

Abstract
Introduction
In vitro human diagnostic (IVD) company recalls are a common practice aimed to either minimize a potential error or eliminate an existing failure. In this case report, we aim to provide a critical analysis of a recent IVD recall and to provide a practical framework about what to do when an IVD company recalls product(s) based on the International Organization for Standardization - ISO 15189:2012 standard.
Case report
In 2014, Abbott Laboratories® (Green Oaks, IL) published an urgent field safety notice regarding a product recall (Architect Intact parathyroid hormone (PTH) Assay List Number 8K25) with immediate action required. The IVD company explained the reasons for the recall as follows: i) Abbott has confirmed that a performance shift in the Architect Intact PTH assay has the potential to generate falsely elevated results on patient samples; ii) results generated with impacted lots may demonstrate a positive shift relative to those generated with previous reagent and/or calibrator lots. This issue may also impact established Architect Intact PTH reference ranges; iii) the magnitude of shift averages approximately 13% to 45%; iv) Abbott Architect Intact PTH controls do not detect the shift; and v) all current reagent, calibrator, and control inventory are impacted. The recall could have resulted in ~40,000 inaccurate laboratory tests reported by 18 laboratories from Italy (Lombardy region).
Conclusion
IVD company recalls have a serious impact on the patient safety and require a thorough investigation and responsible approach to minimize the possible damage. Medical laboratories accredited according to the ISO 15189 standard have procedures in place to manage such situations and ensure that patient safety is maintained when such recalls are issued.
Key words: clinical laboratory techniques, patient safety, laboratory variability, reference standards, quality control, parathyroid hormone
Introduction
Analytical processes in laboratories are error susceptible. However, failures in adhering to best practices of either analytical procedures or quality management can jeopardize patient safety (1, 2). Experts have encouraged and developed productive discussions about this topic and the relevant assumptions of error responsibility (3, 4). With an objective to guarantee patient safety, medical laboratory directors are continuously improving their quality management system through certification and accreditation. Medical laboratory certification (i.e., by the International Organization for Standardization-ISO 9001:2008 standard) aims to assess conformity to a specific documented system without verifying the technical competence, whereas accreditation (i.e., by ISO 15189:2012 standard) is a process aimed at providing an independent appraisal and recognition—by one expert medical laboratory professional—of the specific competence of testing (5, 6). The processes of validation, verification, and non-conformity management are essential for quality system maintenance in accredited laboratories (6). Unfortunately, only few medical laboratories worldwide are accredited (7), and some clinical directors prefer certification to the accreditation process when the laboratories are located inside a hospital.
To verify if each laboratory test is both precise and accurate, quality control (i.e., both internal and external) procedures are performed. Presently, every in vitro human diagnostic (IVD) company provides proprietary internal quality control materials for each product kit. However, sometimes a recall can happen because of issues occurring in the production/control phases. Briefly, according to the European Commission, recall means any measure aimed at achieving the return of a device that has already been made available to the end user (8). Nevertheless, laboratory professionals are aware that IVD company recalls are an usual practice aimed to either minimize a potential error or eliminate an existent failure (9). In this case report we aimed to critically analyze a recent IVD recall and to provide a practical framework about what to do when an IVD company recalls product(s), based on the ISO 15189:2012 standard (6).
Case report
On February 12th 2014, the Abbott Laboratories® (Green Oaks, IL) published one urgent field safety notice (recall) regarding an IVD product kit: Architect Intact parathyroid hormone (PTH) Assay List Number 8K25, requiring immediate action (10).
The IVD company (10) explained the recall as follows:
A performance shift in the assay may generate falsely elevated results on samples;
Consequent results may demonstrate a positive shift in respect to previous reagent/calibrator lots, with a possible impact on established reference intervals;
The shift magnitude is averaging approximately 13% to 45%;
Kit-included controls do not detect the shift;
All kit inventory (i.e., reagent, calibrator, and control) is impacted.
As common practice, the Italian Health Ministry started to disseminate news about this important recall on the website a few days later (February 21th, 2014) (11). Obviously, the recall had an impact only on the laboratories that were using this kit.
Nonetheless, Italian mass media (12, 13) widely propagated the news of the Abbott Laboratories® recall and the consequent impact on the medical laboratory of the hospital involved. Briefly, the following was reported: i) ~3,500 patients went to the hospital laboratory for PTH testing between February 2013 to February 2014 and had their tests recalled due to unreliable PTH results; ii) staff from N.A.S. (Nucleo Antisofisticazioni e Sanitŕ, the Italian health enforcing agency (14) comparable to the United States Food and Drug Administration (FDA)) requested all relevant documents in order to justify the substantial patient recall i.e., the recall report from Abbott, the official business agreement between the IVD company and medical laboratories, the corrective action plan, a list of patients with recalled test results, and a list of physicians contacted by the laboratory professionals; iii) there was a quest by all laboratories that used the same kit (N = 18) for accurate PTH test results (15, 16).
Discussion
Medical errors that jeopardize patient safety have attracted a great deal of public attention, especially when the situation demands the review of a large numbers of patient records or a recall of patient test results, as in this Italian case. Recently, the United States Department of Justice released that another IVD company was penalized to pay to United States $262 million plus interest to resolve malpractices related to the Advantage Intact PTH assay and four other manufactured assays (17).
To the best of our knowledge, the laboratories involved in the Italian recall were not accredited under the ISO 15189 standard; moreover, they were unable to recognize the above problem before the Abbott recall. It is likely that the laboratory managers failed to satisfy the formal method verification and did not provide to their quality managers the essential information for performing the needed preventive/corrective actions regarding this non-conformity.
According to the ISO 15189 standard, laboratory quality managers should: i) select both analyzers and reagents, which have been validated by the producer for their intended use and show preference to procedures specified in the IVD instructions for use (i.e., the datasheet from the intact PTH assay for Architect, Abbott Laboratories®); ii) verify the performance characteristics declared by the producer to confirm the datasheet information before it is introduced into routine use; and iii) plan quality control procedures to verify the quality of the results, e.g., by internal- and external-quality controls (6). Probably Abbott laboratory-customers’ had performed their internal quality control using exclusively proprietary (Abbott) PTH controls. Otherwise, PTH result variability could have been prevented by third-party control materials-independent from calibrator materials-either instead of or in addition to any control materials supplied by the analyzer manufacturer (Abbott). Moreover, the reported variability was undetectable when using Abbott Architect Intact PTH controls (10). However, if this implies that IVD companies provide quality control materials that are unable to detect non-conformity in their IVD diagnostic medical devices, then the same IVD companies should abstain from providing quality control materials (8, 18, 19).
The PTH external quality assessment (EQA) annual report of 2013 (when laboratories used the kit recalled by Abbott in February 2014) showed producers’ inter-method “inaccuracy” for PTH quality samples (i.e., with low-, medium- and high-concentrations of PTH). The inaccuracy of Abbott-Architect for PTH assay were higher than either Siemens-Advia Centaur, Siemens-Immulite, DiaSorin-Liaison, or Tosoh-AIA for samples with both low- and medium-concentration of PTH (20). Laboratory quality managers that used Architect to measure PTH could have assessed the data from the EQA report plus the internal quality control data to investigate the processed sample commutability following CLSI EP14-A3 document (21). With the CLSI EP14-A3 protocol, laboratory quality managers are able to distinguish between the effects caused by measurement procedure malfunctions and those caused by the use of artificial or human-based sample processing (e.g., quality control samples). Moreover, from an analytical point-of-view, the bias due to the Intact PTH assay of Abbott Laboratories on Architect is not a new outcome. Monge et al. compared intact PTH from Architect (Abbott Laboratories®, Green Oaks, IL, USA) with that from Elecsys (Roche Diagnostics, Meylan, France) and the PTH results from the Architect kit were significantly higher (approximately +30%) than those obtained using the kit from Elecsys (22). However, no adjustments were reported by Abbott leaving us to speculate as to whether or not the 18 laboratories involved, belonging to the same Italian region (i.e., Lombardia) and using the same kit on about 40,000 patient samples, undertaken any corrective action. Was the 13% to 45% shift recently claimed by Abbott in addition to the already known +30% (demonstrated by Monge et al. (22))? Should it be regarded as a further correction? In the latter hypothesis, such skewed PTH analyses could undoubtedly jeopardize patient safety.
Clinically, the measure of PTH is a second-level examination, requested by physicians with other tests: e.g., calcium to elucidate a hyperparathyroidism hypothesis. Consequently, most patients with hyperparathyroidism show both elevated PTH and calcium levels (23). Normocalcemic primary hyperparathyroidism, characterized by an increased PTH concentration in the absence of hypercalcemia, has been identified (24). In the present case, dutiful physicians may have abstained from the appropriate patient treatments’ as a consequence of the large average shift reported by Abbott (up to 45%) (10). It is well-known that PTH shows a rather wide biological variation, both within- and between-subjects (25.3% CVI and 43.4% CVG, respectively), and a low index of individuality that advices against the use of the reference interval and rather suggests the longitudinal evaluation of the same individual (25). Therefore, each patient should have increasing PTH levels with time, and relevant feedback to the laboratory should have arrived. However, this was not the case, suggesting that individual PTH data was only compared with the reference interval and not against the patient’s previous results, which is the preferred way for evaluating patient’s data over time, especially for analytes such as PTH (26).
When an IVD company shares their recall, the laboratory quality managers should: i) look for all results reported using either the reagent or instrument affected; ii) classify the results reported based on the potential risk involved in misleading physicians (i.e., borderline results that require possible exchanges regarding “clinical decision limits”); iii) find alternative methods to re-test the patient samples that showed borderline results (the method should be selected based on both similar performance indicated in the EQA report and commutability) and if the patient sample is missing, the patient should be contacted for new sample collection; and iv) formally inform the physicians (or whoever ordered the laboratory test) that the results from the 2nd determination might modify the clinical decision; otherwise, the formal communication to the physician is facultative (if the 2nd result remains like the 1st), but all results should be traceable and maintained in the laboratory (Figure 1).
Figure 1.
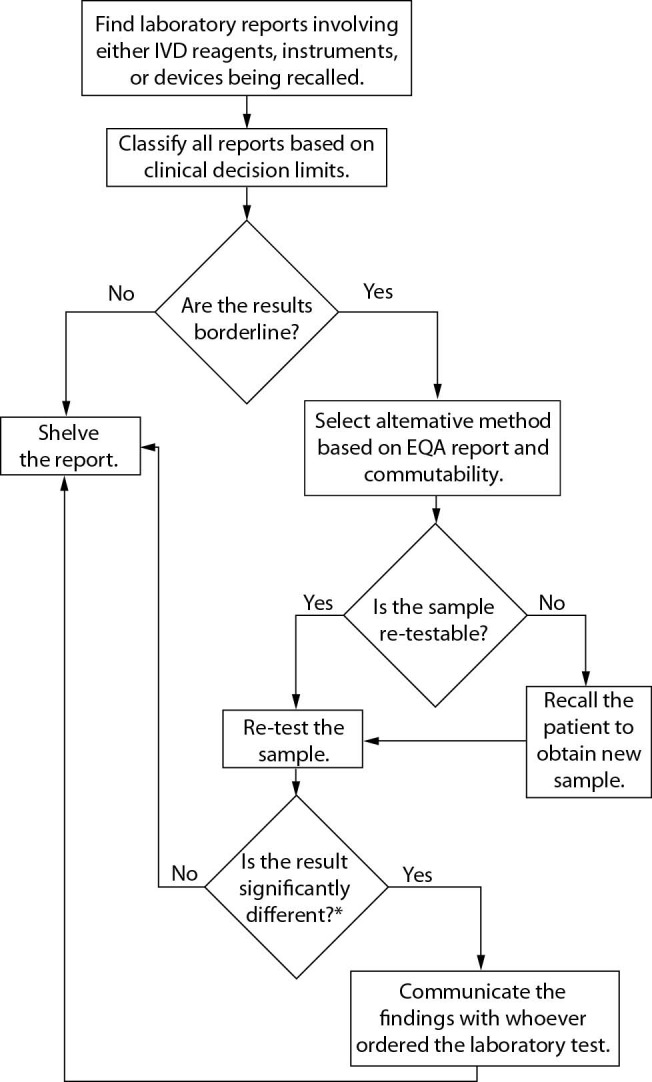
Flow chart outlining the ideal IVD company recall response protocol. *We suggest classifying a potential and clinically significant change in the 2nd determination using the reference change value (RCV) equation as follows: RCV = 21/2 × Z × (CVI2 + CVA2)1/2; where Z is a constant depending on the probability, 1.96 is most often considered significant, that is, P < 0.05, CVI is the within-subject biological variation obtained from the Westgard database (27), and CVA is the analytical coefficient of variation from our internal quality control. Critical Difference (CD) needs to be determined according to the formula: CD = [(determination before IVD company recall – determination after IVD company recall “second method”) / determination before IVD company recall] × 100%. Finally, the CD should be compared to the RCV. Only a CD higher than the RCV should be considered clinically significant. Desirable specification for imprecision (DSI) derived from biologic variation (27) could be used as an acceptance criteria for re-testing patient samples after an IVD company recall.
This case report is by itself an indicator that harmonization among IVD producers is both urgent and necessary. The above laboratories (i.e., 18 laboratories from Northern Italy) needed to replace the Abbott method after the recall. So, quality managers should revise the associated reference intervals and clinical decision values taking into account all causes of inaccuracy (6). Harmonization among IVD producers would enable laboratories to replace their methods from one manufacturer to another, without method verification. Unfortunately, this is not currently possible. Consequently, the agreement between different IVD companies (i.e., Abbott with another IVD company) to guarantee either instrument or reagent replacement when a product’s failure occurs is not enough. Case-by-case evaluation should be performed by laboratory quality managers in order to make the best choice when replacement is needed.
In conclusion, the major criticism of the recent Abbott® recall (10) is about its impact on the patient safety. Medical laboratories accredited according to the ISO 15189 standard (6) are able to guarantee patient safety when IVD companies start a recall procedure. Presently, the main concern among all laboratory professionals, being informed about a company’s recall, is the quality. All the documents requested by the NAS (14) (see above) from the laboratory of the Italian hospital are easily traceable, and such is the case with every laboratory accredited by the ISO 15189:2012 standard (6). In our opinion, the medical laboratory accreditation process is the best way to guarantee patient safety in laboratory diagnostics. We also believe that the procedure for industry recall, viewed as a common practice to minimize a potential error, should be revised, and harmonization among IVD manufacturers should be considered with highest priority.
Footnotes
None declared.
References
- 1.Lippi G, Plebani M, Simundic AM. Quality in laboratory diagnostics: from theory to practice. Biochem Med (Zagreb). 2010;20:126–30. 10.11613/BM.2010.014 [DOI] [Google Scholar]
- 2.Sonntag O. Quality in the analytical phase. Biochem Med (Zagreb). 2010;20:147–53. 10.11613/BM.2010.017 [DOI] [Google Scholar]
- 3.Lippi G, Simundic AM. Total quality in laboratory diagnostics. It’s time to think outside the box. Biochem Med (Zagreb). 2010;20:5–8. 10.11613/BM.2010.001 [DOI] [Google Scholar]
- 4.Gallagher TH, Mello MM, Levinson W, Wynia MK, Sachdeva AK, Snyder Sumalsy L, et al. Talking with patients about other clinicians’ error. N Engl J Med. 2013;369:1752–7. 10.1056/NEJMsb1303119 [DOI] [PubMed] [Google Scholar]
- 5.International Organization for Standardization. Quality management systems - Requirements ISO 9001 document. Geneva, Switzerland: International Organization for Standardization; 2008. [Google Scholar]
- 6.International Organization for Standardization. Medical laboratories - Requirements for quality and competence ISO document 15189. Geneva, Switzerland: International Organization for Standardization; 2012. [Google Scholar]
- 7.Huisman W. European medical laboratory accreditation. Present situation and steps to harmonisation. Clin Chem Lab Med. 2012;50:1147–52. 10.1515/cclm-2011-0586 [DOI] [PubMed] [Google Scholar]
- 8.EUROPEAN COMMISSION. Proposal for a regulation of the european parliament and of the council on in vitro diagnostic medical devices. Brussels: EUROPEAN COMMISSION; 2012; Available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=COM:2012:0541:FIN:EN:PDF. Accessed: 28th March 2014
- 9.Valenstein PN, Alpern A, Keren DF. Responding to large-scale testing errors. Am J Clin Pathol. 2010;133:440–6. 10.1309/AJCPXLZE0YYNID0X [DOI] [PubMed] [Google Scholar]
- 10.Abbott. Urgent Field Safety Notice - Product Recall - Immediate Action Required. Abbott; 2014; Available at: http://www.imb.ie/images/uploaded/documents/SN201405_Abbott_ARCHITECTIntactPTHAssayReagentsCalibratorsControls_V20026_FSN_210214.pdf. Accessed: 14th March 2014.
- 11.Italian Health Ministry. [Avvisi di sicureza 300651]. Italian Health Ministry; 2014; Available at: http://www.salute.gov.it/portale/news/p3_2_1_3_1_1.jsp?menu=notizie&p=avvisi&tipo=dispo&id=2863. Accessed: 14th March 2014. (in Italian)
- 12.De Montis L. [Crema, anali sbagliate. Test da ripetere per 2500 persone]. il Giornale; 2014; Available at: http://www.ilgiornale.it/news/cronache/crema-analisi-sbagliate-test-ripetere-2500-persone-1001469.html. Accessed: 14th March 2014. (in Italian)
- 13.Crema, analisi sbagliate in ospedale. I Nas sequestrano i documenti Corriere Della Sera; 2014; Available at: http://www.corriere.it/cronache/14_marzo_14/crema-analisi-sbagliate-ospedale-nas-sequestrano-documenti-04bc0942-ab3b-11e3-a415-108350ae7b5e.shtml. Accessed: 14th March 2014. (in Italian)
- 14.NAS. Ministero della difesa. 2014; Available at: http://www.carabinieri.it/Internet/Arma/Curiosita/Non+tutti+sanno+che/N/11+N.htm. Accessed: 14th March 2014. (in Italian)
- 15.Corlazzoli A. [Test falsati in 18 ospedali lombardi]. il Fatto - Cronaca; 2014; Available at: http://www.ilfattoquotidiano.it/2014/03/17/test-falsati-18-ospedali-coinvolti-in-lombardia-la-regione-scrisse-ad-asl-un-mese-dopo-lallarme/916905/. Accessed: 17th March 2014. (in Italian)
- 16.Corica A. [Quarentamila esami del sangue effettuati in Lombardia con i kit ritirati]. la Repubblica; 2014; Available at: http://ricerca.repubblica.it/repubblica/archivio/repubblica/2014/03/18/quarantamila-esami-del-sangue-effettuati-in-lombardia.html?ref=search. Accessed: 18th March 2014. (in Italian)
- 17.The United States Department of Justice. Quest Diagnostics to pay U.S. $302 million to resolve allegations that a subsidiary sold misbranded test Kits. 2014; Available at: http://www.justice.gov/opa/pr/quest-diagnostics-pay-us-302-million-resolve-allegations-subsidiary-sold-misbranded-test-kits. Accessed: 7th April 2015.
- 18.Ministero della Salute-dgFDM. [Dispositivi Medici - Aspetti regolatori e operativi]. Roma: Ministero della Salute-dgFDM; 2010; Available at: http://www.salute.gov.it/imgs/C_17_pubblicazioni_1238_allegato.pdf. Accessed: 28th March 2014. (in Italian)
- 19.British Standards Institution. Performance evaluation of in vitro diagnostic medical devices - BS EN 13612:2002. British Standards Institution; 2002. [Google Scholar]
- 20.QUALIMEDLAB. [Valutazione delle prestazioni dei metodi PTH (pg/mL) - Riepilogo cumulativo dal campione IM31a al campione IM36c (ciclo 2013)]. Pisa-Italy 2014. (in Italian)
- 21.Clinical Laboratory Standards Institute. Evaluation of commutability of processed samples. CLSI EP14-A3 document. 3rd ed. Wayne, PA: Clinical Laboratory Standards Institute; 2014. [Google Scholar]
- 22.Monge M, Jean G, Bacri JL, Lemaitre V, Masy E, Joly D, et al. Higher parathyroid hormone (PTH) concentrations with the Architect PTH assay than with the Elecsys assay in hemodialysis patients, and a simple way to standardize these two methods. Clin Chem Lab Med. 2009;47:362–6. 10.1515/CCLM.2009.068 [DOI] [PubMed] [Google Scholar]
- 23.Marcocci C, Cetani F. Primary Hyperparathyroidism. N Engl J Med. 2011;365:2389–97. 10.1056/NEJMcp1106636 [DOI] [PubMed] [Google Scholar]
- 24.Silverberg SJ, Bilezikian JP. “Incipient” primary hyperparathyroidism: a “forme fruste” of an old disease. J Clin Endocrinol Metab. 2003;88:5348–52. 10.1210/jc.2003-031014 [DOI] [PubMed] [Google Scholar]
- 25.Viljoen A, Singh DK, Twomey PJ, Farrington K. Analytical quality goals for parathyroid hormone based on biological variation and on reference change value. Clin Chem Lab Med. 2008;46:1438–42. 10.1515/CCLM.2008.275 [DOI] [PubMed] [Google Scholar]
- 26.Guidi GC, Lippi G, Solero GP, Poli G, Plebani M. Managing transferability of laboratory data. Clin Chim Acta. 2006;374:57–62. 10.1016/j.cca.2006.06.009 [DOI] [PubMed] [Google Scholar]
- 27.Westgard J. Desirable Biological Variation Database Specifications. 2014; Available at: http://www.westgard.com/biodatabase1.htm. Accessed: 28th March 2014.