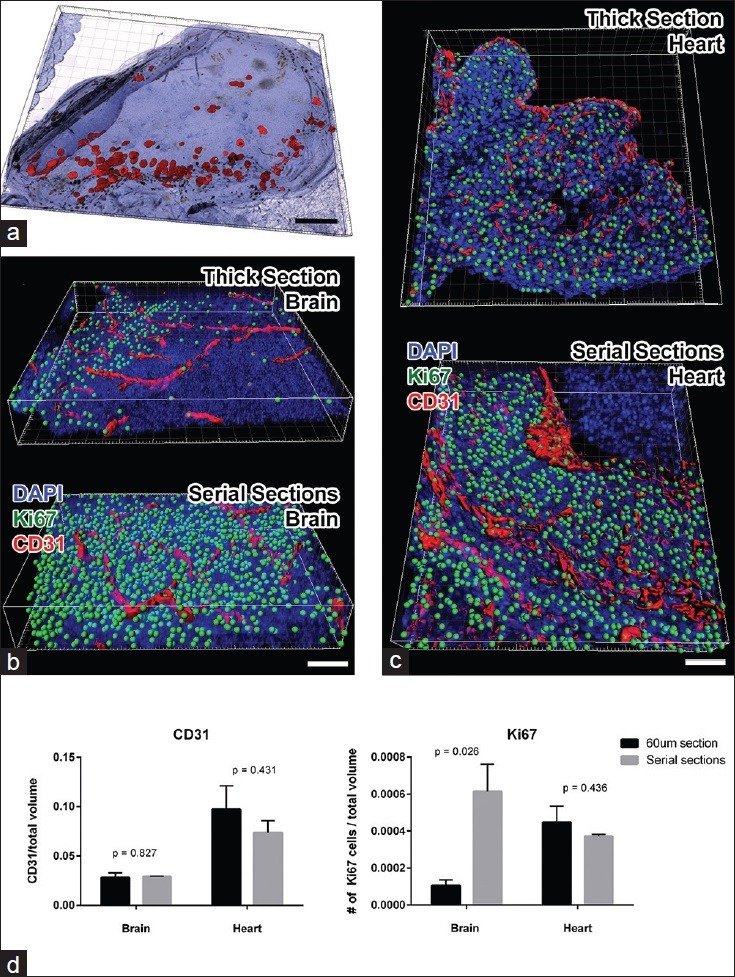
Validation of volumetric analysis of reconstructed serial sections. (a) Imaris snapshot shows reconstructed ovary sections stained for p27 protein with DAB chromogen. The red surfaces show the rendering of the p27 signal using Imaris. In brightfield imaging, folds in the tissue can have similar spectral characteristics as DAB staining; therefore, automated count of p27-positive cells exceeded that of manual counting. (b) Imaris snapshots were taken from a brain region of a mouse embryo stained and imaged as a 60 μm thick section (above) and serially sectioned and reconstructed (below). CD31-positive vasculature is rendered in red and Ki67-positive nuclei were spot detected (green). One sees that staining and detection of Ki67 signal is much more extensive in the serial sections. (c) Similar snapshots were taken from the heart region of the same 60 μm (above) or serially sectioned (below) mouse embryo. In this area of the embryo, similar amounts of CD31 and Ki67 proteins were immunofluorescently detected in both methods. Results suggest that in the heart, antibody penetration in 60 μm thick section was more complete than in the brain. (d) Graphs depict the quantitative analysis of normalized CD31 volume (left panel) and of Ki67-positive cell counts (right panel). Student t-test comparison between thick section values and serial section values was performed, and the P values are indicated above the bars. Scale bars: A =500 μm; B and C = 50 μm
In another attempt to measure the accuracy of the 3D reconstructed stacks, we compared data from two 60 μm sections of E13.5 mouse embryo. One was stained and imaged as an intact 60 μm thick section using the automated stainer. The other was further cut into adjacent 5 μm sections, stained, scanned, and reconstructed. In both cases, we stained with anti-CD31 and anti-Ki67 antibodies. Regions within the brain [Figure 3b] and the heart [Figure 3c] were analyzed for normalized vascular volume and count of proliferating cells using Imaris (n = 3 for each set). As one can see from the comparison [Figure 3d], the normalized volume of CD31-positive blood vessels were similar between the two methods, while the nuclear count of Ki67-positive proliferating cells drastically differed in the brain.
There may be two reasons for this discrepancy. One is that the staining of Ki67 in the thick section was sub-optimal. The level of Ki67 signal is clearly different between the two volumes demonstrating the difficulty of antibodies to penetrate 60 μm of brain tissue. The heart region, on the other hand, had comparable Ki67 staining and subsequent analysis results between the two methodologies. The difference in staining success between two regions of the same embryo demonstrates the difficulty in staining some whole-mount samples while others can be beautifully stained.
Another contributing reason for the discrepancy may be that Ki67 staining is nuclear and, therefore, difficult to align from one serial section to another. In the 3D analysis, one positive nucleus may have been counted as 2 only because they were not well aligned during the reconstruction. With better staining and alignment issues, the serial sections in the brain region contained far more Ki67-positive nuclei than the same region of the thick section. These data emphasize the caution one must employ in deciding which methods to use to analyze what type of immunofluorescent signals.
CONCLUSIONS
The current paper described in detail how one may take advantage of automated technologies and algorithms to significantly increase the reliability, reproducibility, and speed of performing 3D reconstruction of serially sectioned tissues. Because this method of visualizing 3D environment relies heavily on each serial section to be processed and stained in exactly the same way, it is important to minimize and eliminate human errors and variability from the process.
We have also shown here that while chromogen-based immunohistochemical staining such as DAB is well-used for single antigen detection, alignment of immunofluorescently stained sections works just as well and allows researchers to observe relationships between multiple antigens in a 3D environment. In addition, immunofluorescent signals are thought to be more linear, allowing scientists to easily analyze and compare intensities and infer about protein concentration in situ.[8] 3D image analysis software like Imaris makes volumetric quantification of fluorescent signals very easy and streamlined compared to chromogen signals.
Despite advances in automation, there are inherent drawbacks to 3D reconstruction that may encourage scientists to search/develop alternative methods. Imperfect or inconsistent sectioning and distortion of sections during staining and processing will always remain as an obstacle. Because a small amount of destruction is unavoidable during serial sectioning, it is difficult to align small signals that are nuclear or subcellular in localization. Furthermore, when the aligned stack is zoomed into ×20 or higher, the small imperfections in the alignment become more obvious. Small objects may not be aligned well and be counted as two separate objects during 3D analysis, as was the case with Ki67 staining in our study.
Scientists are continually developing new tissue processing methods to circumvent these problems. For example, Parfitt et al.[13] have demonstrated that embedding samples in butyl-methylmethacrylate allows precise sectioning at 0.5–5 μm thickness. Jirkovska et al.[14] have shown that non-deparaffinized sections suffer less distortion during the processing and staining. While minimizing unintended changes in the morphology of the tissue is tempting, deparaffinization is an unavoidable step in immunohistochemical or immunofluorescent staining. Therefore, nondeparaffinized samples can only be visualized if they express endogenous fluorescent proteins or dyes.
Smith and others have developed array tomography[15] in which small tissues are embedded in resin and cut with ultramicrotome much like in electron microscopy. The ribbon of serial sections is transferred onto cover slip and immunofluorescently stained as usual. This sample can then be imaged using optical or electron microscope and reconstructed. While this method still comes with difficulties of any 3D reconstruction of serial sections, the authors claim that resin embedding maintains the integrity of the tissue quite well, even though many rounds of immunostaining. The downside of this method, of course, is the limitation in the size of the tissue that can be cut with the diamond knife of the ultramicrotome.
Alignment software used in this study, Voloom and AutoAligner, perform rotational and translational manipulations to individual images. However, when shrinkage/expansion of tissue sections is an issue, using software that allows nonlinear manipulation of images may be necessary. Such procedure is computationally costly and may take a long time to complete.
An alternative method of immunological staining and imaging of thick sections and whole mount samples has its own demerits. Penetration of antibodies as well as detection of signals is a huge problem especially in densely-packed tissues like tumors. Advances have been made in clearing whole-mount samples that endogenously express fluorophores that attached to proteins of interest.[16,17] However, transfecting animals with these fluorophore-expressing genes may still be financially and temporally costly, not to mention impossible in case of human samples.
Despite these difficulties, it is nevertheless crucial to observe and understand structures and phenomena in 3Ds. We know that cells behave differently in a 3D environment than in a flat monolayer, and scientists have recently been actively trying to recreate the 3D environment in vitro.[18] In order for us to truly understand the mechanism behind the pathological phenomenon and combat it, we must study it in its biological environment. This study describes one of the multiple ways to achieve this goal.
ACKNOWLEDGMENTS
We would like to personally thank the Pathology Department, Dr. Willie Mark and Jing X Liu from the Mouse Genetics Core Facility and the laboratory of Dr. Joan Massague and his postdoctoral fellow Dr. Manuel Valiente for providing tissue samples. We also greatly appreciate Mr. Martin Groher and his staff members at MicroDimensions for the continuing efforts and collaboration in the advancement of features in the Voloom software.
Footnotes
Available FREE in open access from: http://www.jpathinformatics.org/text.asp?2015/6/1/27/158052
REFERENCES
- 1.Wang Y, Xu R, Luo G, Wu J. Three-dimensional reconstruction of light microscopy image sections: Present and future. Front Med. 2015;9:30–45. doi: 10.1007/s11684-014-0337-z. [DOI] [PubMed] [Google Scholar]
- 2.Vestentoft PS, Brøchner CB, Lynnerup N, Andersen CY, Møllgård K. Human embryonic and hepatic stem cell differentiation visualized in two and three dimensions based on serial sections. Methods Mol Biol. 2014 doi: 10.1007/7651_2014_128. [DOI: 10.1007/7651_2014_128] [DOI] [PubMed] [Google Scholar]
- 3.Booth ME, Treanor D, Roberts N, Magee DR, Speirs V, Hanby AM. Three-dimensional reconstruction of ductal carcinoma in situ with virtual slides. Histopathology. 2014 doi: 10.1111/his.12561. [DOI: 10.1111/his.12561] [DOI] [PubMed] [Google Scholar]
- 4.Christensen EI, Grann B, Kristoffersen IB, Skriver E, Thomsen JS, Andreasen A. Three-dimensional reconstruction of the rat nephron. Am J Physiol Renal Physiol. 2014;306:F664–71. doi: 10.1152/ajprenal.00522.2013. [DOI] [PubMed] [Google Scholar]
- 5.Shipitsin M, Small C, Giladi E, Siddiqui S, Choudhury S, Hussain S, et al. Automated quantitative multiplex immunofluorescence in situ imaging identifies phospho-S6 and phospho-PRAS40 as predictive protein biomarkers for prostate cancer lethality. Proteome Sci. 2014;12:40. doi: 10.1186/1477-5956-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biesterfeld S, Kraus HL, Reineke T, Muys L, Mihalcea AM, Rudlowski C. Analysis of the reliability of manual and automated immunohistochemical staining procedures. A pilot study. Anal Quant Cytol Histol. 2003;25:90–6. [PubMed] [Google Scholar]
- 7.Stadler C, Rexhepaj E, Singan VR, Murphy RF, Pepperkok R, Uhlén M, et al. Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat Methods. 2013;10:315–23. doi: 10.1038/nmeth.2377. [DOI] [PubMed] [Google Scholar]
- 8.Mortensen K, Larsson LI. Quantitative and qualitative immunofluorescence studies of neoplastic cells transfected with a construct encoding p53-EGFP. J Histochem Cytochem. 2001;49:1363–7. doi: 10.1177/002215540104901104. [DOI] [PubMed] [Google Scholar]
- 9.Onozato ML, Hammond S, Merren M, Yagi Y. Evaluation of a completely automated tissue-sectioning machine for paraffin blocks. J Clin Pathol. 2013;66:151–4. doi: 10.1136/jclinpath-2011-200205. [DOI] [PubMed] [Google Scholar]
- 10.Fujisawa S, Turkekul M, Barlas A, Fan N, Manova K. Double in situ detection of sonic hedgehog mRNA and pMAPK protein in examining the cell proliferation signaling pathway in mouse embryo. Methods Mol Biol. 2011;717:257–76. doi: 10.1007/978-1-61779-024-9_15. [DOI] [PubMed] [Google Scholar]
- 11.Yarilin D, Xu K, Turkekul M, Fan N, Romin Y, Fijisawa S, et al. Machine-based method for multiplex in situ molecular characterization of tissues by immunofluorescence detection. Sci Rep. 2015;5:9534. doi: 10.1038/srep09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rojo MG, García GB, Mateos CP, García JG, Vicente MC. Critical comparison of 31 commercially available digital slide systems in pathology. Int J Surg Pathol. 2006;14:285–305. doi: 10.1177/1066896906292274. [DOI] [PubMed] [Google Scholar]
- 13.Parfitt GJ, Xie Y, Reid KM, Dervillez X, Brown DJ, Jester JV. A novel immunofluorescent computed tomography (ICT) method to localise and quantify multiple antigens in large tissue volumes at high resolution. PLoS One. 2012;7:e53245. doi: 10.1371/journal.pone.0053245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jirkovská M, Náprstková I, Janácek J, Kucera T, Macásek J, Karen P, et al. Three-dimensional reconstructions from non-deparaffinized tissue sections. Anat Embryol (Berl) 2005;210:163–73. doi: 10.1007/s00429-005-0006-8. [DOI] [PubMed] [Google Scholar]
- 15.Micheva KD, Smith SJ. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calle EA, Vesuna S, Dimitrievska S, Zhou K, Huang A, Zhao L, et al. The use of optical clearing and multiphoton microscopy for investigation of three-dimensional tissue-engineered constructs. Tissue Eng Part C Methods. 2014;20:570–7. doi: 10.1089/ten.tec.2013.0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott GD, Blum ED, Fryer AD, Jacoby DB. Tissue optical clearing, three-dimensional imaging, and computer morphometry in whole mouse lungs and human airways. Am J Respir Cell Mol Biol. 2014;51:43–55. doi: 10.1165/rcmb.2013-0284OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haycock JW 3D cell culture: A review of current approaches and techniques. Methods Mol Biol. 2011;695:1–15. doi: 10.1007/978-1-60761-984-0_1. [DOI] [PubMed] [Google Scholar]