

Abstract
Background
Animal studies show that peripheral inflammatory stimuli may activate microglial cells in the brain implicating an important role for microglia in sepsis-associated delirium. We systematically reviewed animal experiments related to the effects of systemic inflammation on the microglial and inflammatory response in the brain.
Methods
We searched PubMed between January 1, 1950 and December 1, 2013 and Embase between January 1, 1988 and December 1, 2013 for animal studies on the influence of peripheral inflammatory stimuli on microglia and the brain. Identified studies were systematically scored on methodological quality. Two investigators extracted independently data on animal species, gender, age, and genetic background; number of animals; infectious stimulus; microglial cells; and other inflammatory parameters in the brain, including methods, time points after inoculation, and brain regions.
Results
Fifty-one studies were identified of which the majority was performed in mice (n = 30) or in rats (n = 19). Lipopolysaccharide (LPS) (dose ranging between 0.33 and 200 mg/kg) was used as a peripheral infectious stimulus in 39 studies (76 %), and live or heat-killed pathogens were used in 12 studies (24 %). Information about animal characteristics such as species, strain, sex, age, and weight were defined in 41 studies (80 %), and complete methods of the disease model were described in 35 studies (68 %). Studies were also heterogeneous with respect to methods used to assess microglial activation; markers used mostly were the ionized calcium binding adaptor molecule-1 (Iba-1), cluster of differentiation 68 (CD68), and CD11b. After LPS challenge microglial activation was seen 6 h after challenge and remained present for at least 3 days. Live Escherichia coli resulted in microglial activation after 2 days, and heat-killed bacteria after 2 weeks. Concomitant with microglial response, inflammatory parameters in the brain were reviewed in 23 of 51 studies (45 %). Microglial activation was associated with an increase in Toll-like receptor (TLR-2 and TLR-4), tumor necrosis factor alpha (TNF-α), and interleukin 1 beta (IL-1β) messenger ribonucleic acid (mRNA) expression or protein levels.
Interpretation
Animal experiments robustly showed that peripheral inflammatory stimuli cause microglial activation. We observed distinct differences in microglial activation between systemic stimulation with (supranatural doses) LPS and live or heat-killed bacteria.
Keywords: Microglia, Microglia activation, Systemic inflammation, Review, Animal experiments
Introduction
The peripheral immune system has a strong effect on the brain as exemplified by the high incidence of delirium and the strongly increased risk for the development of dementia after systemic infections [1, 2]. In rodent experiments, peripheral challenge with lipopolysaccharide (LPS) caused a steep increase of brain tumor necrosis factor alpha (TNF-α) that can persist for months [3–9]. Peripheral (systemic) LPS challenge activates microglia, the major active immune cells in the central nervous system. Microglia can be in a resting state (morphologically “ramified”) or an activated state (morphologically “amoeboid”) [10]. Resting microglia survey their environment for damage, ready to support endangered neurons or to interfere with a potential threat to the tissue integrity. Danger signals may trigger these surveying microglia and cause transformation to activated states, referred to as the M1 and M2 phenotypes [11]. M1 activated microglia produce pro-inflammatory mediators and are assumed to act as neurotoxic cells [11, 12], while M2 activation is induced by signals from apoptotic cells and have a role in remodeling and repair [11–13].
Sepsis in humans has also been associated with microglial activation [14]. Previously, we postulated that impaired cholinergic inhibitory control of microglia in elderly people, and to a greater extent in patients with (incipient) neurodegenerative disorders, contributes to uncontrolled neuro-inflammation [15]. High concentrations of pro-inflammatory mediators released by M1 activated microglia are potentially neurotoxic and might not only cause acute, reversible, behavioral effects, such as delirium, but also lead to persistent detrimental effects through bystander damage to neighboring neurons [16, 17]. The microglial response drifts out of control and ultimately causes neurodegeneration [1, 18]. This cycle might account for why neurobehavioral occurrences can persist in elderly patients after recovery from sepsis and after systemic cytokine production has fallen. This information inspired the formulation of a neuro-inflammatory hypothesis explaining the association of systemic infection, chronic central nervous system inflammation, and poor outcome, where microglial cells play a key role. Animal studies on systemic inflammation and microglial reaction support this hypothesis, but studies vary widely in setup and interpretation of results. In this review, we summarize available evidence on the effect of different systemic inflammatory stimuli on timing and intensity of the microglial reaction.
Methods
Search strategy
We searched PubMed between January 1, 1950 and December 1, 2013 and Embase between January 1, 1988 and December 1, 2013 for animal studies using peripheral inflammatory stimuli and evaluating the effect of these stimuli on microglia, using search terms “microglia” AND “animal model” NOT “review”. We also searched the reference lists of articles identified by this search strategy and selected those that we judged to be relevant. Two independent observers reviewed articles for inclusion and exclusion criteria, and differences were resolved by discussion.
Selection of articles
Studies were included if they fulfilled the following criteria: (1) the study described an experiment where a peripheral infectious stimulus was administrated in animals, in vivo; (2) the study assessed the effects on microglia in the brain; (3) the effects on microglia were determined with specific microglial markers; (4) a group of control animals was described; (5) the study was an original full paper which presented unique data; and (6) the studies were published in English, French, or German. Reasons for exclusion of an article were as follows: (1) any manipulation in or around the brain before, during, or after the peripheral infectious stimulus; (2) the use of a neurotropic pathogen; (3) the use of a chemical synthetic infectious stimulus; (4) animal models in which the infectious stimulus reached the brain and caused secondary meningitis; (5) the use of transgenic animal models for a specific (brain) disease; and (6) animal models where the infectious stimulus was given intra-uterine. Full text articles of selected studies were obtained for further evaluation. Two independent observers extracted data and resolved differences by discussion.
Data extraction
Each study was scored for key issues, such as animal species, gender, age, and genetic background; number of animals in treated and control groups; method of dosage, site of inoculation, and kind of infectious stimulus that was administrated; the effect of peripheral infectious stimulus on microglial cells in the brain; and methods of how this effect was determined on which time point after inoculation and in which region of the brain this effect was examined. The quality of studies was judged by a risk of bias assessment, scoring external and internal validity for each study [19].
Definition of microglial activation
Microglial cells were defined as activated based on the following criteria: (1) microglia showed an activated morphology based on immunohistochemical staining; (2) there was a significant increase in number and/or size of microglia compared to the control group; and (3) there was a significant increase in expression of a microglial marker. When all three criteria were negative, microglia were inactive. If one or more criteria were positive, microglia were activated. If results were contradictory (e.g., increased expression of microglial marker but morphology was negative) microglia were judged as moderately activated.
Results
Description of studies
In total, 2950 publications were identified and 149 were selected for further review (Fig. 1); 29 publications met inclusion criteria and 22 additional articles were identified in a reference search, so 51 publications were included in this systemic review. There was considerable variation in animal species and stimuli. The majority of studies was performed in mice (n = 30) or in rats (n = 19). LPS was used as a peripheral infectious stimulus in 39 studies (76 %), and live or heat-killed pathogens were used in 12 studies (24 %).
Fig. 1.
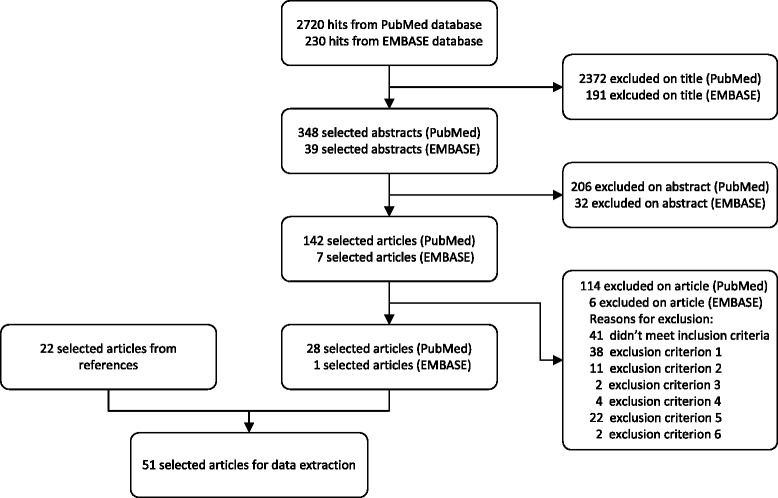
Study selection process
Information about animal characteristics such as species, strain, sex, age, and weight was defined in 41 studies (80 %), and methods of the disease model were described in 35 studies (68 %). Four studies lacked description of animal characteristics and disease model. Just one study described treatment allocation as randomized and evaluation in a blinded fashion [20]. None of the studies provided power calculations for animal group sizes, reported baseline measurements of animals between groups, or handling of outlined or missing data.
Outcome parameters
Microglial response was the main outcome parameter in all studies; the state of microglia was defined by immunohistochemistry in 36 studies (70 %), by the combination of immunohistochemistry, and respectively, quantitative polymerase chain reaction (qPCR) in two studies (4 %), Western blot in two studies (4 %), or in situ hybridization in one study (2 %); six studies used qPCR (12 %), three flow cytometry (6 %), and one study Western blot (2 %) to define microglial activation.
The most commonly used marker of microglial activation was the ionized calcium-binding adaptor molecule 1 (Iba-1), either as sole marker (n = 20), or in combination with cluster of differentiation 68 (CD68; n = 2), Griffonia symplicifolia isolectin B4 (IB4; n = 1), or macrophage receptor with collagenous structure (MARCO; n = 1). CD11b was used in 14 studies, and 5 of these studies combined CD11b with CD68, major histocompatibility complex II (MCHII), Toll-like receptors 2 and 4 (TLR-2, TLR-4), and F4/80. In three other studies, TLR-2 was stated as an activation marker of microglial cells after identifying the cells with CD45 and CD11b antibodies by flow cytometry or in combination with Iba-1. One study used TLR-2 with qPCR while MHCII expression between groups was similar [21]. The markers CD68 (n = 5), F4/80 (n = 2,) and IB4 (n = 2) were also used as sole markers.
The brain region of interest in the majority of studies was the hippocampus. Several studies restricted their interest to the hippocampal area (n = 17). Eight studies evaluated the hippocampal area in combination with other brain regions: cortex (n = 8), substantia nigra (n = 2), cerebellum (n = 2), thalamus (n = 1), striatum (n = 1), midbrain (n = 1). Five studies were limited to the cortical areas. Four studies homogenized the hemispheres for flow cytometry analysis or qPCR. The remainder 16 studies (25 %) evaluated other brain regions, and brain regions were not specified in one study.
Secondary outcomes were inflammatory mediators in the brain, for example cytokines, chemokines, Toll-like receptors (TLRs), or markers for damage or death, and were evaluated in 37 studies (73 %). Behavioral studies were performed in 14 studies (27 %).
Infectious stimuli
Lipopolysaccharide
A single-dose of LPS was evaluated in wild-type mice in 20 studies (Table 1). LPS was administered intraperitoneally in 19 studies and subcutaneously in one study. Mice were male in 16 of 20 studies (80 %) and varied with respect to age and genetic background. The majority of studies used LPS derived from Escherichia coli (E. coli) (12 of 20 studies [60 %]), four studies used LPS from Salmonella ssp., and four studies did not report the LPS origin. The dose of LPS ranged between 0.33 and 200 mg/kg, with 1 and 5 mg/kg both used in seven studies. Microglial response was evaluated 1 h to 1 year after LPS injection.
Table 1.
Single challenge with lipopolysaccharide (LPS) in mice and rats
| Study | Genetic background | N | Age/weight | Sex | Type of LPS | Site of LPS | Dose (mg/kg) | Time of termination | Microglial activation |
|---|---|---|---|---|---|---|---|---|---|
| Mice | |||||||||
| Henry [33] | BALB/c | 6 | 3 m | M | E. coli (O127:B8) | ip | 0.33 | 1 day | Yes |
| Henry [21] | BALB/c | 7 | 3–4 m | M | E. coli (O127:B8) | ip | 0.33 | 4 h | Yes |
| 18–20 m | 4 h | Yes | |||||||
| Carnavale [58] | C57BL/6 | 4 | 12–15 w | M | S. equine abortus | ip | 0.5 | 1 day | Yes |
| 5 weeks | No | ||||||||
| Terrando [22] | C57BL/6 | 4 | 12–14 w | M | E. coli (O111:B4) | ip | 1 | 1 day | Yes |
| 3 days | Yes | ||||||||
| 1 week | No | ||||||||
| Chung [36] | ICR | 7 | 6 w | M | U | ip | 1 | 6 h | Yes |
| 12 h | Yes | ||||||||
| 1 day | Yes | ||||||||
| Chen [25] | C57BL/6 | 4 | 8–12 w | M | E. coli (O55:B5) | ip | 1 | 1 day | No |
| Laflamme [3] | CD1 | U | 20–25 gr | M | E. coli (O55:B5) | ip | 1 | U | Yes |
| Gao [59] | B6C3F1/J | U | 7 m | M | E. coli (O111:B4) | ip | 1 | 1 day | Yes |
| 5 months | No | ||||||||
| Okuyama [41] | ICR | 10 | 6 w | M | S. typhimurium | ip | 1 | 3 days | Yes |
| Kaushik [60] | BALB/c | U | 6–8 w | U | S. enterica | ip | 5 | 1 day | Yes |
| Hwang [61] | C57BL/6 | U | 11 w | M | E. coli (O55:B5) | ip | 5 | 3 days | Yes |
| Qin [7] | B6:129SF2 | U | 8 w | M | E. coli (O111:B4) | ip | 5 | 2 h | Yes |
| C57BL/6 | U | 3 h | Yes | ||||||
| Sierra [6] | p7.2fms-EGFP | U | 2 m | B | S. typhimurium | ip | 5 | 1 day | Yes |
| Masocha [62] | C57BL/6 | 8 | 8–12 w | U | E. coli (O111:B4) | ip | 5 | 4 h | No |
| 1 day | Yes | ||||||||
| 1 year | Yes | ||||||||
| Ha [63] | C57BL/6 | 5 | 7 w | M | U | ip | 5 | 3 h | Yes |
| O’Callaghan [40] | C57BL/6 | 4 | 8–10 w | M | E. coli (O111:B4) | ip | 5 | 1 day | Yes |
| 3 months | Moderate | ||||||||
| Bhaskar [23] | C57BL/6 | U | 2 m | U | U | ip | 1 | 1 day | Moderate |
| 10 | 1 day | Yes | |||||||
| Nishioku [45] | ICR | 4 | 8 w | M | E. coli (O55:B5) | ip | 20 | 1 h | No |
| 3 h | No | ||||||||
| 6 h | Yes | ||||||||
| 1 day | Yes | ||||||||
| Smithason [20] | C57BL/6 | 6 | 10–12 w | M | U | ip | 200 | 2 days | Moderate |
| Sehgal [8] | C57BL/6 | 15 | 2–3 m | M | E. coli (O55:B5) | sc | 3 | 12 h | Yes |
| Rats | |||||||||
| Monje [64] | Fisher 344 | 3 | 160–180 gr | F | U | ip | 1 | 1 week | Yes |
| Fan [39] | Sprangue-Dawley | 6 | 5 d | B | E. coli (O55:B5) | ip | 2 | 1 day | Yes |
| Wang [65] | Sprangue-Dawley | 3 | 280–300 gr | M | E. coli (O55:B5) | ip | 5 | 1 day | Yes |
| Semmler [66] | Wistar | 4 | 250–300 gr | M | E. coli (O127:B8) | ip | 10 | 4 h | No |
| 8 h | No | ||||||||
| 1 day | Moderate | ||||||||
| Semmler [5] | Wistar | 5 | 250–300 gr | M | E. coli (O127:B8) | ip | 10 | 1 day | Yes |
| Garcia-Bueno [67] | Sprangue-Dawley | 5 | 260–340 gr | M | E. coli (O55:B5) | iv | 0.002 | 1–3 h | Moderate* |
| Jiang-Shieh [28] | Wistar | 10 | 200–250 gr | M | E. coli (O55:B5) | iv | 0.05 | 2 days | Yes |
| Buttini [44] | Sprangue-Dawley | 2 | 180–200 gr | M | E. coli (O55:B5) | iv | 1 | 1 day | No* |
| 2.5 and 5 | 1 h | No* | |||||||
| 3 h | Moderate* | ||||||||
| 6 h | Moderate* | ||||||||
| 8 h | Yes* | ||||||||
| 1 day | Yes* | ||||||||
| 3 day | Moderate* | ||||||||
| 1 week | No* |
Column time of termination is the time from the (first) LPS challenge
Abbreviations: N number of animals per group, m months, wk weeks, d days, gr gram, U unknown, M male, F female, B both sexes, ip intraperitoneal, sc subcutaneous, iv intravenous, iv intravenous
*Did not express data in statistical values, no statistical information
Two studies described activation of microglia 6 h after inoculation, 11 of 12 studies (92 %) showed microglial activation 1 day after inoculation, and three studies found microglial activation 3 days after challenge. Findings implicate that a single LPS challenge activates microglia 6 h after challenge and that the activation remains for at least 3 days. Five studies evaluated microglial response after this period: three studies showed no microglial activation after 1 week, 5 weeks, and 5 months, while another study reported moderately activated microglia after 3 months. One study showed microglial activation 1 year after LPS challenge (Table 1). Importantly, overall, the interpretation of the results was hampered by lack of information. The number of animals was noted in only 13 of 20 studies (65 %), and a statistical test comparing microglial response in LPS challenged and control groups were provided in 10 studies (50 %). LPS challenges in experiments using knockout mice were described in four studies, all using intraperitoneal injection, and are discussed below in the separate sections (Appendix) [22–25].
A single LPS challenge in rats was evaluated in eight studies. LPS was given intraperitoneally (63 %) or intravenously (37 %; Table 1), and seven studies used LPS from E. coli. LPS origin was not mentioned in one study. Dose of LPS ranged between 0.002 and 10 mg/kg. In general, microglia start to become moderately active 3 h after LPS challenge, reaching their activation state after 8 h to 2 days, and return to their normal resting state after 7 days.
Sequential LPS challenges were evaluated in nine studies (Table 2); seven studies used mice and two studies rats. Five of nine studies (56 %) were done in mice with a C57BL/6 genetic background, and age of these animals varied between 5 weeks and 10 months. LPS from E. coli was used in six of the nine studies; origin of LPS was not mentioned in three studies. Dose of LPS ranged from 0.05 to 4 mg/kg. Animals were challenged between 2 and 48 times over a period of 1 day to 6 months with total LPS dose ranging from 0.1 to 56 mg/kg. Eight of these nine studies showed microglial activation (89 %); however, the majority of time points evaluated after 1 month showed only moderate or no microglial activation.
Table 2.
Multiple intraperitoneal challenges with lipopolysaccharide (LPS) in mice and rats
| Study | Genetic background | N | Age | Sex | Type of LPS | Dose (mg/kg) | Number of hits | Total dose (mg/kg) | Time of termination | Microglial activation |
|---|---|---|---|---|---|---|---|---|---|---|
| Mice | ||||||||||
| Frank-Cannon [38] | C57BL/6 | 3 | 6–13 w | U | E. coli (O111:B4) | 0.1 b.i.w. | 16× | 1.6 | 2 months | No |
| 0.1 b.i.w. | 24× | 2.4 | 3 months | Yes* | ||||||
| 0.1 b.i.w. | 48× | 4.8 | 6 months | No | ||||||
| Katafuchi [68] | C57BL/6 | 8 | 10 m | M | U | 0.25 q.d. | 7× | 1.75 | 7 days | Yes |
| Lee [52] | ICR | 5 | 5 w | M | U | 0.25 q.d. | 7× | 1.75 | 18 days | Yes |
| Nguyen [34] | C57BL/6 | U | 6 m | U | E. coli (O55:B5) | 1 q.2wk. | 6× | 6 | 15 weeks | No* |
| Franciosi [46] | FVB/N | 5 | 5 m | B | U | 1 q.wk. | 4× | 4 | 4 weeks | Yes |
| 8 m | 16× | 16 | 17 weeks | Yes | ||||||
| Chen [25] | C57BL/6 | 4 | 8–12 w | M | E. coli (O55:B5) | 1 q.d. | 2× | 2 | 3 days | Moderate* |
| 4× | 4 | 5 days | Yes | |||||||
| Shankaran [69] | C57BL/6 | 5 | 10–15 w | F | E. coli (O111:B4) | 0.3 q.a.d. | 4× | 1.2 | 7 days | No |
| 1 q.a.d. | 4× | 4 | 7 days | Yes | ||||||
| 4 q.a.d. | 4× | 16 | 7 days | Yes | ||||||
| 4 q.a.d. | 14× | 56 | 4 weeks | Yes | ||||||
| Rats | ||||||||||
| Yin [70] | Sprangue-Dawley | 4 | 3 d | M | E. coli (O55:B5) | 0.05 q.a.d | 2× | 0.1 | 6 days | Yes |
| 18 days | Yes | |||||||||
| 42 days | No | |||||||||
| Wu [9] | Sprangue-Dawley | 8 | 10 w | M | E. coli (U) | 1.2 q.d. | 14× | 16.8 | 1 week | Yes |
Column time of termination is the time from the (first) LPS challenge. All challenges were intraperitoneal
Abbreviations: N number of animals per group, U unknown, M male, F female, B both sexes, m months, w weeks, d days, gr gram, q.d. every day, q.2wk once every 2 weeks, q.wk once a week, q.a.d. every other day, b.i.w. 2 times a week
*Did not express data in statistical values, no statistical information
One study administered LPS 1 mg/kg intraperitoneally in 6-month-old male gerbils, prompting a moderate microglial response at day 4 [26]. Another study evaluated the effect of serial intraperitoneal injections of 0.2 mg/kg LPS at different stages of brain development in opossums (on postnatal day [P] P14, P35, and P42) [27]. At day 10 after the first LPS injection, immunohistochemistry revealed microglial activation in younger age groups (P14 and P35) but not in older animals (P42) [27].
Lipoteichoic acid
One study evaluated intravenous administration of 20 mg/kg lipoteichoic acid (LTA) from Staphylococcus aureus (strain L2515) in rats [28]. Two days after challenge, microglial cells were activated as shown by enhanced immunoreactivity for CD11b and MHCII as compared to the unchallenged group, 2 days after LTA challenge. In these experiments, CD68 immunoreactivity of the pineal microglia appeared unaltered, while challenge with LPS (0.05 mg/kg) induced enhancement of CD68 immunoreactivity in addition to response for morphology, CD11b, and MHCII expression.
Bacteria
Bacteria were used as systemic challenge in 12 of 51 studies (24 %; Table 3). Animal species were rats in the majority of studies (75 %). Live bacteria were used in seven studies (58 %) and heat-killed bacteria in five studies (42 %). E. coli (American Type Culture Collection (ATCC) 15746) was most commonly used as systemic challenge with live bacteria (86 %); other live bacteria administered were Salmonella typhimurium. The heat-killed bacteria that were used were Mycobacterium tuberculosis and Mycobacterium butyricum, also referred to as complete Freund’s adjuvant (CFA).
Table 3.
Challenge with pathogens in mice and rats
| Study | Genetic background | N | Age/weight | Sex | Pathogen | Site of challenge | Dose | ||
|---|---|---|---|---|---|---|---|---|---|
| Mice | |||||||||
| Püntener [43] | C57BL/6 | 3 | >8 w | F | S. typhimurium (SL3261) | ip | 1 × 106 CFU | 1 day | No |
| 7 days | Moderate | ||||||||
| 21 days | No | ||||||||
| Rabchevsky [31] | C57BL/6 | 6 | 6 w | F | CFA | sc + ipl | 150 ug | 14 days | No |
| 21 days | No | ||||||||
| Di Filippo [32] | Biozzi ABH | 4 | 6–8 w | F | CFA | sc + ipl | 100 ug | U | Yes |
| Rats | |||||||||
| Bland [71] | Sprangue-Dawley | 8 | 4 d | M | E. coli (ATCC 15746) | sc | 1 × 105 CFU/g | 2 days | Yes |
| 70 days | Yes | ||||||||
| 98 days | Yes | ||||||||
| Williamson [72] | Sprangue-Dawley | 9 | 4 d | M | E. coli (ATCC 15746) | sc | 1 × 105 CFU/g | 2–3 months | Yes |
| Bilbo [73] | Sprangue-Dawley | 6 | 4 d | M | E. coli (ATCC 15746) | sc | 1 × 105 CFU/gram | 2 months | No* |
| 16 months | Yes | ||||||||
| Bilbo [42] | Sprangue-Dawley | 8 | 4 d | M | E. coli (ATCC 15746) | sc | 1 × 105 CFU/g | 56 days | Yes |
| Bilbo [35] | Sprangue-Dawley | 6 | 4 d | M | E. coli (ATCC 15746) | sc | 1 × 105 CFU/g | 56 days | No |
| Bilbo [37] | Sprangue-Dawley | 8 | 4 d | M | E. coli (ATCC 15746) | sc | 1 × 106 CFU | 2 h | No |
| 8 h | No | ||||||||
| 1 day | Yes | ||||||||
| 2 days | No | ||||||||
| 3 days | Yes | ||||||||
| 2-3 months | Yes | ||||||||
| Raghavendra [4] | Sprangue-Dawley | 4 | 175–200 gr | M | CFA | ipl | 100 ul | 4 h | Moderate |
| 4 days | Yes | ||||||||
| 2 weeks | Yes | ||||||||
| Liu [30] | Lewis | 6 | 2 m | F | Heat-killed M. butyricum | id | 1.5 mg | 3 weeks | Moderate* |
| 12 m | Yes | ||||||||
| Wu [29] | Lewis | 6 | 100–110 gr | F | Heat-killed M. butyricum | id | 25 mg/kg | 2 weeks | Yes |
| 3 weeks | Yes | ||||||||
| 4 weeks | Yes |
Column time of termination is the time from the (first) LPS challenge
Abbreviations: U unknown, F female, m months, d days, ip intraperitoneal, ipl intraplantar, M male, B both sexes, w weeks, gr gram, sc subcutaneous, id intradermal, N number of animals per group, CFA complete Freund’s adjuvant
*Did not express data in statistical values, no statistical information
Six studies from one research group focused on the effect of systemic infectious challenge in early life. In these studies, infant rats (P4) were challenged with E. coli 1x 105 colony-forming units (CFU)/g subcutaneously. Immunohistochemistry with Iba-1 revealed activated microglia in the hippocampus 2 days after challenge and increased CD11b expression in the hippocampus area up to 3 months after infection. Another study using S. typhimurium (SL3261; 106 CFU) showed increased expression of CD11b and CD68 in the thalamus 7 days after the challenge. Interestingly, CD11b and CD68 expression returned to baseline levels 3 weeks after challenge. At all time points during this experiment, microglia were morphological ramified with fine processes.
Studies evaluating challenge with heat-killed bacteria used CFA, a solution composed of heat-killed and dried mycobacteria (usually M. butyricum and/or M. tuberculosis). Injecting CFA intradermal or intraplantar induces adjuvant arthritis, a model of chronic peripheral inflammation. One study showed CD11b messenger ribonucleic acid (mRNA) expression in brainstem and forebrain 4 h, 4 days, and 2 weeks after CFA challenge [4]. Another study challenging rats with 25 mg/kg CFA showed cortical microglial activation immunohistochemistry with Iba-1 2, 3, and 4 weeks after inoculation [29]. One study evaluated the role of age and compared rats of two and 12 months old, using 1.5 mg challenge of CFA, showing morphologically activated microglia revealed in the CA1 region of the hippocampus for both age groups, three weeks after challenge [30]. However, no CD68 or IL-1β-positive microglial cells were detected in the brains of 2-month-old rats, while the expression of CD68 and IL-1β was significantly increased in hippocampal CA1 region of 12-month-old rats compared to control rats.
CFA is also used to induce experimental allergic encephalomyelitis (the EAE model), an animal model for multiple sclerosis (MS), by peripheral injections of CNS tissue homogenized with these heat-inactivated mycobacteria. Only the control groups (CFA) could be included in the current review. One mouse study showed no microglial activation in the brainstem up to 3 weeks after inoculation with 150 ug CFA [31]. Another mouse study using 100 ug CFA on days 1 and 7 showed activated microglia in the CA1 region of the hippocampus 22 days after the last challenge [32].
Inflammatory parameters
Inflammatory parameters were reviewed if they were evaluated concomitantly with the microglial response. This was done in 23 of 51 studies (45 %).
Toll-like receptors
Seven studies evaluated TLRs on microglia, by qPCR, immunohistochemistry, in situ hybridization, or flow cytometry: four evaluated TLR-2 expression [3, 21, 33, 34] and three TLR-4 expression [4, 35, 36]. Three studies described TLR-2 upregulation [3, 21, 33] and two TLR-4 upregulation [4, 36]. Microglial activation was associated with TLR upregulation independent of type of challenge (LPS, E. coli, or CFA) or time point of evaluation. In those studies showing resting microglia after challenge, TLR expression was not different from the control group. One study challenged TLR-4 knockout mice with serial LPS injections of 1 mg/kg every day for 4 days, showing decreased microglial activation in the knockout mice as compared to wild-type animals (Appendix) [25]. Although TLR-2 is known for its recognition of lipopeptides, peptidoglycans (PGN), and LTA, all of which are cell wall components of gram-positive bacteria, TLR-2 upregulation was found after LPS challenge [3, 21, 33]. One study using a head-to-head comparison between LPS, PGN, and LTA challenges showed a profound transcriptional activation of TLR-2 only after LPS.
Cytokines and chemokines
TNF-α protein levels or mRNA expression were determined in 14 studies [3–9, 29, 32, 34, 37–40], by qPCR, ELISA, western blot, in situ hybridization, and immunohistochemistry, at 23 different time points after the challenge. Microglial activation was associated with increased expression of TNF-α in the brain, as compared to controls, at 12 time points: ranging from 3 h to 1 day after single LPS (n = 5), after multiple LPS (n = 1), or after a CFA challenge (n = 6). At four time points, microglial activation was described without increased TNF-α protein levels: 1 day after single LPS (n = 1), 1 and 3 days after E. coli (n = 2), or 22 h after CFA challenge (n = 1). One study observed an elevation of TNF-α mRNA and protein levels 30 min after a single LPS challenge [7], and this effect remained up to 10 months after the LPS administration. Interestingly, the studies evaluating E. coli challenge showed no differences in TNF-α concentration as compared to the control group, independent of whether or not microglia were activated [37].
Interleukin 1 beta (IL-1β) protein levels or mRNA expression were determined in 16 studies [4–6, 8, 9, 21, 22, 30, 32, 35, 37, 39–43], at 24 different time points after challenge. At 13 time points microglial activation was present in combination with increased IL-1β protein levels or mRNA expression patterns, from 4 h to 1 week after LPS challenge (n = 7), from 1 and 56 days after E. coli challenge (n = 2), and from 4 h to 3 weeks after CFA challenge (n = 4). At three time points, microglia were activated but no IL-1β increase was measured; 1 day after LPS (n = 1), 2 days after E. coli (n = 1), and 22 h after CFA challenge (n = 1). In studies showing microglia in resting or moderately activated states, brain IL-1β levels were comparable to that of the control group (n = 7). IL-1 receptor (IL-1R) knockout mice were used in one study, evaluating a single dose of 1 mg/kg of LPS (Appendix) [22], and IL-1R knockouts had no microglial activation in the CA1 region of the hippocampal area, in contrast to the wild-type group. IL-1R antagonist (IL-1Ra) injection in wild-type mice just before the LPS challenge also prevented microglial activation, suggesting an important role of IL-1 in the activation of microglia.
Interleukin 6 (IL-6) protein levels or mRNA expression were determined in seven studies [4–6, 8, 9, 22, 37], at 17 different time points after challenge. At five time points, microglial activation was present in combination with increased protein levels or mRNA expression patterns, at 12 h and 1 week after a single LPS (n = 2) and at 4 h, 4 days, and 2 weeks after CFA challenge (n = 3). At five time points, microglia were activated but brain IL-6 levels were similar to controls: 1 day after single LPS (n = 3) and 1 and 3 days after E. coli challenge (n = 2). Animals challenged with E. coli had similar IL-6 concentration as compared to the controls, independent of whether microglia were activated (n = 2) or not (n = 3).
Interleukin 10 (IL-10) protein levels or mRNA expression were determined in seven studies [5, 6, 8, 21, 29, 37], at 12 different time points after challenge. At four time points, microglia were activated in combination with increased protein levels or mRNA expression patterns: 4 h and 1 day after systemic LPS (n = 2) and 3 and 4 weeks after CFA challenge (n = 2). At four time points, microglia were activated but brain IL-10 levels were similar to controls: 1 day after LPS (n = 1), 1 and 3 days after E. coli (n = 2), and 2 weeks after CFA challenge (n = 1). One study showed microglial activation after LPS challenge but decreased levels of IL-10 in the brain [8].
Two studies measured transforming growth factor beta (TGF-β) mRNA expression with qPCR 1 day after a single LPS challenge [5, 6]. Microglial cells were activated in both studies. One study measured brain TGF-β mRNA expression and found increased concentrations [5]. The other study measured TGF-β in isolated microglia and found no differences between challenged and control groups [6]. Two studies measured monocyte chemotactic protein 1 (MCP-1) mRNA expression in the brain [5, 8], showing that microglial activation was associated with MCP-1 upregulation. Fractalkine receptor (CX3CR1) knockout mice were used in two studies evaluating a single dose of 10 mg/kg LPS [23] and multiple LPS challenges (20 ug for 4 days) (Appendix) [24]. One day after the (last) LPS challenge, immunohistochemistry with Iba-1 revealed enhanced microglial activation in the hippocampal area of the CX3CR1 knockout mice as compared to wild-type animals.
Blood–brain barrier
The blood–brain barrier (BBB) was examined in eight studies [27, 28, 31, 36, 37, 44–46]. Four studies showed disruption of the BBB (challenge was LPS in two studies, either intraperitoneal or intravenous, E. coli subcutaneously in one study and CFA intraperitoneal in the other study) [28, 31, 37, 45], three studies showed intact BBB (challenge was LPS in all three studies, either intraperitoneal [n = 2] or intravenous [n = 1]) [36, 44, 46], and one study showed inconclusive results (challenge was LPS) [27]. Studies were highly variable with respect to methods used to define the integrity of the BBB (fluorescent sodium, protein and fibrinogen extravasation, exogenous horseradish peroxidase, Evans blue dye, ribosomal ribonucleic acid (rRNA) of E. coli, fluorescent LPS, and the invasion by blood monocytes or macrophages).
Discussion
Experimental studies have shown that peripheral inflammatory stimuli, such as LPS, cause a profound immunological response in the brain resulting in microglial activation. After a single challenge of LPS, microglia were moderately active within 3 h after administration, reaching their profound activation state after 8 h to 2 days and subsequently return to their normal resting state after 7 days. Interestingly, cytokine expression levels in the brain and activation markers may remain elevated for months after a single LPS challenge. These experiments also showed that systemic challenge with live bacteria, mainly the gram-negative bacteria E. coli and S. typhimurium, causes microglial activation. A gram-negative bacterium contains approximately 10−15 g of LPS, which implicates that the LPS dosages used in the included animal studies are supernatural. Consequently, the microglial response of challenge with bacteria is less profound as compared to that found in the experiments using a challenge with supernatural LPS doses. LPS as a peripheral inflammatory challenge is much easier to use than live bacteria: there is no waiting for bacteria to be cultured, no monitoring of bacteria to grow in midlog phase, and no time is lost with harvesting, wash and dilute the bacteria in the right amount. In addition, there is no danger of contamination, and therefore, the laboratory and animal facility do not have to comply with special safety matters. However, because of the differences in microglial response between a peripheral challenge with LPS or live bacteria, the clinical relevance of using LPS in these animal models is questionable. Experimental studies using live bacteria suggest an important role of age in the process of microglial activation after systemic challenge, although further research is needed to confirm this. The observed effects support the central role of microglial response in the development of sepsis-associated delirium and poor functional outcome after sepsis, in particular in the elderly population [15].
The mechanisms connecting systemic inflammatory challenge and microglial activation remain unclear. Several pathophysiological mechanisms have shown to play a role in this process. Microglia may be activated through primary autonomic afferents—in particular the vagal nerve—by active BBB transport of pro-inflammatory chemo- and/or cytokines, passive transport of pro-inflammatory products via the circumventricular organs, or by signaling the epithelial cells of the blood–brain barrier [47]. Microglial activation was associated with upregulation of TLRs, independent of type of challenge or time point of evaluation. In studies in which microglial activation was not observed, TLR expression was not different from the control group [34, 35]. Moreover, microglia could not be activated in TLR receptor knocked-out mice [25]. Microglial activation was associated with elevated levels of pro-inflammatory mediators in the brain.
Age is an important intrinsic factor determining the level of microglial activation after a systemic inflammatory challenge. The normal aging process induces changes in microglial phenotype, and these age-related changes are also called “priming” [48]. Two studies compared the effect of age on microglia after a peripheral challenge [21, 30]. Systemic LPS challenges caused a hyperactive microglial response in the brain of aged mice, associated with higher induction of inflammatory IL-1β and anti-inflammatory IL-10 [21]. Peripheral CFA injection induced hippocampal microglial activation in middle-aged rats and moderate activation in young rats. In these experiments, microglial activation in middle-aged rats was associated with neurocognitive deficits [30]. Aging-induced immune senescence occurs in the brain as age-associated microglial senescence, which renders microglia to function abnormally and may eventually promote neurodegeneration.
Evidence suggests that microglia act neurotoxic when fully activated (M1 phenotype) [49], whereas other studies show that activated microglia show more diversity and can have a role in remodeling and repair as well (M2 phenotype) [50, 51]. The M1 immune response of microglia is triggered by the activation of TLRs via pathogen-associated molecular patterns (PAMPs) or intracellular proteins released from damaged neurons; other M1 triggers are complement 1q (C1q) and adenosine triphosphate (ATP) released from astrocytes in response to neuronal injury [11, 12]. These activated M1 microglia produce the pro-inflammatory mediators TNF-α, IL-1β, and IL-6 [11, 12]. M2 activation is induced by signals from apoptotic cells that activate triggering receptor expressed by myeloid cells-2 (TREM2) such as heat shock protein 60 (Hsp60), or by anti-inflammatory cytokines, such as interleukin 14 (IL-14) and interleukin 13 (IL-13). M2-activated microglia have a role in remodeling and repair, triggering anti-inflammatory responses via release of TGF-β and IL-10 [11–13]. While beyond the scope of this review, several studies show an association between systemic LPS challenge, microglial activation, and cognitive deficits in mice [52–55]. These studies demonstrate that systemic LPS challenge causes cyclooxygenase-1 (COX-1), COX-2, and inducible nitric oxide synthase (iNOS) expression in the brain, which is hypothesized to cause susceptibly to cognitive deficits in mice [52, 53, 55]. Regarding these facts, it is imperative—when it comes to defining microglial activation—to focus not only on proliferation and morphology but also examine pro- and anti-inflammatory markers on or around microglia. If more homogeneous data on these inflammatory markers, in relation to microglial activation, would be available, then bigger steps can be made in understanding the pathogenesis in why neuro-inflammation occurs when a systemic challenge is administrated. However, less than half of the studies (45 %) included in this review contain data on inflammatory mediators. When microglia were activated, an increase in TLR-2, TLR-4, TNF-α, and IL-1β mRNA expression or protein levels in the brain was seen in most studies. A few studies examined anti-inflammatory markers IL-10 (n = 6), TGF-β (n = 2), and MCP-1 (n = 2).
Studies have shown that microglial cells express various neurotransmitter receptors [56], and neurotransmitters could also exert pro- and anti-inflammatory effects on microglial cells. For example, ionotropic glutamate receptors (iGluRs) can modulate TNF-α release, gamma-amino-butytric acid (GABA) receptors modulate IL release (IL-6 and IL-12) and adrenergic, dopaminergic, and cholintergic receptors exhibit anti-inflammatory effects [57]. These receptors add another challenge on the pathogenesis on neuro-inflammation and should not be ignored.
Heterogeneity among the included 51 studies hampered the opportunity for a synthesis, e.g., quantitatively, in this systematic review. Studies investigated several animal species and inflammatory stimuli at different time points. Lack of adequate experimental description, power calculations for animal group sizes, reported baseline measurements of animals between groups, or handling of outlined or missing data further limited generalization of the results. Nevertheless, this review provides a valuable overview of current knowledge.
Conclusion
Experimental studies have shown that peripheral challenge with LPS causes a profound immunological response in the brain resulting in microglial activation, but systemic challenge with live bacteria causes microglial activation as well. However, the microglial response of challenge with bacteria is less profound as compared to that found in the experiments using a challenge with supernatural LPS doses. The mechanisms connecting systemic inflammatory challenge and microglial activation remain unclear, but age is an important intrinsic factor determining the level of microglial activation after a systemic inflammatory challenge. Heterogeneity among the included studies hampered the opportunity for a synthesis, e.g. quantitatively, in this systematic review. Future experimental studies should opt for mouse models, use live bacteria as well as standardized and quantitative measurements of microglial activation, for example, with flow cytometry, and focus on the role of the aging brain. These studies should apply with the current standards of animal experiments [19]. For optimal external validation, experimental studies should investigate the role of aging on microglial activation following a systemic infection with live bacteria, analogous to the human situation: the clinical problem of long-term poor outcome of sepsis-associated delirium in elderly patients (Table 4).
Table 4.
Keypoints review
| Keypoints |
| • Systemic challenge with LPS and live bacteria cause a profound immunological response in the brain resulting in microglial activation. |
| • Microglial response after challenge with bacteria is less profound as compared to challenge with LPS, which makes the clinical relevance of using LPS in these animal models questionable. |
| • When defining microglial activation, researcher should not only focus on proliferation and morphology, but also examine pro-and anti-inflammatory markers indicating M1/M2 responses. |
| • Mechanisms connecting systemic inflammatory challenge and microglial activation remain unclear, but age is an important intrinsic factor. |
| • Future experimental studies on studying systemic inflammation and microglial response should do the following: −use mouse models −use live bacteria −use standardized and quantitative measurements of microglial activation −focus on the role of the aging brain −apply with the current standards of animal experiments |
Abbreviations
- ATCC
American Type Culture Collection
- ATP
adenosine triphosphate
- BBB
blood–brain barrier
- C1q
complement 1q
- CD11b
cluster of differentiation 11b
- CD45
cluster of differentiation 45
- CD68
cluster of differentiation 68
- CFA
complete Freund’s adjuvant
- CFU
colony-forming units
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- CX3CR1
fractalkine receptor
- E. coli
Escherichia coli
- GABA
gamma-amino-butytric acid
- Hsp60
heat shock protein 60
- IB4
Griffonia symplicifolia isolectin B4
- Iba-1
ionized calcium-binding adaptor molecule 1
- iGluRs
ionotropic glutamate receptors
- IL-10
interleukin 10
- IL-13
interleukin 13
- IL-14
interleukin 14
- IL-1R
interleukin-1 receptor
- IL-1Ra
interleukin-1 receptor antagonist
- IL-1β
interleukin 1 beta
- IL-6
interleukin 6
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- M. butyricum
Mycobacterium butyricum
- M. tuberculosis
Mycobacterium tuberculosis
- MARCO
macrophage receptor with collagenous structure
- MHCII
major histocompatibility complex II
- MCP-1
monocyte chemotactic protein 1
- mRNA
messenger ribonucleic acid
- MS
multiple sclerosis
- P
postnatal day
- PAMPs
pathogen-associated molecular patterns
- PGN
peptidoglycans
- qPCR
quantitative polymerase chain reaction
- rRNA
ribosomal ribonucleic acid
- S. typhimurium
Salmonella typhimurium
- TGF-β
transforming growth factor beta
- TLR
Toll-like receptor
- TLR-2
Toll-like receptor 2
- TLR-4
Toll-like receptor 4
- TLRs
Toll-like receptors
- TNF-α
tumor necrosis factor alpha
- TREM2
triggering receptor expressed by myeloid cells-2
Appendix
Table 5.
Intraperitoneal challenge with lipopolysaccharide (LPS) in knock-out mice
| Study | Knock out gene | N | Age | Sex | Type of LPS | Dose | Time of termination | Microglial activation | Compared to wild-type |
|---|---|---|---|---|---|---|---|---|---|
| Bhaskar [23] | CX3CR1−/− | U | 2 m | U | U | 10 mg/kg | 1 d | Yes* | More* |
| Terrando [22] | IL-1R−/− | 4 | 12–14 w | M | E. coli (O111:B4) | 1 mg/kg | 1 d | No | Less* |
| 3 d | No | Less* | |||||||
| 1 w | No | Same* | |||||||
| Cardona [24] | CX3CR1−/− | U | U | U | U | 20 ug q.d. 2× | 4 d | Yes* | More* |
| Chen [25] | TLR4−/− | 4 | 8–12 w | M | E. coli (O55:B5) | 1 mg/kg q.d. 2× | 4 d | No* | Less* |
Column time of termination is the time from the (first) LPS challenge. All challenges were intraperitoneal
Abbreviations: U unkown, m months, d days, M male, w weeks, q.a.d every other day, N number of animals per group
*Did not express data in statistical values, no statistical information
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ICMH and CH searched PubMed and Embase database, reviewed articles for inclusion and exclusion criteria, and extracted and analyzed data. All authors participated in its design and coordination. ICMH and DB wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Financial support
This work was supported by ZonMW (WAvG, TOP grant #40-00812-98-10017). DvdB is supported by The European Research Council (ERC Starting Grant #281156) and ZonMW (Vidi grant #016.116.358).
Contributor Information
Inge C.M. Hoogland, Email: i.c.hoogland@amc.uva.nl
Carin Houbolt, Email: carin.houbolt@gmail.com.
David J. van Westerloo, Email: D.J.van_Westerloo@lumc.nl
Willem A. van Gool, Email: w.a.vangool@amc.uva.nl
Diederik van de Beek, Phone: +31205663842, Email: d.vandebeek@amc.uva.nl.
References
- 1.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- 2.Witlox J, Eurelings LS, de Jonghe JF, Kalisvaart KJ, Eikelenboom P, van Gool WA. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304(4):443–51. doi: 10.1001/jama.2010.1013. [DOI] [PubMed] [Google Scholar]
- 3.Laflamme N, Soucy G, Rivest S. Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J Neurochem. 2001;79(3):648–57. doi: 10.1046/j.1471-4159.2001.00603.x. [DOI] [PubMed] [Google Scholar]
- 4.Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20(2):467–73. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 5.Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, et al. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation. 2008;5:38. doi: 10.1186/1742-2094-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55(4):412–24. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- 7.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55(5):453–62. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sehgal N, Agarwal V, Valli RK, Joshi SD, Antonovic L, Strobel HW, et al. Cytochrome P4504f, a potential therapeutic target limiting neuroinflammation. Biochem Pharmacol. 2011;82(1):53–64. doi: 10.1016/j.bcp.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 9.Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9(1):212. doi: 10.1186/1742-2094-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 11.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2015;20. [DOI] [PubMed]
- 12.Moehle MS, West AB. M1 and M2 immune activation in Parkinson's disease: foe and ally? Neuroscience. 2014 doi: 10.1016/j.neuroscience.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 14.Lemstra AW, Woud JCG. i't, Hoozemans JJ, van Haastert ES, Rozemuller AJ, Eikelenboom P, et al. Microglia activation in sepsis: a case–control study. J Neuroinflammation. 2007;4:4. doi: 10.1186/1742-2094-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. 2010;375(9716):773–5. doi: 10.1016/S0140-6736(09)61158-2. [DOI] [PubMed] [Google Scholar]
- 16.Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18(5):407–13. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Teeling JL, Perry VH. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience. 2009;158(3):1062–73. doi: 10.1016/j.neuroscience.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 18.MacLullich AM, Beaglehole A, Hall RJ, Meagher DJ. Delirium and long-term cognitive impairment. Int Rev Psychiatry. 2009;21(1):30–42. doi: 10.1080/09540260802675031. [DOI] [PubMed] [Google Scholar]
- 19.Hooijmans CR, Rovers MM, de Vries RB, Leenaars M, Ritskes-Hoitinga M, Langendam MW. SYRCLE’s risk of bias tool for animal studies. BMC Med Res Methodol. 2014;14:43. doi: 10.1186/1471-2288-14-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smithason S, Moore SK, Provencio JJ. Systemic administration of LPS worsens delayed deterioration associated with vasospasm after subarachnoid hemorrhage through a myeloid cell-dependent mechanism. Neurocrit Care. 2012;16(2):327–34. doi: 10.1007/s12028-011-9651-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23(3):309–17. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terrando N, Rei FA, Vizcaychipi M, Cibelli M, Ma D, Monaco C, et al. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care. 2010;14(3):R88. doi: 10.1186/cc9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–24. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32(34):11706–15. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu JT, Lee CH, Yoo KY, Choi JH, Li H, Park OK, et al. Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal CA1 region preconditioned with lipopolysaccharide. J Neurol Sci. 2010;296(1–2):69–78. doi: 10.1016/j.jns.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Stolp HB, Ek CJ, Johansson PA, Dziegielewska KM, Bethge N, Wheaton BJ, et al. Factors involved in inflammation-induced developmental white matter damage. Neurosci Lett. 2009;451(3):232–6. doi: 10.1016/j.neulet.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 28.Jiang-Shieh YF, Wu CH, Chien HF, Wei IH, Chang ML, Shieh JY, et al. Reactive changes of interstitial glia and pinealocytes in the rat pineal gland challenged with cell wall components from gram-positive and -negative bacteria. J Pineal Res. 2005;38(1):17–26. doi: 10.1111/j.1600-079X.2004.00170.x. [DOI] [PubMed] [Google Scholar]
- 29.Wu Z, Zhang J, Nakanishi H. Leptomeningeal cells activate microglia and astrocytes to induce IL-10 production by releasing pro-inflammatory cytokines during systemic inflammation. J Neuroimmunol. 2005;167(1–2):90–8. doi: 10.1016/j.jneuroim.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Wu Z, Hayashi Y, Nakanishi H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience. 2012;216:133–42. doi: 10.1016/j.neuroscience.2012.04.050. [DOI] [PubMed] [Google Scholar]
- 31.Rabchevsky AG, Degos JD, Dreyfus PA. Peripheral injections of Freund’s adjuvant in mice provoke leakage of serum proteins through the blood–brain barrier without inducing reactive gliosis. Brain Res. 1999;832(1–2):84–96. doi: 10.1016/S0006-8993(99)01479-1. [DOI] [PubMed] [Google Scholar]
- 32.Di FM, Chiasserini D, Gardoni F, Viviani B, Tozzi A, Giampa C, et al. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol Dis. 2013;52:229–36. doi: 10.1016/j.nbd.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 33.Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, et al. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2004;24(6):1340–9. doi: 10.1523/JNEUROSCI.4786-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bilbo SD, Wieseler JL, Barrientos RM, Tsang V, Watkins LR, Maier SF. Neonatal bacterial infection alters fever to live and simulated infections in adulthood. Psychoneuroendocrinology. 2010;35(3):369–81. doi: 10.1016/j.psyneuen.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 36.Chung DW, Yoo KY, Hwang IK, Kim DW, Chung JY, Lee CH, et al. Systemic administration of lipopolysaccharide induces cyclooxygenase-2 immunoreactivity in endothelium and increases microglia in the mouse hippocampus. Cell Mol Neurobiol. 2010;30(4):531–41. doi: 10.1007/s10571-009-9477-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. 2005;25(35):8000–9. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28(43):10825–34. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka S, Numazawa S, et al. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience. 2013;240:27–38. doi: 10.1016/j.neuroscience.2013.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Callaghan EK, Anderson ST, Moynagh PN, Coogan AN. Long-lasting effects of sepsis on circadian rhythms in the mouse. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0047087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okuyama S, Makihata N, Yoshimura M, Amakura Y, Yoshida T, Nakajima M, et al. Oenothein B suppresses lipopolysaccharide (LPS)-induced inflammation in the mouse brain. Int J Molecular Sci. 2013;14(5). [DOI] [PMC free article] [PubMed]
- 42.Bilbo SD, Newsum NJ, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Differential effects of neonatal handling on early life infection-induced alterations in cognition in adulthood. Brain Behav Immun. 2007;21(3):332–42. doi: 10.1016/j.bbi.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Puntener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buttini M, Limonta S, Boddeke HW. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem Int. 1996;29(1):25–35. doi: 10.1016/0197-0186(95)00141-7. [DOI] [PubMed] [Google Scholar]
- 45.Nishioku T, Dohgu S, Takata F, Eto T, Ishikawa N, Kodama KB, et al. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood–brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol. 2009;29(3):309–16. doi: 10.1007/s10571-008-9322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franciosi S, Ryu JK, Shim Y, Hill A, Connolly C, Hayden MR, et al. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2012;45(1):438–49. doi: 10.1016/j.nbd.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21(6):727–35. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39(1):19–34. doi: 10.1111/j.1365-2990.2012.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 50.Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147(4):867–83. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 51.Simard AR, Rivest S. Neuroprotective effects of resident microglia following acute brain injury. J Comp Neurol. 2007;504(6):716–29. doi: 10.1002/cne.21469. [DOI] [PubMed] [Google Scholar]
- 52.Lee YJ, Choi DY, Choi IS, Kim KH, Kim YH, Kim HM, et al. Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappaB in vitro and in vivo models. J Neuroinflammation. 2012;9:35. doi: 10.1186/1742-2094-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weberpals M, Hermes M, Hermann S, Kummer MP, Terwel D, Semmler A, et al. NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J Neurosci. 2009;29(45):14177–84. doi: 10.1523/JNEUROSCI.3238-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murray CL, Skelly DT, Cunningham C. Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1beta and IL-6. J Neuroinflammation. 2011;8:50. doi: 10.1186/1742-2094-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffin EW, Skelly DT, Murray CL, Cunningham C. Cyclooxygenase-1-dependent prostaglandins mediate susceptibility to systemic inflammation-induced acute cognitive dysfunction. J Neurosci. 2013;33(38):15248–58. doi: 10.1523/JNEUROSCI.6361-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farber K, Kettenmann H. Purinergic signaling and microglia. Pflugers Arch. 2006;452(5):615–21. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- 57.Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30(10):527–35. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Carnevale D, Mascio G, Ajmone-Cat MA, D’Andrea I, Cifelli G, Madonna M, et al. Role of neuroinflammation in hypertension-induced brain amyloid pathology. Neurobiol Aging. 2012;33(1):205–29. doi: 10.1016/j.neurobiolaging.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 59.Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect. 2011;119(6):807–14. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaushik DK, Mukhopadhyay R, Kumawat KL, Gupta M, Basu A. Therapeutic targeting of Kruppel-like factor 4 abrogates microglial activation. J Neuroinflammation. 2012;9:57. doi: 10.1186/1742-2094-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang J, Hwang H, Lee HW, Suk K. Microglia signaling as a target of donepezil. Neuropharmacology. 2010;58(7):1122–9. doi: 10.1016/j.neuropharm.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 62.Masocha W. Systemic lipopolysaccharide (LPS)-induced microglial activation results in different temporal reduction of CD200 and CD200 receptor gene expression in the brain. J Neuroimmunol. 2009;214(1–2):78–82. doi: 10.1016/j.jneuroim.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 63.Ha SK, Moon E, Lee P, Ryu JH, Oh MS, Kim SY. Acacetin attenuates neuroinflammation via regulation the response to LPS stimuli in vitro and in vivo. Neurochem Res. 2012;37(7):1560–7. doi: 10.1007/s11064-012-0751-z. [DOI] [PubMed] [Google Scholar]
- 64.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302(5651):1760–5. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 65.Wang Q, van Hoecke H, Tang XN, Lee H, Zheng Z, Swanson RA, et al. Pyruvate protects against experimental stroke via an anti-inflammatory mechanism. Neurobiol Dis. 2009;36(1):223–31. doi: 10.1016/j.nbd.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat. 2005;30(2–3):144–57. doi: 10.1016/j.jchemneu.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 67.Garcia-Bueno B, Serrats J, Sawchenko PE. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J Neurosci. 2009;29(41):12970–81. doi: 10.1523/JNEUROSCI.2373-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katafuchi T, Ifuku M, Mawatari S, Noda M, Miake K, Sugiyama M, et al. Effects of plasmalogens on systemic lipopolysaccharide-induced glial activation and beta-amyloid accumulation in adult mice. Ann N Y Acad Sci. 2012;1262:85–92. doi: 10.1111/j.1749-6632.2012.06641.x. [DOI] [PubMed] [Google Scholar]
- 69.Shankaran M, Marino ME, Busch R, Keim C, King C, Lee J, et al. Measurement of brain microglial proliferation rates in vivo in response to neuroinflammatory stimuli: application to drug discovery. J Neurosci Res. 2007;85(11):2374–84. doi: 10.1002/jnr.21389. [DOI] [PubMed] [Google Scholar]
- 70.Yin P, Li Z, Wang YY, Qiao NN, Huang SY, Sun RP, et al. Neonatal immune challenge exacerbates seizure-induced hippocampus-dependent memory impairment in adult rats. Epilepsy Behav. 2013;27(1):9–17. doi: 10.1016/j.yebeh.2012.12.015. [DOI] [PubMed] [Google Scholar]
- 71.Bland ST, Beckley JT, Young S, Tsang V, Watkins LR, Maier SF, et al. Enduring consequences of early-life infection on glial and neural cell genesis within cognitive regions of the brain. Brain Behav Immun. 2010;24(3):329–38. doi: 10.1016/j.bbi.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. J Neurosci. 2011;31(43):15511–21. doi: 10.1523/JNEUROSCI.3688-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bilbo SD. Early-life infection is a vulnerability factor for aging-related glial alterations and cognitive decline. Neurobiol Learn Mem. 2010;94(1):57–64. doi: 10.1016/j.nlm.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]