

Abstract
PURPOSE
To critically assess the status of molecular prognostic testing and its use for individualized patient care in uveal melanoma.
DESIGN
Perspective, literature review, evidence assessment, and commentary.
METHODS
Evaluation of selected articles from the literature and the authors’ clinical and laboratory studies.
RESULTS
The most accurate molecular tests for predicting metastatic death in patients with uveal melanoma currently involve automated techniques for assessing deoxyribonucleic acid (DNA) copy number alterations and gene expression profiling. Most tests reported in the literature to date do not provide adequate scientific and statistical validation to be used outside of an ethically supervised investigational environment.
CONCLUSIONS
Many cytogenetic and molecular prognostic tests for uveal melanoma have been reported, yet few have reached the standards required for routine clinical testing. Clinicians must understand the statistical and scientific limitations of the tests they are using, and appropriate ethical oversight is essential until such time that validated testing instruments are available that are performed in a standardized clinical testing environment. Well-controlled prospective studies are necessary to identify the most accurate, widely accessible, and affordable tests for routine clinical use.
Uveal melanoma is the most common primary cancer of the eye and remains one of the most deadly diseases encountered in ophthalmology.1 Uveal melanoma occurs in about 4 to 7 individuals per million in the United States and the incidence is similar in other countries with large White populations.2,3 Despite steady improvements in diagnosis and treatment of the primary tumor, a corresponding decrease in metastatic death has not been documented.4 A large proportion of uveal melanoma patients develop metastatic disease, particularly to the liver, which is uniformly fatal.5 Various hypotheses have been put forward to explain the substantial rate of metastasis despite ocular treatment,6 but most experts now agree that this problem is attributable to micro-metastasis that occurs prior to primary treatment and remains dormant for prolonged periods of time before emerging as clinically detectable macro-metastatic disease.7 Consequently, improving survival will most likely require preemptive, adjuvant systemic therapy for patients who are likely to harbor micro-metastasis in order to delay or prevent the progression of preclinical micro-metastases to untreatable macro-metastatic disease. To implement these strategies, uveal melanoma patients who are at high-risk of harboring undetectable micro-metastases must be identified and distinguished from low-risk patients so that the latter are not unnecessarily treated with systemic therapies with their attendant risks and side effects. This will require the development and validation of highly accurate biomarkers. The primary goal of this Perspective is to assess the relative merits of existing biomarkers and to explore future directions in this field.
CLINICAL AND HISTOPATHOLOGIC FEATURES
Several clinical and histopathologic features of primary uveal melanomas have been associated with metastatic death, including increased patient age, increased tumor diameter and thickness, ciliary body involvement, epithelioid cytology, extraocular extension, extravascular matrix patterns, and lymphocytic/monocytic infiltration.8–11 Despite the statistical association of these features with metastasis, their ability to predict metastasis in an individual patient is limited.12 Detection of protein biomarkers, including cyclin D1, Mdm2, nibrin, Ddef1, Igfr1, myc, human leukocyte antigen (HLA) molecules, and others, by immunohistochemical methods has been assessed for prognostic value.13–18 While the prognostic accuracy of some of these markers appears to exceed that of the morphologic features described previously,12 they have proved to be of limited value for prognostic purposes. The expression of a single protein or gene product does not seem to account adequately for the immense tumor complexity that determines metastatic potential. Further, immunohistochemistry is not quantitative, such that the interpretation of such assays is subject to considerable variability from observer to observer, lab to lab, and antibody to antibody. From a practical standpoint, most uveal melanomas are now treated by globe-sparing procedures, so only the occasional cases requiring enucleation result in sufficient tumor tissue for immunohistochemistry.
CYTOGENETIC MARKERS
Several cytogenetic or chromosomal abnormalities, including loss of 1p, loss of chromosome 3 (monosomy 3), gain of 6p, loss of 6q, loss of 8p, and gain of 8q, have been linked statistically to metastatic death in uveal melanoma.19–22 Among these markers, monosomy 3 is by far the most significant predictor of metastatic risk.23 Isodisomy 3, the circumstance in which one copy of chromosome 3 is lost and the remaining, ostensibly faulty copy is duplicated, occurs in 5% to 10% of cases and is prognostically equivalent to monosomy 3.24 This is important for prognostic testing, as some techniques are unable to identify isodisomy 3. As cytogenetic information has become increasingly accessible to clinicians, the risk of misinterpreting or misapplying such information has also increased. It is critical to understand the techniques used to obtain this information, along with the limitations of these techniques, before attempting to apply this information to clinical patient management.
Initially, cytogenetic analysis in uveal melanoma was performed with standard karyotyping,19–21 in which metaphase spreads are directly visualized and chromosomal abnormalities identified by morphologic changes in chromosome size and banding pattern. The main problems with this technique are the need for a highly trained cytogeneticist, sampling error attributable to analysis of only a few tumor cells, the inability to detect small changes, and the inability to detect isodisomy. Other techniques that rely on direct analysis of intact chromosomes include fluorescence in situ hybridization (FISH), spectral karyotyping (SKY), and earlier forms of comparative genomic hybridization (CGH) (Figure).25–30 These techniques use labeled probes that detect specific chromosomal regions, thereby providing the advantage over karyotyping of not relying on morphology to identify chromosomal alterations. However, like traditional karyotyping, these techniques may be prone to sampling error attributable to analysis of a small subset of tumor cells,31 and they are unable to detect isodisomy. For FISH, additional errors can be introduced by a failure to detect probes outside the plane of section, thereby detecting artifactual chromosomal losses or failing to detect chromosomal gains.31
FIGURE.
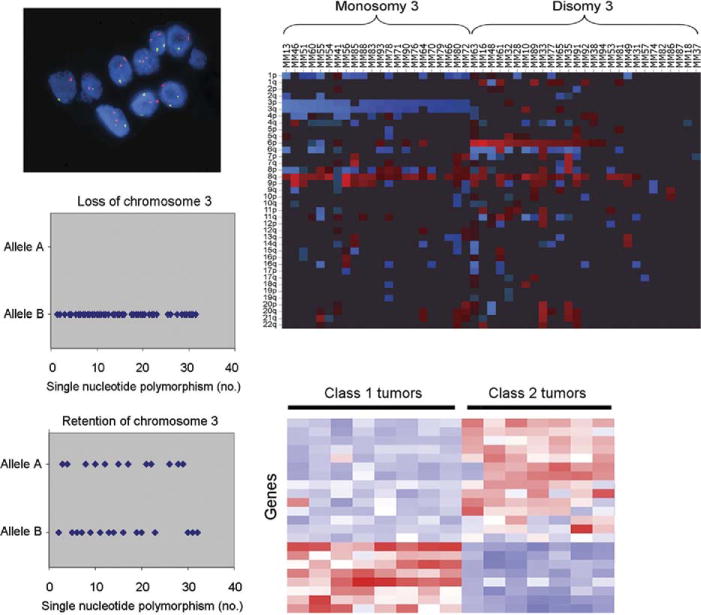
Representative examples of major types of molecular and cytogenetic prognostic tests currently in use for uveal melanoma. (Top left) Fluorescence in situ hybridization. Red dots represent control chromosomal probes; green dots represent probes from chromosome 3. (Top right) Array-based comparative genomic hybridization. Rows represent chromosomal arms; columns represent individual tumor deoxyribonucleic acid (DNA) samples. Red represents DNA gain, blue represents loss, and black represents normal DNA copy number. (Bottom left) Single-nucleotide polymorphism (SNP)-based detection of loss of heterozygosity for chromosome 3. Diamonds represent individual SNPs across chromosome 3. Tumors that have lost one copy of chromosome 3 will have only one type of SNP allele (Top panel), whereas tumors that retain both parental copies of chromosome 3 will show two different alleles at multiple SNPs (Bottom panel). (Bottom right) Heat-map summary of gene expression profile. Rows represent genes; columns represent individual tumor messenger ribonucleic acid samples obtained by fine-needle aspiration biopsy. Red bars represent overexpressed genes; blue bars represent underexpressed genes.
More recent techniques, such as array-based CGH,24 microsatellite analysis (MSA),32,33 single-nucleotide polymorphism (SNP) analysis,24 and multiplex ligation-dependent probe amplification (MLPA),34 are more automated and evaluate multiple regions across individual chromosomes using specific molecular probes (Figure). These techniques allow larger numbers of tumor cells to be examined, thereby reducing the risk of sampling error, and they increase the ability to detect smaller abnormalities compared to karyotyping and FISH. Additionally, MSA and SNP can render a correct classification in the presence of isodisomy 3 because these tests distinguish between the two copies of a particular chromosome that are inherited from each parent. SNP has been shown to be superior to techniques that do not detect isodisomy 3, such as FISH and CGH.24 Except for this article, there have been very few reported attempts to compare available cytogenetic markers for prognostic accuracy using statistically valid methods, and there have been few efforts for any of the mentioned cytogenetic marker studies to calculate their prognostic accuracy (eg, sensitivity, specificity, true positives, false positives, true negatives, false negatives, and positive and negative predictive value). Until such time that these data have been firmly established, such testing should be considered investigational and not for routine clinical testing.
GENE EXPRESSION PROFILING
Gene expression profiling (GEP) involves the simultaneous measurement of messenger ribonucleic acid (mRNA) expression of multiple genes, allowing for a dynamic, multi-dimensional, and physiologic “snapshot” of tumor biology. GEP has identified two major subgroups of uveal melanoma, which are now referred to as class 1 and class 2, with about half of tumors reported to date falling into one or the other subgroup (Figure).35,36 This GEP is highly accurate for predicting patient outcome; class 1 tumors have a very low-risk and class 2 tumors a very high-risk of metastasis. The class 2 gene expression profile is closely associated with historical prognostic features, such as larger tumor size, epithelioid cytology, extravascular looping matrix patterns, and monosomy 3,35–38 but the prognostic accuracy of the class 2 expression profile is much greater than for any of these other features individually or in combination. The superiority of GEP over monosomy 3 has been verified by multiple groups.39–41
In the past, the major drawback to GEP was the expense and limited availability. Once the predictive power of GEP became clear, however, considerable effort was devoted to optimizing it for clinical applications. By using sophisticated software to eliminate unnecessary, redundant genes, the number of genes can be reduced to less than 20 without any loss in prognostic accuracy.42 This small number of genes has allowed us to transition this refined uveal melanoma GEP, or “UM-GEP,” to an inexpensive, custom-designed, polymerase chain reaction (PCR)-based platform. This change has not only retained but actually improved the prognostic accuracy of UM-GEP because of the greater dynamic range of the PCR-based test. Further, the PCR-based platform is highly sensitive and can detect an expression signature from a very small number of cells from fine-needle aspiration biopsy (FNAB).42 The current PCR-based UM-GEP can even be used to analyze formalin-fixed, paraffin-embedded samples, which are notorious for performing poorly on most GEP-based platforms. Most importantly, the new platform has reduced the cost of the UM-GEP by about ten-fold and allows the test to be performed in a standard clinical testing environment.
WHICH TESTS TO USE?
There is now a wide array of molecular prognostic tests being employed by different groups around the world, and these tests vary significantly in their methodology (Table). Surprisingly, there have been very few studies performed to determine which of these tests is superior. Previously, the cost and restricted availability of more technically complex tests limited their widespread availability, and some tests could not be performed on FNAB or fixed tissue. With the recent development of sophisticated, rapid, inexpensive, and accessible tests for both chromosomal changes (eg, MLPA) and gene expression profiles (eg, UM-GEP), cost and availability should no longer represent a barrier to their use. Further, these new platforms can be used on very small FNAB samples and archival fixed tissue. Therefore, the overriding consideration for determining the preferred tests for clinical use is now the quality of the test, based on its reproducibility, reliability, and validity.
TABLE.
Comparison of Major Types of Molecular and Cytogenetic Prognostic Tests Currently in Use for Uveal Melanoma
| Type of Analysis | Molecular Basis | Quantitative | Susceptibility to Sampling Error & Tumor Heterogeneity | Technical Expertise Required | Degree of Automation | Cost | Reference |
|---|---|---|---|---|---|---|---|
| Karyotype | Gross gain, loss, or alteration of chromosomes | + | +++ | +++ | + | + | 19 to 21 |
| FISH | Gain or loss of small number of chromosome segments labeled with specific probes | + | +++ | ++ | + | ++ | 25 to 28 |
| CGH | Gain or loss of large number of chromosome segments labeled with specific probes | ++ | ++ | ++ | ++ | +++ | 24,29,30 |
| MSA | Loss of heterozygosity of a small number of highly polymorphic DNA segments | + | ++ | ++ | ++ | ++ | 32,33 |
| SNP | Loss of heterozygosity of a large number of moderately polymorphic DNA segments | + | ++ | + | +++ | + | 24 |
| MLPA | Gain or loss of a multiple chromosome segments | +++ | ++ | + | +++ | ++ | 34 |
| UM-GEP | Simultaneous measurement of mRNA expression of multiple genes | +++ | + | + | +++ | + | 35 to 38 |
CGH = comparative genomic hybridization; DNA = deoxyribonucleic acid; FISH = fluorescence in situ hybridization; MLPA = multiplex ligation-dependent probe amplification; mRNA = messenger ribonucleic acid; MSA = microsatellite analysis; SNP = single-nucleotide polymorphism; UM-GEP = uveal melanoma gene expression profiling.
+ = low; ++ = medium; +++ = high.
REPRODUCIBILITY
The degree to which a test generates the same result on the same patient when repeated by the same or another examiner is influenced by the complexity of testing procedures, the degree to which the test is automated, and the degree of advanced training, experience, and monitoring required of the examiner. Reproducibility generally increases with decreasing complexity and increasing automation. For example, karyotyping is less reproducible than MLPA and UM-GEP because the former requires an experienced cytogeneticist, whereas the latter can be performed on routine clinical laboratory equipment by a relatively inexperienced technician.
RELIABILITY
The ability of the test to perform and maintain its functions is enhanced by standardized methodology and strict quality control measures to detect sampling errors, mishandling of specimens, and ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) degradation, all of which have a profound impact on reliability. For example, spectrophotometry should routinely be used to confirm the quality and quantity of RNA and DNA from tumor samples prior to molecular testing. Reliability generally increases with increasing automation.
VALIDITY
The degree to which a test measures what you think that it measures (internal validity) and to which it is generalizable to circumstances other than those in which the test was developed (external validity) are critical for determining how a test can be applied in the clinical setting.
Internal Validity
Is the degree to which a test measures what it is purported to measure, and is determined by technical variables, such as the quality of the test methodology, and biological variables, such as tissue heterogeneity. Most prognostic tests in uveal melanoma have been reported in the literature as if they yield simple, categorical answers, such as “disomy 3” vs “monosomy 3.” In reality, these tests yield quantitative data that must be converted in some manner to a categorical test result, and it is critically important to have statistically validated cut-offs for converting quantitative data into categorical test results. For example, what percentage of cells in a tumor must exhibit monosomy 3 before the tumor is “diagnosed” as monosomy 3? The answer to this must be derived empirically based on actual patient data.
Another important aspect of internal validation is to confirm that the specimen being analyzed is in fact what the clinician believes it to be. This is a particularly salient concept in ocular oncology, where many clinicians are accustomed to substituting their “clinical experience” as a surrogate for true tissue confirmation. In tumor samples obtained from enucleation or local resection, the identity of the tissue can easily be confirmed. However, the vast majority of uveal melanomas are treated by radiotherapy, in which tumor tissue is usually obtained by FNAB. In these cases, cytologic examination affords the opportunity to confirm the presence of melanoma cells, to identify necrosis that can adversely affect the accuracy of molecular testing, and to ensure that the sample is not inadvertently contaminated with large numbers of non-neoplastic cells, such as retinal cells, pigment epithelium, and peripheral blood lymphocytes. By analogy, cytologic examination is routinely performed on cerebrospinal fluid specimens, so that the clinician can distinguish between pathologically elevated cerebrospinal protein levels and artifact introduced by contaminating red blood cells as a result of traumatic lumbar puncture. Since none of the tests described herein is designed to distinguish between melanoma and other non-melanoma tissues, inadvertent testing of non-melanoma tissue will lead to an erroneous test result. Too many normal cells in the sample would lead, for example, to an incorrect result of disomy 3 in a tumor that is actually monosomy 3. This may well be an important source of false negatives that limit the utility of monosomy 3 as a prognostic marker.34 A related issue is tumor heterogeneity, where different regions of the tumor may yield different molecular results. This has been a subject of recent concern and controversy for monosomy 3.31 Tests that rely not on a single marker but on multiple independent markers, such as MLPA and UM-GEP, appear to be less susceptible to this source of error.
External Validity
Is the degree to which one may safely generalize the results of a given test from the patient group studied to the whole population of patients with uveal melanoma. Thus, external validity addresses the predictive accuracy of the test, the ability to prospectively identify individual patients who will likely develop metastatic disease (positive predictive value) and those who will not (negative predictive value). External validity is established by evaluating a test on multiple independent datasets, preferably in a prospective manner. One example of an inappropriate generalization is to extrapolate the results of cytogenetic and molecular markers developed from testing on medium to large primary uveal melanomas to small tumors—many of which may actually be nevi—without validating that the results predict metastasis accurately in this group. Since the endpoint of interest in uveal melanoma is metastasis, meaningful conclusions about the external validity of a prognostic test require long follow-up.
In order to determine which of the available tests has superior validity and predictive accuracy, they must be compared in a statistically valid manner. Only a few studies have attempted to do this, including comparisons of GEP and monosomy 3,39,40 and SNP-based assay for chromosome 3 loss vs CGH and FISH.24 Published studies to date have shown that the predictive accuracy of GEP is superior to that for chromosome 3 status.39,40 Importantly, virtually all published articles to date on molecular prognostic testing in uveal melanoma have been retrospective in nature. Therefore, a prospective, multicenter study is now underway to compare the UM-GEP and a SNP-based chromosome 3 assay. Such prospective validation is critical before moving towards routine clinical testing.
THE LEAP FROM RESEARCH TO PATIENT CARE
Traditionally, the use and interpretation of molecular/genetic tests, and the associated patient counseling, have been the responsibility of specially trained clinical geneticists. However, recent technological advances have put highly sophisticated molecular information into the hands of clinicians who may not understand the scientific, statistical, and ethical differences between the investigational use of molecular testing and the routine use of such testing for individualized patient care. Understanding these differences is critical for the informed application of molecular prognostic tests.
MOLECULAR TESTING AS A RESEARCH TOOL
Currently, there is a great deal of confusion and controversy in the ocular oncology field concerning the proper use of investigational prognostic tests in the clinical management of uveal melanoma. None of the prognostic tests published to date has reached the status of a fully validated clinical testing instrument approved by appropriate regulatory agencies. For example, the package insert for the commercially available MLPA uveal melanoma kit (SALSA MLPA KIT P027-B1; description version 08; 26-06-2008; MRC-Holland, Amsterdam, the Netherlands) states that it is not Communauté Européenne/Food and Drug Administration certified for use in diagnostic procedures and that it is for research purposes only. As such, all such tests must be considered to be investigational in nature. This has important ethical, statistical, and scientific implications.
Ethical considerations include patient safety and informed consent. Investigational testing requires the oversight of an institutional review board or ethics committee. The patient should be informed of the investigational nature of the test and should understand that the test is not a routine clinical test and has not been proven to be of benefit in guiding management or prolonging survival. For patients requiring FNAB, the purpose of the procedure, along with its potential complications, must be explained to the patient. Importantly, patients ethically cannot be billed for such investigational testing.
In terms of statistical and scientific considerations, investigational prognostic tests should not be used to guide patient management and should not be generalized to clinical settings for which they were not designed and externally validated. For example, it has been speculated that cytogenetic markers can be used to determine which small suspicious uveal melanocytic tumors should be treated; those with monosomy 3 would presumably be treated promptly, whereas those with disomy 3 would be observed.43 However, one could make the opposite argument. Perhaps monosomy 3 tumors have already metastasized so treating the eye is futile, whereas treatment of disomy 3 tumors may prevent them from converting to monosomy 3 tumors. Sufficient evidence is not yet available to know the correct use of molecular testing in this setting, so that any such clinical activities should be performed in the context of clinical trials with appropriate ethical oversight. Similarly, patients with high-risk molecular markers should not be offered different clinical management than their low-risk counterparts, except in the setting of approved clinical trials, until such time that it has been confirmed that adjuvant systemic therapy improves survival in these patients. As several centers prepare to enroll high-risk patients into clinical trials for adjuvant therapy, it is critical that these centers use the most accurate and contemporary prognostic tests available so that their results will be generalizable to future patients.
MOLECULAR TESTING AS A ROUTINE CLINICAL TEST
In order for a molecular test to achieve the status of a validated instrument to be used as a criterion for entering patients into interventional clinical trials and, eventually, to be used in routine patient care, it must meet several key standards. First, the test must demonstrate high reproducibility, reliability, and validity, as described previously. Second, the testing protocol must be standardized and widely accessible, with quality control measures in place. Ideally, the test should be conducted in a controlled testing facility such as a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory. It is important that the test report include the test result (eg, monosomy 3 or class 2) along with the statistical implications of the test result and an indication of the confidence in the test result. As a theoretical example, analysis of a tumor specimen using the UM-GEP may produce a test result of class 2, which has been shown to convey a 50% risk of metastatic death over a 3-year period, with a confidence level of 95%. It is critical that these parameters be established and validated by each individual testing facility, as they cannot simply be inferred from the published data of other centers.
FUTURE TRENDS
As research into molecular prognostic testing in uveal melanoma continues to mature, the plethora of tests currently in use around the world, which vary greatly in methodology and quality, need to be critically assessed and compared to identify the most accurate, accessible, and affordable tests with which to move forward in future research and clinical trials. Timely and efficient progress toward this goal will require more multi-institutional cooperation than has occurred in the past. Toward that end, we have established a Collaborative Ocular Oncology Group that is currently conducting a prospective study to validate the prognostic value of the UM-GEP vs chromosome 3 status. Further, this group is planning a multicenter clinical trial of systemic adjuvant therapy for high-risk patients, defined as class 2 by the UM-GEP. In accordance with the standard for other forms of medical genetic testing, centralized testing facilities should soon be established that can achieve high quality-control standards and provide worldwide access to this technology.
Acknowledgments
This Study was Supported By The National Cancer Institute (R01 CA125970), Bethesda, Maryland; Barnes-Jewish Hospital Foundation, St. Louis, Missouri; Tumori Foundation, San Francisco, California; Kling Family Foundation, St. Louis, Missouri; Horncrest Foundation, Ossining, New York; Research to Prevent Blindness Inc, New York, New York; and New England Retina Research & Education Foundation, Hamden, Connecticut. Washington University has filed a provisional patent on the UM-GEP assay that is mentioned in the article. The author has received no remuneration for this patent. The author was involved in design and conduct of study; data collection; management, analysis, and interpretation of data; and preparation, review, and approval of the manuscript. Institutional Review Board approval was not needed for this study.
Biography

J. William Harbour, MD, is the Paul Cibis Distinguished Professor and Director of Ocular Oncology at Washington University in St. Louis. His practice is limited to patients with ocular tumors and simulating lesions. His research laboratory utilizes scientific discoveries to improve the care of patients with uveal melanoma and other ocular tumors. His leadership in ocular oncology has been recognized by the ARVO Cogan Award, Macula Society Rosenthal Award, Best Doctors in America and America’s Top Doctors.
References
- 1.Harbour JW. Clinical overview of uveal melanoma: introduction to tumors of the eye. In: Albert DM, Polans A, editors. Ocular Oncology. New York, New York: Marcel Dekker; 2003. pp. 1–18. [Google Scholar]
- 2.Inskip PD. Frequent radiation exposures and frequency-dependent effects: the eyes have it. Epidemiology. 2001;12:1–4. doi: 10.1097/00001648-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18:75–84. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Singh AD, Topham A. Survival rates with uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110:962–965. doi: 10.1016/S0161-6420(03)00077-0. [DOI] [PubMed] [Google Scholar]
- 5.Gragoudas ES, Egan KM, Seddon JM, et al. Survival of patients with metastases from uveal melanoma. Ophthalmology. 1991;98:383–389. doi: 10.1016/s0161-6420(91)32285-1. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerman LE, McLean IW, Foster WD. Does enucleation of the eye containing a malignant melanoma prevent or accelerate the dissemination of tumour cells. Br J Ophthalmol. 1978;62:420–425. doi: 10.1136/bjo.62.6.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eskelin S, Pyrhonen S, Summanen P, Hahka-Kemppinen M, Kivela T. Tumor doubling times in metastatic malignant melanoma of the uvea: tumor progression before and after treatment. Ophthalmology. 2000;107:1443–1449. doi: 10.1016/s0161-6420(00)00182-2. [DOI] [PubMed] [Google Scholar]
- 8.McLean IW, Foster WD, Zimmerman LE. Prognostic factors in small malignant melanomas of choroid and ciliary body. Arch Ophthalmol. 1977;95:48–58. doi: 10.1001/archopht.1977.04450010050004. [DOI] [PubMed] [Google Scholar]
- 9.Shammas HF, Blodi FC. Prognostic factors in choroidal and ciliary body melanomas. Arch Ophthalmol. 1977;95:63–69. doi: 10.1001/archopht.1977.04450010065005. [DOI] [PubMed] [Google Scholar]
- 10.Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Cruz PO, Jr, Specht CS, McLean IW. Lymphocytic infiltration in uveal malignant melanoma. Cancer. 1990;65:112–115. doi: 10.1002/1097-0142(19900101)65:1<112::aid-cncr2820650123>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 12.Chang SH, Worley LA, Onken MD, Harbour JW. Prognostic biomarkers in uveal melanoma: evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008;18:191–200. doi: 10.1097/CMR.0b013e3283005270. [DOI] [PubMed] [Google Scholar]
- 13.Coupland SE, Bechrakis N, Schuler A, et al. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br J Ophthalmol. 1998;82:961–970. doi: 10.1136/bjo.82.8.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brantley MA, Jr, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000;157:1795–1801. doi: 10.1016/s0002-9440(10)64817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehlers JP, Harbour JW. NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res. 2005;11:1849–1853. doi: 10.1158/1078-0432.CCR-04-2054. [DOI] [PubMed] [Google Scholar]
- 16.Ehlers JP, Worley L, Onken MD, Harbour JW. DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res. 2005;11:3609–3613. doi: 10.1158/1078-0432.CCR-04-1941. [DOI] [PubMed] [Google Scholar]
- 17.Royds JA, Sharrard RM, Parsons MA, et al. C-myc oncogene expression in ocular melanomas. Graefes Arch Clin Exp Ophthalmol. 1992;230:366–371. doi: 10.1007/BF00165947. [DOI] [PubMed] [Google Scholar]
- 18.Blom DJ, Mooy CM, Luyten GP, et al. Inverse correlation between expression of HLA-B and C-myc in uveal melanoma. J Pathol. 1997;181:75–79. doi: 10.1002/(SICI)1096-9896(199701)181:1<75::AID-PATH724>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 19.Horsman DE, Sroka H, Rootman J, White VA. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet Cytogenet. 1990;45:249–253. doi: 10.1016/0165-4608(90)90090-w. [DOI] [PubMed] [Google Scholar]
- 20.Sisley K, Rennie IG, Cottam DW, Potter AM, Potter CW, Rees RC. Cytogenetic findings in six posterior uveal melanomas: involvement of chromosomes 3, 6, and 8. Genes Chromosomes Cancer. 1990;2:205–209. doi: 10.1002/gcc.2870020307. [DOI] [PubMed] [Google Scholar]
- 21.Prescher G, Bornfeld N, Becher R. Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst. 1990;82:1765–1769. doi: 10.1093/jnci/82.22.1765. [DOI] [PubMed] [Google Scholar]
- 22.Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:313–317. [PubMed] [Google Scholar]
- 23.Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–1225. doi: 10.1016/s0140-6736(96)90736-9. [DOI] [PubMed] [Google Scholar]
- 24.Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW. Loss of heterozygosity of chromosome 3 detected with single-nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma. Clin Cancer Res. 2007;13:2923–2927. doi: 10.1158/1078-0432.CCR-06-2383. [DOI] [PubMed] [Google Scholar]
- 25.Naus NC, van Drunen E, de Klein A, et al. Characterization of complex chromosomal abnormalities in uveal melanoma by fluorescence in situ hybridization, spectral karyotyping, and comparative genomic hybridization. Genes Chromosomes Cancer. 2001;30:267–273. [PubMed] [Google Scholar]
- 26.McNamara M, Felix C, Davison EV, Fenton M, Kennedy SM. Assessment of chromosome 3 copy number in ocular melanoma using fluorescence in situ hybridization. Cancer Genet Cytogenet. 1997;98:4–8. doi: 10.1016/s0165-4608(96)00405-0. [DOI] [PubMed] [Google Scholar]
- 27.Parrella P, Caballero OL, Sidransky D, Merbs SL. Detection of C-myc amplification in uveal melanoma by fluorescent in situ hybridization. Invest Ophthalmol Vis Sci. 2001;42:1679–1684. [PubMed] [Google Scholar]
- 28.Midena E, Bonaldi L, Parrozzani R, Tebaldi E, Boccassini B, Vujosevic S. In vivo detection of monosomy 3 in eyes with medium-sized uveal melanoma using transscleral fine-needle aspiration biopsy. Eur J Ophthalmol. 2006;16:422–425. doi: 10.1177/112067210601600310. [DOI] [PubMed] [Google Scholar]
- 29.Gordon KB, Thompson CT, Char DH, et al. Comparative genomic hybridization in the detection of DNA copy number abnormalities in uveal melanoma. Cancer Res. 1994;54:4764–4768. [PubMed] [Google Scholar]
- 30.Speicher MR, Prescher G, du Manoir S, et al. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994;54:3817–3823. [PubMed] [Google Scholar]
- 31.Maat W, Jordanova ES, van Zelderen-Bhola SL, et al. The heterogeneous distribution of monosomy 3 in uveal melanomas: implications for prognostication based on fine-needle aspiration biopsies. Arch Pathol Lab Med. 2007;131:91–96. doi: 10.5858/2007-131-91-THDOMI. [DOI] [PubMed] [Google Scholar]
- 32.Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999;59:3032–3037. [PubMed] [Google Scholar]
- 33.Tschentscher F, Prescher G, Zeschnigk M, Horsthemke B, Lohmann DR. Identification of chromosomes 3, 6, and 8 aberrations in uveal melanoma by microsatellite analysis in comparison to comparative genomic hybridization. Cancer Genet Cytogenet. 2000;122:13–17. doi: 10.1016/s0165-4608(00)00266-1. [DOI] [PubMed] [Google Scholar]
- 34.Damato BE, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland S. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest Ophthalmol Vis Sci. 2009;50:3048–3055. doi: 10.1167/iovs.08-3165. [DOI] [PubMed] [Google Scholar]
- 35.Tschentscher F, Husing J, Holter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578–2584. [PubMed] [Google Scholar]
- 36.Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006;66:4602–4609. doi: 10.1158/0008-5472.CAN-05-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onken MD, Lin AY, Worley LA, Folberg R, Harbour JW. Association between microarray gene expression signature and extravascular matrix patterns in primary uveal melanomas. Am J Ophthalmol. 2005;140:748–749. doi: 10.1016/j.ajo.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 39.Worley LA, Onken MD, Person E, et al. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007;13:1466–1471. doi: 10.1158/1078-0432.CCR-06-2401. [DOI] [PubMed] [Google Scholar]
- 40.Petrausch U, Martus P, Tonnies H, et al. Significance of gene expression analysis in uveal melanoma in comparison to standard risk factors for risk assessment of subsequent metastases. Eye. 2007;22:997–1007. doi: 10.1038/sj.eye.6702779. [DOI] [PubMed] [Google Scholar]
- 41.van Gils W, Lodder EM, Mensink HW, et al. Gene expression profiling in uveal melanoma: two regions on 3p related to prognosis. Invest Ophthalmol Vis Sci. 2008;49:4254–4262. doi: 10.1167/iovs.08-2033. [DOI] [PubMed] [Google Scholar]
- 42.Onken MD, Worley LA, Davila RM, Char DH, Harbour JW. Prognostic testing in uveal melanoma by transcriptomic profiling of fine-needle biopsy specimens. J Mol Diagn. 2006;8:567–573. doi: 10.2353/jmoldx.2006.060077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shields JA, Shields CL, Materin M, Sato T, Ganguly A. Role of cytogenetics in management of uveal melanoma. Arch Ophthalmol. 2008;126:416–419. doi: 10.1001/archopht.126.3.416. [DOI] [PubMed] [Google Scholar]