

Summary
Uveal melanoma is the second most common form of melanoma and the most common primary intraocular malignancy. Until recently, very little was known about the genetics of this aggressive cancer. Mutations in oncogenes and tumor suppressors that are common in other cancers are conspicuously absent in uveal melanoma. In recent years, however, uveal melanoma has begun to yield its secrets, and a fascinating picture is emerging of how it develops and progresses. Mutations in the Gq alpha subunits, encoded by GNAQ and GNA11, appear to be early or perhaps initiating events that require further mutations for malignant transformation. On the other hand, mutations in the BRCA1-associated protein-1 (BAP1) appear to occur later and demarcate a molecular brink beyond which metastasis becomes highly likely. BAP1 mutations can also occur in the germline, leading to a distinctive cancer predisposition syndrome. These mutations appear to be key events that provide the potential for targeted therapy. This article will review the genetic findings in uveal melanoma over the past two decades and suggest important areas for future work.
Keywords: melanoma, eye, BAP1, GNAQ, GNA11, metastasis
Epidemiology
Uveal melanoma (UM) is the most common primary malignancy of the eye, with an incidence of about 1200–1500 new cases per year in the United States, and it accounts for about 5% of all melanomas (Egan et al., 1988; Ramaiya and Harbour, 2007). UMs can arise anywhere in the uveal tract, comprising the iris, ciliary body, and choroid, and they often involve more than one of these structures. About 5% of UMs are isolated to the iris, and these are often considered separately because of their less aggressive clinical behavior and distinct genetic alterations. Risk factors for UM include light skin color, red or blonde hair, blue eyes, and cutaneous freckles and nevi (Gallagher et al., 1985; Seddon et al., 1990; Tucker et al., 1985; Van Hees et al., 1994). There is a slight male preponderance (Singh et al., 2005a). Some studies have shown an association between UM and increased sun exposure, sunlamp use, and southern latitude (Seddon et al., 1990; Tucker et al., 1985). Unlike cutaneous melanoma, however, the rates of UM have not increased over recent decades (Singh et al., 2011). Thus, the role of ultraviolet light exposure in UM is less clear than for cutaneous melanoma.
Clinical considerations
UM is similar to other forms of melanoma in its cellular morphology, expression of melanocytic lineage markers, propensity for metastatic spread, and resistance to therapy (Ramaiya and Harbour, 2007). However, UM differs in important ways from other types of melanoma, owing at least in part to its anatomic location within the uveal layer of the eye. UMs are not located within an epithelium, so downregulation of E-cadherin, epithelial-to-mesenchymal transition, and basement membrane invasion are not important steps in UM progression (Onken et al., 2006a). Likewise, the lymphatic structures in the eye are too small for the passage of cells (Yucel et al., 2009), so regional lymphatic spread of UM is extremely rare. Instead, UMs metastasize by hematogenous dissemination. The most common sites of involvement include liver (93%), lung (24%), and bone (16%), with the overwhelming majority presenting initially in the liver (Diener-West et al., 2005). The cause of this marked tropism for the liver remains unknown. Clinical features associated with poor prognosis include larger tumor diameter, ciliary body involvement, and advanced patient age (Augsburger and Gamel, 1990). Histopathologic prognostic factors include epithelioid cell type, inflammatory infiltration, and extracellular matrix patterning (De La Cruz et al., 1990; Folberg et al., 1992; Gamel et al., 1978; Makitie et al., 2001). The mortality rate at 15 years of diagnosis of the primary tumor is about 50% (Kujala et al., 2003), and median survival after detection of metastatic disease is about 9 months (Kath et al., 1993).
Chromosomal alterations
Most UMs exhibit a relatively low degree of genomic instability and aneuploidy compared with many other cancer types. One study found that UMs exhibited less than half the genomic instability of breast cancers (Papadopoulos et al., 2002). In another study of 52 UMs, only one tumor showed microsatellite instability (Cross et al., 2003). In two independent studies that examined a total of 180 primary UMs, about two-thirds were diploid, only one-third demonstrated aneuploidy (which was usually limited to a few specific chromosomal changes), and only 2% were tetraploid (Coleman et al., 1995; Karlsson et al., 1995). Thus, the recurring chromosomal abnormalities in UM discussed below are likely to be specific to tumor progression rather than random events.
The most common of these abnormalities include loss on 1p, 3, 6q, 8p, and 9p and gain on 1q, 6p, and 8q. These were initially identified by standard karyotypic analyses (Griffin et al., 1988; Horsman et al., 1990; Prescher et al., 1990, 1995; Singh et al., 1994; Sisley et al., 1990; Wiltshire et al., 1993), but have since been confirmed by fluorescence in situ hybridization (FISH; McNamara et al., 1997; Patel et al., 2001), comparative genomic hybridization (CGH; Aalto et al., 2001; Ehlers et al., 2008; Ghazvini et al., 1996; Gordon et al., 1994; Hughes et al., 2005; Kilic et al., 2006; Speicher et al., 1994), spectral karyotyping (Naus et al., 2001), microsatellite analysis (MSA; Scholes et al., 2003; Tschentscher et al., 2000), multiplex ligation-dependent probe amplification (MLPA; Damato et al., 2010), and single-nucleotide polymorphisms (SNPs; Onken et al., 2007). These abnormalities are discussed in greater detail later.
Chromosome 1
Loss of part or all of chromosome 1p occurs in about a quarter of UMs (Figure 1) and more often occurs in the context of monosomy 3 (Hausler et al., 2005). 1p loss is one of the few chromosomal abnormalities that provides prognostic information that is independent of chromosome 3 status, with its presence portending decreased disease-free survival (Kilic et al., 2005). Microsatellite analysis of 70 UMs identified the smallest common region of loss on 1p to about 55 Mb at 1p31 (Hausler et al., 2005). No mutations in this region have been identified, but there are several potential candidates such as the Notch pathway members HES2 and HES5, and the p53 homologue TP73 (Kilic et al., 2008).
Figure 1.

Common chromosomal gains and losses in cutaneous and uveal melanomas. This represents a summary of data published by Hoglund and colleagues (Hoglund et al., 2004). Data are presented for all chromosomal arms in which the indicated alteration was present in at least 20% of either cutaneous or uveal melanomas. (−) indicates loss and (+) indicates gain of the indicated chromosomal arm of whole chromosome.
Chromosome 6
Gain of 6p and loss of 6q occur in about a quarter to a third of UMs (Figure 1). Both abnormalities are often present in the same tumor, suggesting the formation of an isochromosome 6p (Aalto et al., 2001). 6p gain has received much more attention than has 6q loss, but it is unclear which chromosomal arm is pathogenetically more significant or whether both are important. Overall, 6p gain is associated with a better prognosis than monosomy 3, which has led some investigators to speculate that 6p gain is somehow ‘protective’ against metastasis (Damato et al., 2009; White et al., 1998). However, it seems more likely that 6p gain is associated with better prognosis simply because it tends to occur in the absence of monosomy 3 (Ehlers et al., 2008; Parrella et al., 1999; Prescher et al., 1995). This relative mutual exclusivity of 6p gain and monosomy 3 may represent alternative evolutionary pathways that are available during tumor progression, the former being less likely to eventuate in metastasis than the latter (Landreville et al., 2008; Parrella et al., 1999). Using conventional karyotyping, CGH and FISH, the common region of 6p gain has been narrowed to 6pter–6p21 and of 6q loss to 6q16.1–6q22 (Bott et al., 2011; Speicher et al., 1994). However, no pathogenic mutations in these regions have yet been reported.
Chromosome 8
8p loss occurs in about a quarter and 8q gain in almost 40% of UMs (Figure 1). 8q gain is statistically associated with metastasis (Sisley et al., 1997) and has attracted a great deal more attention than 8p loss, yet its significance remains elusive. The smallest region of common gain on 8q has been narrowed to the large region 8q23–24 → qter (Prescher et al., 1995; Speicher et al., 1994), which contains many potential oncogenes such as MYC, which is amplified in about 30% of UMs (Parrella et al., 2001). Other potential oncogenes in this region include DDEF1 and NBS1 (now referred to as ASAP1 and NBN, respectively), which are overexpressed in UMs with poor prognosis (Ehlers and Harbour, 2005; Ehlers et al., 2005). However, a pathogenetic significance for any of these observations has not been established, and no specific oncogenic mutations on 8q have been reported in UM. Using MLPA, which interrogates a limited number of loci (Van Dijk et al., 2005), 8q status purportedly yielded prognostic information that was independent of chromosome 3 status (Damato et al., 2009). However, this finding has not been corroborated by other investigators using higher-resolution genome-wide techniques such as array CGH (Kilic et al., 2005; Onken et al., 2008a). It is perhaps notable that 8q gain is a poor prognostic factor mainly when it occurs in the context of 8p loss (Bernstein et al., 2005; Onken et al., 2008a), suggesting the formation of an isochromosome 8q (Prescher et al., 1995). This accounts for about a quarter of UMs with 8q gain and occurs almost exclusively in poor prognosis monosomy 3 tumors (Figure 2). Our group found that 8p loss was a more important prognostic factor than 8q gain (Onken et al., 2008a), and we identified LZTS1, located within a minimal deleted region on 8p, as a potential metastasis suppressor gene (Onken et al., 2008a). Thus, it may be that 8p loss rather than 8q gain is more significant, both prognostically and pathogenetically. Further work is needed to determine the role of chromosome 8 abnormalities in UM progression.
Figure 2.

Summary of the combinations of alterations observed on chromosomal arms 8p and 8q in 240 primary uveal melanomas. These data were compiled from 10 published studies that used karyotype analysis, FISH or CGH (Aalto et al., 2001; Hughes et al., 2005; Kilic et al., 2006; Naus et al., 2001; Prescher et al., 1995; Sisley et al., 1997; Speicher et al., 1994; Tschentscher et al., 2000; White et al., 1998; Wiltshire et al., 1993).
Chromosome 9
Cytogenetically detectable loss on chromosome 9p occurs in almost a quarter of UMs (Figure 1), and smaller regions of LOH around 9p21, including the CDKN2A locus, are found in up to a third of UMs (Merbs and Sidransky, 1999; Ohta et al., 1996). Methylation of the CDKN2A promoter occurs in 24–31% of cases (Merbs and Sidransky, 1999; Van Der Velden et al., 2001). These findings suggest that inactivation of CDKN2A may play a role in UM progression. However, germline CDKN2A mutations are very rare in patients with UM (Ohta et al., 1996; Singh et al., 1996a; Soufir et al., 2000).
Chromosome 3 and BAP1
Loss of one copy of chromosome 3 (monosomy 3) occurs in almost half of UMs (Figure 1) and is by far the most prognostically significant chromosomal marker in UM. Monosomy 3 is strongly associated with clinical and histopathologic prognostic factors and with metastatic death (Prescher et al., 1996; Scholes et al., 2003). Chromosome 3 has attracted enormous attention, with the expectation that it may harbor one or more tumor suppressor genes important to UM progression (Horsthemke et al., 1992). In the past, many groups attempted to narrow the minimal deleted region(s) on chromosome 3 and implicated various loci, including 3p11–14 (Blasi et al., 1999; Cross et al., 2006), 3p25–26 (Cross et al., 2006; Tschentscher et al., 2001), 3p25.1–3p25.2 (Parrella et al., 2003), and less consistent regions on 3q (Cross et al., 2006; Tschentscher et al., 2001). Unfortunately, many of these studies did not consider partial deletions in the context of patient outcome. It is now known that partial deletions of chromosome 3 in UM are quite common, but are usually not prognostically relevant (Toyota et al., 2000), such that the significance of partial deletions in the absence of metastasis is not clear. Many studies have analyzed candidate genes on chromosome 3, but have been unsuccessful in identifying the specific mutations needed to establish pathogenetic relevance (Myatt et al., 2000; Sisley et al., 1993; Zeschnigk et al., 2003).
We approached this problem using exome capture followed by next-generation sequencing (Harbour et al., 2010). Initially, two UMs were interrogated, both of which were known to be monosomic for chromosome 3 and to have given rise to metastasis. BRCA1-associated protein-1 (BAP1), located at chromosome 3p21.1, was the only gene on chromosome 3 that was mutated in both tumors. Using Sanger sequencing, we went on to find inactivating mutations in BAP1 in 27 of 57 (47%) UMs. These mutations occurred almost exclusively in metastasizing tumors that had also lost the other copy of chromosome 3, consistent with the ‘two-hit’ model for recessive cancer genes.
BAP1 mutations had previously been identified in a small number of breast and lung cancer cell lines (Jensen et al., 1998) and more recently in malignant pleural mesotheliomas (Bott et al., 2011; Testa et al., 2011), cutaneous melanoma (Wiesner et al., 2011), and possibly other cancers such as meningioma (Abdel-Rahman et al., 2011). BAP1 was identified through a screen for proteins that interact with BRCA1 and has been shown to cooperate with BRCA1 in tumor suppression in cultured cells (Jensen et al., 1998). BAP1 has also been shown to be involved in cell cycle regulation through interaction with host cell factor-1 (Machida et al., 2009), which functions as a transcriptional coactivator with E2F proteins during cell division (Tyagi et al., 2007). More recently, it was shown that calypso, the Drosophila homologue of BAP1, as well as human Bap1 protein, removes monoubiquitin moieties from histone H2A in a manner dependent on interaction with ASX (ASXL1 in humans) (Scheuermann et al., 2010). This activity regulates Hox gene expression, suggesting that BAP1 plays a role in transcriptional regulation during development. While the relative importance of these various interactions remains unclear, a crucial role for BAP1’s deubiquinating activity is strongly suggested by several lines of evidence: (i) the requirement for this activity for tumor suppression in cell culture experiments (Ventii et al., 2008) and (ii) most missense mutations directly target the deubiquitinating catalytic domain (Harbour et al., 2010).
Gene expression profiling
Cytogenetic alterations have provided important insights into the pathobiology of UM, but for use in clinical prognostication such markers are susceptible to sampling error owing to significant intratumoral heterogeneity (Damato et al., 2009). Thus, several groups have explored the use of gene expression profiling (GEP) as a potentially more robust prognostic method, as well as for its potential insights into UM pathobiology. In an early study using nylon filter arrays, several genes were found to be differentially expressed in 12 UM cell lines compared with 3 normal melanocyte cultures (Zuidervaart et al., 2003). Using high-density microarrays, another group found that UMs with disomy 3 exhibited a different GEP than those with monosomy 3 (Tschentscher et al., 2003).
Our group went a step further and showed that GEP could classify UMs into two prognostically significant groups using unsupervised clustering techniques without regard to cytogenetic status (Onken et al., 2004). Class 1 tumors had a low risk, and class 2 tumors had a high risk of metastasis. Notably, the prognostic accuracy of this GEP classification outperformed clinical, pathological, and cytogenetic prognostic indicators (Worley et al., 2007), and this has been confirmed by several independent groups (Petrausch et al., 2008; Van Gils et al., 2008). A likely reason for the superiority of GEP over cytogenetic methods for prognostication is that cytogenetic markers are often distributed heterogeneously throughout the tumor and are thus prone to sampling error. In contrast, GEP represents a functional ‘snapshot’ of the tumor’s microenvironment that is less variable across the tumor (Onken et al., 2010). We migrated the GEP to an assay comprising 12 discriminating genes and 3 control genes performed on a microfluidics platform that could be used on a routine clinical basis on very small samples from fine-needle biopsies (Onken et al., 2006b, 2010). The prognostic accuracy of this assay, and its superiority over chromosome 3 status for clinical prognostic testing, was recently validated in a prospective study involving ten centers across North America (Onken et al., in press).
Aside from its clinical value, gene expression profiling has provided important insights into the pathobiology of UM. The GEP of class 1 tumors closely resembles that of normal uveal melanocytes and low-grade uveal melanocytic tumors, whereas the GEP of class 2 tumors shows reduced expression of melanocytic genes and instead resembles the transcriptome of primitive neural/ectodermal cells (Chang et al., 2008; Onken et al., 2006a). Notably, depletion of BAP1 in cultured class 1 UM cells induced a change in cell morphology to class 2-like epithelioid phenotype and a shift in gene expression to resemble the class 2 GEP (Harbour et al., 2010). Taken together, these findings suggest that BAP1 may play a role in maintaining key aspects of melanocytic differentiation that, when lost, allow malignant progression.
MicroRNA expression
MicroRNA expression profiling can cluster UMs into prognostically significant groups, with one study identifying let-7b and miR-199a as the most significant discriminators (Worley et al., 2008). In other studies, miR-34a inhibited UM cell proliferation and migration through downregulation of c-Met (McGarvey et al., 2008), and miR-137 was found to exhibit tumor suppressor activity through downregulation of MITF and CDK6 (Chen et al., 2011). The pathogenetic relevance of these microRNA alterations in vivo is yet to be determined.
Molecular pathway defects
The Rb and p53 pathways are functionally inhibited in most UMs, although mutations in the RB1 and TP53 genes are rare (Brantley and Harbour, 2000a,b; Chana et al., 1999; Scholes et al., 2001; Sun et al., 2005). The Rb protein is constitutively hyperphosphorylated and functionally inactivated in most UMs, possibly as result of cyclin D1 overexpression or CDKN2A promoter methylation, which occur in about two-thirds and one-third of cases, respectively (Brantley and Harbour, 2000a; Coupland et al., 1998; Van Der Velden et al., 2001). The p53 pathway is inhibited downstream of p53 in many UMs (Sun et al., 2005), and this may be a consequence of MDM2 overexpression, which is common in UM (Brantley and Harbour, 2000a; Coupland et al., 2000).
The PI3K/AKT pathway is constitutively activated in a majority of UMs, and phosphorylated AKT correlates with poor prognosis in UM (Saraiva et al., 2005). In a study of nine UM cell lines, mutations in PTEN were not observed (Naus et al., 2000). However, in a much larger study of 75 primary UMs, LOH of the PTEN locus was found in 76% of tumors, and actual mutations within the PTEN coding region were found in 11% of tumors (Abdel-Rahman et al., 2006). PTEN inactivation was also found to be associated with increased aneuploidy and decreased survival in UM (Abdel-Rahman et al., 2006; Ehlers et al., 2008). Taken together, these findings implicate a role for PTEN in UM progression and warrant further work on this subject.
Most UMs demonstrate constitutive activation of the mitogen-activated protein kinase (MAPK) pathway, suggesting the presence of upstream activating mutations (Weber et al., 2003; Zuidervaart et al., 2005). Mutations in KIT and the three RAS family members, which can activate the MAPK pathway, have proven to be exceedingly rare in UM (Cruz et al., 2003; Mooy et al., 1991; Pache et al., 2003; Soparker et al., 1993; Zuidervaart et al., 2005). BRAF (V600E) mutations have been reported in a few UMs (Malaponte et al., 2006), but such mutations are rare (Cruz et al., 2003; Rimoldi et al., 2003; Weber et al., 2003; Zuidervaart et al., 2005). Interestingly, however, BRAF mutations may occur in up to 47% of iris melanomas (Henriquez et al., 2007), which are more anterior and more strongly linked to ultraviolet light exposure than the more common posterior UMs of the ciliary body and choroid. Mutations in the other two members of the RAF family, ARAF and CRAF, have not been found in UM (Onken et al., 2008b). A systemic interrogation of 21 other candidate oncogenes in the MAPK pathway identified no mutations in UM (Onken et al., 2008b).
GNAQ/11 mutations
This curious absence of MAPK pathway mutations persisted until the recent discovery of mutations in GNAQ, which encodes the Gαq subunit, in almost half of UMs (Jensen and Rauscher, 1999). Mutant GNAQ was shown to activate the MAPK pathway, although it may have also important effects on other pathways such as the phosphatidylinositol–calcium second messenger system. Attention was drawn to this gene as a result of a forward genetic screen in mice that identified hypermorphic mutations in Gnaq or its paralog Gna11, which act through the melanocyte lineage factor Ednrb, as a cause of increased numbers of intradermal melanocytes (Van Raamsdonk et al., 2004). A subsequent study found that 83% of UMs contained mutations in either GNAQ or GNA11 affecting either Q209 or R183 in a mutually exclusive pattern (Van Raamsdonk et al., 2010). These mutations lead to constitutive activation of the Gαq and Gα11 subunits by abrogating their intrinsic GTPase activity required to return them to an inactive state.
GNAQ/11 mutations are found in benign uveal nevi and in the vast majority of UMs regardless of cytogenetic status, GEP class, or BAP1 status (Bauer et al., 2009; Jensen and Rauscher, 1999; Onken et al., 2008b). Further, these mutations are not sufficient for full malignant transformation to melanoma (Van Raamsdonk et al., 2009). This would seem to place GNAQ/11 mutations as early or perhaps initiating events in UM progression. On the other hand, BAP1 mutations are seen almost exclusively in metastasizing class 2 tumors with monosomy 3 (Harbour et al., 2010), suggesting that this mutation occurs relatively late in the primary tumor and may represent a rate-limiting step in metastasis. Either BAP1 mutation or loss of chromosome 3 can occur first, but both events appear to be necessary for the tumor to acquire the metastasizing class 2 phenotype (Harbour et al., 2010). These early and late mutational events allow a tentative outline of UM progression to be constructed (Figure 3).
Figure 3.
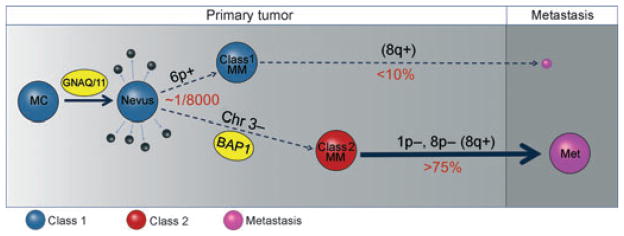
Summary of major molecular events in uveal melanoma progression. The earliest known and perhaps initiating event is an activating mutation in GNAQ or GNA11, presumably in a normal uveal melanocyte (MC), which may function primarily to trigger inappropriate cell cycle re-entry through activation of the MAPK and perhaps other pathways. Usually the mutant cell clone does not progress to melanoma, but rather undergoes senescence resulting in a nevus, or is eliminated by apoptosis or immune surveillance (small black spheres). Less than one in 8000 nevi progress beyond this stage (Singh et al., 2005b). The rare tumor that progresses does so along one of the two pathways characterized by distinct gene expression profiles (GEP). The GEPs of normal uveal melanocytes and nevi are very similar to that of class 1 uveal melanomas (blue spheres) (Chang et al., 2008), which have a low risk of metastasis (small purple sphere). Melanomas that acquire the class 2 GEP (class 2 MM) have a very high risk of metastasis (large purple sphere). Class 1 tumors often exhibit 6p gain and 8q gain, but have less overall aneuploidy than class 2 tumors, which often exhibit 1p loss, 8p loss, and 8q gain. 8q gain is more common in class 2 tumors, but is also seen in class 1 tumors, so this may be a late event (Parrella et al., 1999). The class 2 GEP is strongly associated with mutation of BAP1, located at 3p21, and loss of the other copy of chromosome 3, suggesting that bi-allelic loss of BAP1 is a key step in uveal metastasis (Harbour et al., 2010). Metastatic tumors (purple spheres) have a distinct gene expression profile that is more similar to that of class 2 than that of class 1 primary tumors (author’s unpublished data). MC, melanocyte; MM, malignant melanoma; Met, metastasis.
Genetic comparison of uveal and cutaneous melanomas
Several groups have compared the cytogenetic alterations in uveal and cutaneous melanomas (Bastian et al., 1998; Curtin et al., 2005; Hoglund et al., 2004). Some of the most common chromosomal alterations in UM – 1p loss, 1q gain, 6p gain, 6q loss, 8p loss, 8q gain, and 11q loss – are also common in cutaneous melanoma (Figure 1). Monosomy 3, the most common change in UM, is also seen in cutaneous melanoma, although at a lower frequency. Likewise, loss on 9p and 10, which are very common in cutaneous melanoma, are also seen in UM, albeit not as often. Greater differences arise at the level of individual gene mutations. Activating mutations in BRAF and NRAS are common in some types of cutaneous melanoma, but are distinctively rare in UM (Cruz et al., 2003; Curtin et al., 2005; Davies et al., 2002; Mooy et al., 1991; Rimoldi et al., 2003; Soparker et al., 1993; Weber et al., 2003; Zuidervaart et al., 2005). Likewise, activating mutations in GNAQ/GNA11 occur in the vast majority of UMs, but are rare in cutaneous melanoma (Van Raamsdonk et al., 2010). Nevertheless, all of these mutations appear to have in common the constitutive activation of the MAPK pathway (Weber et al., 2003). BAP1 mutations, which are strongly linked to metastasis in UM (Harbour et al., 2010), also occur in cutaneous melanoma (Wiesner et al., 2011), but it is unclear whether these mutations play the same role in the latter as they do in the former. Taken together, there are clearly genetic differences between uveal and cutaneous melanoma, but there are also remarkable similarities, with many seemingly divergent features potentially having similar effects at the molecular and cellular level.
Familial uveal melanoma
Traditionally, it has been thought that familial UM is extremely rare (Singh et al., 1996b). A few studies have suggested a link between UM and breast cancer, possibly as a consequence of germline BRCA2 mutations (Iscovich et al., 2002; Scott et al., 2002; Sinilnikova et al., 1999). In one study, constitutional DNA samples were analyzed for BRCA2 mutations in 62 patients with UM who were selected primarily on the basis of a family history of breast cancer or UM harbored; three (4.8%) patients harbored BRCA2 sequence variants that were judged to be potentially deleterious (Sinilnikova et al., 1999). An Israeli study identified 4/143 (2.8%) patients with UM who carried a germline 6174delT BRCA2 mutation (Iscovich et al., 2002). However, as this alteration is prevalent in the Israeli population, the pathogenetic relevance of this finding is unclear. An Australian study found germline BRCA2 mutations in 2/71 (2.8%) patients with UM, but neither of these patients had a positive family history, thus leaving open the possibility that these were silent polymorphisms (Scott et al., 2002).
Given the rarity of familial UM in the literature, we were surprised to find that one of the patients with UM in our original study carried a germline BAP1 mutation that was reduced to homozygosity in the tumor by loss of the other copy of chromosome 3 (Harbour et al., 2010). We have identified another family from our ocular oncology center in which UM and cutaneous melanoma occurred in multiple family member in association with a germline BAP1 mutation (author’s unpublished data). The scope of tumors associated with this emerging BAP1 cancer predisposition syndrome has now been expanded to include malignant mesothelioma, cutaneous melanocytic tumors, and other cancers (Testa et al., 2011; Wiesner et al., 2011).
Thus, while familial UM is indeed uncommon, representing perhaps 2–5% of patients with new UM, it is not as rare as once believed. The reason that familial UM was not recognized more commonly in the past may be because of a reduced penetrance for UM in these families (Testa et al., 2011; Wiesner et al., 2011). This would be consistent with previous studies that found that familial UM rarely involved more than 2–3 family members and was more likely to occur in larger families (where reduced penetrance would more likely be recognized; Abdel-Rahman et al., 2011; Canning and Hungerford, 1988; Singh et al., 1996b; Young et al., 1994). A possible explanation for the reduced penetrance is that BAP1 inactivation appears to be a relatively late event in melanoma progression and requires an initiating event, such as an activating mutation GNAQ/11 in UM or BRAF in cutaneous melanoma, and loss of the other copy of BAP1 in order for the germline BAP1 mutation to become manifest. In support of this idea, a cutaneous melanocytic tumor associated with germline BAP1 mutation also harbored mutant BRAF, and only the portion of the tumor that had lost the other copy of BAP1 progressed to melanoma (Wiesner et al., 2011). Similarly, our reported case of UM associated with a germline BAP1 mutation harbored a GNAQ mutation and had lost the other copy of chromosome 3 (Harbour et al., 2010).
Implications for targeted therapy
While excellent local therapies exist for treating the primary ocular tumor, there are no consistently effective therapies for metastatic UM (Augsburger et al., 2009). The discovery of GNAQ/11 and BAP1 mutations in UM provides an unprecedented opportunity for targeted therapy of metastatic disease. Nevertheless, molecular targeting of these mutations will pose significant challenges.
For GNAQ/11 mutations, the therapeutic goal is to inhibit oncogenic downstream signaling resulting from these mutations. Direct inhibition of mutant Gαq or Gα11 may prove difficult, however, because these mutations abrogate the intrinsic GTPase activity that would normally allow these proteins to return to their GDP-bound, inactive state. This is similar to the long-standing problem targeting mutations in RAS family oncogenes, which also rely on intrinsic GTPase activity to terminate signaling (Diaz-Flores and Shannon, 2007). An alternative strategy is to inhibit downstream signaling molecules that are activated by GNAQ/11 mutations. One such target is MEK (Mitsiades et al., 2011), a key component of the MAPK mitogenic pathway that is activated by GNAQ/11 mutations (Jensen and Rauscher, 1999). Other potential targets that are currently being investigated include phospholipase C (PLC), which is activated by Gq, and protein kinase C (PKC), which is activated downstream of PLC (Patel et al., 2011).
Therapeutic targeting of BAP1 mutations poses different challenges. First, as BAP1 acts as a recessive cancer gene, the goal of therapy is to restore one or more functions of BAP1 that are lost when it is inactivated. This is technically more challenging than inhibiting an overactive oncogene. Second, it remains unclear which function(s) of BAP1 is responsible for its anticancer role. Nevertheless, it seems likely that this role is dependent on BAP1’s deubiquinating activity, as discussed in a previous section. Further, a major function of this deubiquinating activity appears to be the removal of monoubiquitin moieties from histone H2A, which alters local chromatin structure to regulate transcription. Thus, a compound that inhibits H2A monoubiquitination may at least in part offset the biochemical, and consequently cellular, effects of BAP1 loss. Histone deacetylase (HDAC) inhibitors represent one such class of compounds. Through inhibition of the Bmi1/Ring1 complex, which monoubiquitinates H2A (Bommi et al., 2010), HDAC inhibitors such as valproic acid, trichostatin A, LBH-589, and suberoylanilide hydroxamic acid can reverse the H2A hyperubiquitination that occurs in cultured UM cells depleted of BAP1, and this is accompanied by increased melanocytic differentiation, cell cycle exit, and a shift from class 1 to class 2 GEP (Landreville et al., 2012). This study also showed antitumor activity of valproic acid in vivo in an animal model of UM. Thus, HDAC inhibitors may have therapeutic potential in UM, and clinical trials are currently being planned.
Conclusions
In recent years, there has been tremendous progress in understanding the genetics of melanoma in general and UM in particular. Activating mutations in GNAQ/11 appear to represent a very early or initiating event, whereas inactivating mutations in BAP1 appear to demarcate a threshold in tumor progression beyond which metastasis and death await. The opportunities for targeted therapy afforded by the discovery of GNAQ/11 and BAP1 mutations are being explored. Therapies based on GNAQ/11 status are underway for metastatic UM. A greater understanding of the normal function of BAP1 in the melanocyte lineage and the adverse effects of BAP1 loss are needed to develop agents that specifically target this mutation. Further insights to guide therapy may derive from future research into the genetic events that occur after metastasis.
Acknowledgments
The author would like to acknowledge Michael D. Onken and Lori A. Worley for critical review of the manuscript, and Justis Ehlers for compiling the chromosomal data presented in Figure 2. Dr. Harbour is funded by grants from the National Cancer Institute (R01 CA125970 and R01 CA16187001), Melanoma Research Foundation, Melanoma Research Alliance, Research to Prevent Blindness Senior Investigator Award, Barnes-Jewish Hospital Foundation, Kling Family Foundation, and Tumori Foundation. Dr. Harbour and Washington University may receive royalties based on a license of related technology by the University to Castle Biosciences, Inc.
References
- Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:313–317. [PubMed] [Google Scholar]
- Abdel-Rahman MH, Yang Y, Zhou XP, Craig EL, Davidorf FH, Eng C. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J Clin Oncol. 2006;24:288–295. doi: 10.1200/JCO.2005.02.2418. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, Hovland P, Davidorf FH. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–859. doi: 10.1136/jmedgenet-2011-100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augsburger JJ, Gamel JW. Clinical prognostic factors in patients with posterior uveal malignant melanoma. Cancer. 1990;66:1596–1600. doi: 10.1002/1097-0142(19901001)66:7<1596::aid-cncr2820660726>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Augsburger JJ, Correa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009;148:119–127. doi: 10.1016/j.ajo.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Bastian BC, Leboit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–2175. [PubMed] [Google Scholar]
- Bauer J, Kilic E, Vaarwater J, Bastian BC, Garbe C, De Klein A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br J Cancer. 2009;101:813–815. doi: 10.1038/sj.bjc.6605226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Blasi MA, Roccella F, Balestrazzi E, Del Porto G, De Felice N, Roccella M, Rota R, Grammatico P. 3p13 region: a possible location of a tumor suppressor gene involved in uveal melanoma. Cancer Genet Cytogenet. 1999;108:81–83. doi: 10.1016/s0165-4608(98)00114-9. [DOI] [PubMed] [Google Scholar]
- Bommi PV, Dimri M, Sahasrabuddhe AA, Khandekar J, Dimri GP. The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle. 2010;9:2663–2673. doi: 10.4161/cc.9.13.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley MA, Jr, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000a;157:1795–1801. doi: 10.1016/s0002-9440(10)64817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley MA, Jr, Harbour JW. Inactivation of retinoblastoma protein in uveal melanoma by phosphorylation of sites in the COOH-terminal region. Cancer Res. 2000b;60:4320–4323. [PMC free article] [PubMed] [Google Scholar]
- Canning CR, Hungerford J. Familial uveal melanoma. Br J Ophthalmol. 1988;72:241–243. doi: 10.1136/bjo.72.4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chana JS, Wilson GD, Cree IA, Alexander RA, Myatt N, Neale M, Foss AJ, Hungerford JL. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. Br J Ophthalmol. 1999;83:110–114. doi: 10.1136/bjo.83.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Worley LA, Onken MD, Harbour JW. Prognostic biomarkers in uveal melanoma: evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008;18:191–200. doi: 10.1097/CMR.0b013e3283005270. [DOI] [PubMed] [Google Scholar]
- Chen X, Wang J, Shen H, Lu J, Li C, Hu DN, Dong XD, Yan D, Tu L. Epigenetics, microRNAs, and carcinogenesis: functional role of microRNA-137 in uveal melanoma. Invest Ophthalmol Vis Sci. 2011;52:1193–1199. doi: 10.1167/iovs.10-5272. [DOI] [PubMed] [Google Scholar]
- Coleman K, Baak JP, Van Diest PJ, Curran B, Mullaney J, Fenton M, Leader M. DNA ploidy status in 84 ocular melanomas: a study of DNA quantitation in ocular melanomas by flow cytometry and automatic and interactive static image analysis. Hum Pathol. 1995;26:99–105. doi: 10.1016/0046-8177(95)90121-3. [DOI] [PubMed] [Google Scholar]
- Coupland SE, Bechrakis N, Schuler A, Anagnostopoulos I, Hummel M, Bornfeld N, Stein H. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br J Ophthalmol. 1998;82:961–970. doi: 10.1136/bjo.82.8.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupland SE, Anastassiou G, Stang A, Schilling H, Anagnostopoulos I, Bornfeld N, Stein H. The prognostic value of cyclin D1, p53, and MDM2 protein expression in uveal melanoma. J Pathol. 2000;191:120–126. doi: 10.1002/(SICI)1096-9896(200006)191:2<120::AID-PATH591>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Cross NA, Murray AK, Rennie IG, Ganesh A, Sisley K. Instability of microsatellites is an infrequent event in uveal melanoma. Melanoma Res. 2003;13:435–440. doi: 10.1097/00008390-200310000-00001. [DOI] [PubMed] [Google Scholar]
- Cross NA, Ganesh A, Parpia M, Murray AK, Rennie IG, Sisley K. Multiple locations on chromosome 3 are the targets of specific deletions in uveal melanoma. Eye. 2006;20:476–481. doi: 10.1038/sj.eye.6701906. [DOI] [PubMed] [Google Scholar]
- Cruz F, 3rd, Rubin BP, Wilson D, Town A, Schroeder A, Haley A, Bainbridge T, Heinrich MC, Corless CL. Absence of BRAF and NRAS Mutations in Uveal Melanoma. Cancer Res. 2003;63:5761–5766. [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Damato BE, Dopierala J, Klaasen A, Van Dijk M, Sibbring J, Coupland S. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest Ophthalmol Vis Sci. 2009;50:3048–3055. doi: 10.1167/iovs.08-3165. [DOI] [PubMed] [Google Scholar]
- Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010;16:6083–6092. doi: 10.1158/1078-0432.CCR-10-2076. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- De La Cruz PO, Jr, Specht CS, Mclean IW. Lymphocytic infiltration in uveal malignant melanoma. Cancer. 1990;65:112–115. doi: 10.1002/1097-0142(19900101)65:1<112::aid-cncr2820650123>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Diaz-Flores E, Shannon K. Targeting oncogenic Ras. Genes Dev. 2007;21:1989–1992. doi: 10.1101/gad.1587907. [DOI] [PubMed] [Google Scholar]
- Diener-West M, Reynolds SM, Agugliaro DJ, et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005;123:1639–1643. doi: 10.1001/archopht.123.12.1639. [DOI] [PubMed] [Google Scholar]
- Egan KM, Seddon JM, Glynn RJ, Gragoudas ES, Albert DM. Epidemiologic aspects of uveal melanoma. Surv Ophthalmol. 1988;32:239–251. doi: 10.1016/0039-6257(88)90173-7. [DOI] [PubMed] [Google Scholar]
- Ehlers JP, Harbour JW. NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res. 2005;11:1849–1853. doi: 10.1158/1078-0432.CCR-04-2054. [DOI] [PubMed] [Google Scholar]
- Ehlers JP, Worley L, Onken MD, Harbour JW. DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res. 2005;11:3609–3613. doi: 10.1158/1078-0432.CCR-04-1941. [DOI] [PubMed] [Google Scholar]
- Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res. 2008;14:115–122. doi: 10.1158/1078-0432.CCR-07-1825. [DOI] [PubMed] [Google Scholar]
- Folberg R, Pe’er J, Gruman LM, Woolson RF, Jeng G, Montague PR, Moninger TO, Yi H, Moore KC. The morphologic characteristics of tumor blood vessels as a marker of tumor progression in primary human uveal melanoma: a matched case–control study. Hum Pathol. 1992;23:1298–1305. doi: 10.1016/0046-8177(92)90299-i. [DOI] [PubMed] [Google Scholar]
- Gallagher RP, Elwood JM, Rootman J, Spinelli JJ, Hill GB, Threlfall WJ, Birdsell JM. Risk factors for ocular melanoma: Western Canada Melanoma Study. J Natl Cancer Inst. 1985;74:775–778. [PubMed] [Google Scholar]
- Gamel JW, Mclean IW, Foster WD, Zimmerman LE. Uveal melanomas: correlation of cytologic features with prognosis. Cancer. 1978;41:1897–1901. doi: 10.1002/1097-0142(197805)41:5<1897::aid-cncr2820410534>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Ghazvini S, Char DH, Kroll S, Waldman FM, Pinkel D. Comparative genomic hybridization analysis of archival formalin-fixed paraffin-embedded uveal melanomas. Cancer Genet Cytogenet. 1996;90:95–101. doi: 10.1016/s0165-4608(96)00076-3. [DOI] [PubMed] [Google Scholar]
- Gordon KB, Thompson CT, Char DH, O’brien JM, Kroll S, Ghazvini S, Gray JW. Comparative genomic hybridization in the detection of DNA copy number abnormalities in uveal melanoma. Cancer Res. 1994;54:4764–4768. [PubMed] [Google Scholar]
- Griffin CA, Long PP, Schachat AP. Trisomy 6p in an ocular melanoma. Cancer Genet Cytogenet. 1988;32:129–132. doi: 10.1016/0165-4608(88)90319-6. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausler T, Stang A, Anastassiou G, Jockel KH, Mrzyk S, Horsthemke B, Lohmann DR, Zeschnigk M. Loss of heterozygosity of 1p in uveal melanomas with monosomy 3. Int J Cancer. 2005;116:909–913. doi: 10.1002/ijc.21086. [DOI] [PubMed] [Google Scholar]
- Henriquez F, Janssen C, Kemp EG, Roberts F. The T1799A BRAF mutation is present in iris melanoma. Invest Ophthalmol Vis Sci. 2007;48:4897–4900. doi: 10.1167/iovs.07-0440. [DOI] [PubMed] [Google Scholar]
- Hoglund M, Gisselsson D, Hansen GB, White VA, Sall T, Mitelman F, Horsman D. Dissecting karyotypic patterns in malignant melanomas: temporal clustering of losses and gains in melanoma karyotypic evolution. Int J Cancer. 2004;108:57–65. doi: 10.1002/ijc.11558. [DOI] [PubMed] [Google Scholar]
- Horsman DE, Sroka H, Rootman J, White VA. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet Cytogenet. 1990;45:249–253. doi: 10.1016/0165-4608(90)90090-w. [DOI] [PubMed] [Google Scholar]
- Horsthemke B, Prescher G, Bornfeld N, Becher R. Loss of chromosome 3 alleles and multiplication of chromosome 8 alleles in uveal melanoma. Genes Chromosom Cancer. 1992;4:217–221. doi: 10.1002/gcc.2870040305. [DOI] [PubMed] [Google Scholar]
- Hughes S, Damato BE, Giddings I, Hiscott PS, Humphreys J, Houlston RS. Microarray comparative genomic hybridisation analysis of intraocular uveal melanomas identifies distinctive imbalances associated with loss of chromosome 3. Br J Cancer. 2005;93:1191–1196. doi: 10.1038/sj.bjc.6602834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iscovich J, Abdulrazik M, Cour C, Fischbein A, Pe’er J, Goldgar DE. Prevalence of the BRCA2 6174 del T mutation in Israeli uveal melanoma patients. Int J Cancer. 2002;98:42–44. doi: 10.1002/ijc.10155. [DOI] [PubMed] [Google Scholar]
- Jensen DE, Rauscher FJ., 3rd BAP1, a candidate tumor suppressor protein that interacts with BRCA1. Ann N Y Acad Sci. 1999;886:191–194. doi: 10.1111/j.1749-6632.1999.tb09414.x. [DOI] [PubMed] [Google Scholar]
- Jensen DE, Proctor M, Marquis ST, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16:1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Boeryd B, Carstensen J, Kagedal B, Wingren S. DNA ploidy and S-phase fraction as prognostic factors in patients with uveal melanomas. Br J Cancer. 1995;71:177–181. doi: 10.1038/bjc.1995.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kath R, Hayungs J, Bornfeld N, Sauerwein W, Hoffken K, Seeber S. Prognosis and treatment of disseminated uveal melanoma. Cancer. 1993;72:2219–2223. doi: 10.1002/1097-0142(19931001)72:7<2219::aid-cncr2820720725>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Kilic E, Naus NC, Van Gils W, et al. Concurrent loss of chromosome arm 1p and chromosome 3 predicts a decreased disease-free survival in uveal melanoma patients. Invest Ophthalmol Vis Sci. 2005;46:2253–2257. doi: 10.1167/iovs.04-1460. [DOI] [PubMed] [Google Scholar]
- Kilic E, Van Gils W, Lodder E, Beverloo HB, Van Til ME, Mooy CM, Paridaens D, De Klein A, Luyten GP. Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci. 2006;47:3703–3707. doi: 10.1167/iovs.06-0101. [DOI] [PubMed] [Google Scholar]
- Kilic E, Bruggenwirth HT, Meier M, Naus NC, Beverloo HB, Meijerink JP, Luyten GP, De Klein A. Increased expression of p73Deltaex2 transcript in uveal melanoma with loss of chromosome 1p. Melanoma Res. 2008;18:208–213. doi: 10.1097/CMR.0b013e3283036aa1. [DOI] [PubMed] [Google Scholar]
- Kujala E, Makitie T, Kivela T. Very Long-Term Prognosis of Patients with Malignant Uveal Melanoma. Invest Ophthalmol Vis Sci. 2003;44:4651–4659. doi: 10.1167/iovs.03-0538. [DOI] [PubMed] [Google Scholar]
- Landreville S, Agapova OA, Harbour JW. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008;4:629–636. doi: 10.2217/14796694.4.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreville S, Agapova OA, Matatall KA, Kneass ZT, Onken MD, Lee RS, Bowcock AM, Harbour JW. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012;18:408–416. doi: 10.1158/1078-0432.CCR-11-0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem. 2009;284:34179–34188. doi: 10.1074/jbc.M109.046755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makitie T, Summanen P, Tarkkanen A, Kivela T. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:1414–1421. [PubMed] [Google Scholar]
- Malaponte G, Libra M, Gangemi P, Bevelacqua V, Mangano K, D’amico F, Mazzarino MC, Stivala F, Mccubrey JA, Travali S. Detection of BRAF gene mutation in primary choroidal melanoma tissue. Cancer Biol Ther. 2006;5:225–227. doi: 10.4161/cbt.5.2.2429. [DOI] [PubMed] [Google Scholar]
- McGarvey KM, Van Neste L, Cope L, Ohm JE, Herman JG, Van Criekinge W, Schuebel KE, Baylin SB. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008;68:5753–5759. doi: 10.1158/0008-5472.CAN-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara M, Felix C, Davison EV, Fenton M, Kennedy SM. Assessment of chromosome 3 copy number in ocular melanoma using fluorescence in situ hybridization. Cancer Genet Cytogenet. 1997;98:4–8. doi: 10.1016/s0165-4608(96)00405-0. [DOI] [PubMed] [Google Scholar]
- Merbs SL, Sidransky D. Analysis of p16 (CDKN2/MTS-1/INK4A) alterations in primary sporadic uveal melanoma. Invest Ophthalmol Vis Sci. 1999;40:779–783. [PubMed] [Google Scholar]
- Mitsiades N, Chew SA, He B, Riechardt AI, Karadedou T, Kotoula V, Poulaki V. Genotype-dependent sensitivity of uveal melanoma cell lines to inhibition of B-Raf, MEK, and Akt kinases: rationale for personalized therapy. Invest Ophthalmol Vis Sci. 2011;52:7248–7255. doi: 10.1167/iovs.11-7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooy CM, Van Der Helm MJ, Van Der Kwast TH, De Jong PT, Ruiter DJ, Zwarthoff EC. No N-ras mutations in human uveal melanoma: the role of ultraviolet light revisited. Br J Cancer. 1991;64:411–413. doi: 10.1038/bjc.1991.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt N, Aristodemou P, Neale MH, Foss AJ, Hungerford JL, Bhattacharya S, IAC Abnormalities of the transforming growth factor-beta pathway in ocular melanoma. J Pathol. 2000;192:511–518. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH778>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Naus NC, Zuidervaart W, Rayman N, Slater R, Van Drunen E, Ksander B, Luyten GP, Klein A. Mutation analysis of the PTEN gene in uveal melanoma cell lines. Int J Cancer. 2000;87:151–153. doi: 10.1002/1097-0215(20000701)87:1<151::aid-ijc23>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Naus NC, Van Drunen E, De Klein A, Luyten GP, Paridaens DA, Alers JC, Ksander BR, Beverloo HB, Slater RM. Characterization of complex chromosomal abnormalities in uveal melanoma by fluorescence in situ hybridization, spectral karyotyping, and comparative genomic hybridization. Genes Chromosom Cancer. 2001;30:267–273. [PubMed] [Google Scholar]
- Ohta M, Berd D, Shimizu M, Nagai H, Cotticelli MG, Mastrangelo M, Shields JA, Shields CL, Croce CM, Huebner K. Deletion mapping of chromosome region 9p21-p22 surrounding the CDKN2 locus in melanoma. Int J Cancer. 1996;65:762–767. doi: 10.1002/(SICI)1097-0215(19960315)65:6<762::AID-IJC9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006a;66:4602–4609. doi: 10.1158/0008-5472.CAN-05-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Davila RM, Char DH, Harbour JW. Prognostic testing in uveal melanoma by transcriptomic profiling of fine needle biopsy specimens. J Mol Diagn. 2006b;8:567–573. doi: 10.2353/jmoldx.2006.060077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW. Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma. Clin Cancer Res. 2007;13:2923–2927. doi: 10.1158/1078-0432.CCR-06-2383. [DOI] [PubMed] [Google Scholar]
- Onken MD, Worley L, Harbour JW. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res. 2008a;14:3737–3745. doi: 10.1158/1078-0432.CCR-07-5144. [DOI] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, Harbour JW. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008b;49:5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010;12:461–468. doi: 10.2353/jmoldx.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken M, Worley L, Char D, et al. Collaborative Ocular Oncology Group Report No. 1: prospective Validation of a Multi-Gene Prognostic Assay in Uveal Melanoma. Ophthalmology. doi: 10.1016/j.ophtha.2012.02.017. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pache M, Glatz K, Bosch D, et al. Sequence analysis and high-throughput immunohistochemical profiling of KIT (CD 117) expression in uveal melanoma using tissue microarrays. Virchows Arch. 2003;443:741–744. doi: 10.1007/s00428-003-0883-2. [DOI] [PubMed] [Google Scholar]
- Papadopoulos S, Benter T, Anastassiou G, Pape M, Gerhard S, Bornfeld N, Ludwig WD, Dorken B. Assessment of genomic instability in breast cancer and uveal melanoma by random amplified polymorphic DNA analysis. Int J Cancer. 2002;99:193–200. doi: 10.1002/ijc.10297. [DOI] [PubMed] [Google Scholar]
- Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999;59:3032–3037. [PubMed] [Google Scholar]
- Parrella P, Caballero OL, Sidransky D, Merbs SL. Detection of c-myc Amplification in Uveal Melanoma by Fluorescent In Situ Hybridization. Invest Ophthalmol Vis Sci. 2001;42:1679–1684. [PubMed] [Google Scholar]
- Parrella P, Fazio VM, Gallo AP, Sidransky D, Merbs SL. Fine mapping of chromosome 3 in uveal melanoma: identification of a minimal region of deletion on chromosomal arm 3p25.1–p25.2. Cancer Res. 2003;63:8507–8510. [PubMed] [Google Scholar]
- Patel KA, Edmondson ND, Talbot F, Parsons MA, Rennie IG, Sisley K. Prediction of prognosis in patients with uveal melanoma using fluorescence in situ hybridisation. Br J Ophthalmol. 2001;85:1440–1444. doi: 10.1136/bjo.85.12.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Smyth E, Chapman PB, Wolchok JD, Schwartz GK, Abramson DH, Carvajal RD. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin Cancer Res. 2011;17:2087–2100. doi: 10.1158/1078-0432.CCR-10-3169. [DOI] [PubMed] [Google Scholar]
- Petrausch U, Martus P, Tonnies H, et al. Significance of gene expression analysis in uveal melanoma in comparison to standard risk factors for risk assessment of subsequent metastases. Eye. 2008;22:997–1007. doi: 10.1038/sj.eye.6702779. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Becher R. Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst. 1990;82:1765–1769. doi: 10.1093/jnci/82.22.1765. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Friedrichs W, Seeber S, Becher R. Cytogenetics of twelve cases of uveal melanoma and patterns of nonrandom anomalies and isochromosome formation. Cancer Genet Cytogenet. 1995;80:40–46. doi: 10.1016/0165-4608(94)00165-8. [DOI] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–1225. doi: 10.1016/s0140-6736(96)90736-9. [DOI] [PubMed] [Google Scholar]
- Ramaiya KJ, Harbour JW. Current management of uveal melanoma. Expert Rev Ophthalmol. 2007;2:939–946. [Google Scholar]
- Rimoldi D, Salvi S, Lienard D, Lejeune FJ, Speiser D, Zografos L, Cerottini JC. Lack of BRAF Mutations in Uveal Melanoma. Cancer Res. 2003;63:5712–5715. [PubMed] [Google Scholar]
- Saraiva VS, Caissie AL, Segal L, Edelstein C, Burnier MN., Jr Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res. 2005;15:245–250. doi: 10.1097/00008390-200508000-00003. [DOI] [PubMed] [Google Scholar]
- Scheuermann JC, De Ayala Alonso AG, Oktaba K, Ly-Hartig N, Mcginty RK, Fraterman S, Wilm M, Muir TW, Muller J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes AG, Liloglou T, Maloney P, Hagan S, Nunn J, Hiscott P, Damato BE, Grierson I, Field JK. Loss of heterozygosity on chromosomes 3, 9, 13, and 17, including the retinoblastoma locus, in uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:2472–2477. [PubMed] [Google Scholar]
- Scholes AG, Damato BE, Nunn J, Hiscott P, Grierson I, Field JK. Monosomy 3 in uveal melanoma: correlation with clinical and histologic predictors of survival. Invest Ophthalmol Vis Sci. 2003;44:1008–1011. doi: 10.1167/iovs.02-0159. [DOI] [PubMed] [Google Scholar]
- Scott RJ, Vajdic CM, Armstrong BK, Ainsworth CJ, Meldrum CJ, Aitken JF, Kricker A. BRCA2 mutations in a population-based series of patients with ocular melanoma. Int J Cancer. 2002;102:188–191. doi: 10.1002/ijc.10693. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Gragoudas ES, Glynn RJ, Egan KM, Albert DM, Blitzer PH. Host factors, UV radiation, and risk of uveal melanoma. A case–control study. Arch Ophthalmol. 1990;108:1274–1280. doi: 10.1001/archopht.1990.01070110090031. [DOI] [PubMed] [Google Scholar]
- Singh AD, Boghosian-Sell L, Wary KK, Shields CL, De Potter P, Donoso LA, Shields JA, Cannizzaro LA. Cytogenetic findings in primary uveal melanoma. Cancer Genet Cytogenet. 1994;72:109–115. doi: 10.1016/0165-4608(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Singh AD, Croce CM, Wary KK, Shields JA, Donoso LA, Shields CL, Huebner K, Ohta M. Familial uveal melanoma: absence of germline mutations involving the cyclin-dependent kinase-4 inhibitor gene (p16) Ophthalmic Genet. 1996a;17:39–40. doi: 10.3109/13816819609057868. [DOI] [PubMed] [Google Scholar]
- Singh AD, Shields CL, De Potter P, Shields JA, Trock B, Cater J, Pastore D. Familial uveal melanoma. Clinical observations on 56 patients. Arch Ophthalmol. 1996b;114:392–399. doi: 10.1001/archopht.1996.01100130388005. [DOI] [PubMed] [Google Scholar]
- Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005a;18:75–84. viii. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Singh AD, Kalyani P, Topham A. Estimating the risk of malignant transformation of a choroidal nevus. Opthalmology. 2005b;112:1784–1789. doi: 10.1016/j.ophtha.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881–1885. doi: 10.1016/j.ophtha.2011.01.040. [DOI] [PubMed] [Google Scholar]
- Sinilnikova OM, Egan KM, Quinn JL, Boutrand L, Lenoir GM, Stoppa-Lyonnet D, Desjardins L, Levy C, Goldgar D, Gragoudas ES. Germline brca2 sequence variants in patients with ocular melanoma. Int J Cancer. 1999;82:325–328. doi: 10.1002/(sici)1097-0215(19990730)82:3<325::aid-ijc3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Sisley K, Rennie IG, Cottam DW, Potter AM, Potter CW, Rees RC. Cytogenetic findings in six posterior uveal melanomas: involvement of chromosomes 3, 6, and 8. Genes Chromosom Cancer. 1990;2:205–209. doi: 10.1002/gcc.2870020307. [DOI] [PubMed] [Google Scholar]
- Sisley K, Curtis D, Rennie IG, Rees RC. Loss of heterozygosity of the thyroid hormone receptor B in posterior uveal melanoma. Melanoma Res. 1993;3:457–461. doi: 10.1097/00008390-199311000-00009. [DOI] [PubMed] [Google Scholar]
- Sisley K, Rennie IG, Parsons MA, Jacques R, Hammond DW, Bell SM, Potter AM, Rees RC. Abnormalities of chromosomes 3 and 8 in posterior uveal melanoma correlate with prognosis. Genes Chromosom Cancer. 1997;19:22–28. doi: 10.1002/(sici)1098-2264(199705)19:1<22::aid-gcc4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Soparker CN, O’brien JM, Albert DM. Investigation of the role of the ras protooncogene point mutation in human uveal melanomas. Invest Ophthalmol Vis Sci. 1993;34:2203–2209. [PubMed] [Google Scholar]
- Soufir N, Bressac-De Paillerets B, Desjardins L, Levy C, Bombled J, Gorin I, Schlienger P, Stoppa-Lyonnet D. Individuals with presumably hereditary uveal melanoma do not harbour germline mutations in the coding regions of either the P16INK4A, P14ARF or cdk4 genes. Br J Cancer. 2000;82:818–822. doi: 10.1054/bjoc.1999.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speicher MR, Prescher G, Du Manoir S, Jauch A, Horsthemke B, Bornfeld N, Becher R, Cremer T. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994;54:3817–3823. [PubMed] [Google Scholar]
- Sun Y, Tran BN, Worley LA, Delston RB, Harbour JW. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46:1561–1564. doi: 10.1167/iovs.04-1362. [DOI] [PubMed] [Google Scholar]
- Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci U S A. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschentscher F, Prescher G, Zeschnigk M, Horsthemke B, Lohmann DR. Identification of chromosomes 3, 6, and 8 aberrations in uveal melanoma by microsatellite analysis in comparison to comparative genomic hybridization. Cancer Genet Cytogenet. 2000;122:13–17. doi: 10.1016/s0165-4608(00)00266-1. [DOI] [PubMed] [Google Scholar]
- Tschentscher F, Prescher G, Horsman DE, et al. Partial deletions of the long and short arm of chromosome 3 point to two tumor suppressor genes in uveal melanoma. Cancer Res. 2001;61:3439–3442. [PubMed] [Google Scholar]
- Tschentscher F, Husing J, Holter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578–2584. [PubMed] [Google Scholar]
- Tucker MA, Shields JA, Hartge P, Augsburger J, Hoover RN, Fraumeni JF., Jr Sunlight exposure as risk factor for intraocular malignant melanoma. N Engl J Med. 1985;313:789–792. doi: 10.1056/NEJM198509263131305. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- Van Der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H, Hurks H, Frants RR, Gruis NA, Jager MJ. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61:5303–5306. [PubMed] [Google Scholar]
- Van Dijk MC, Rombout PD, Boots-Sprenger SH, Straatman H, Bernsen MR, Ruiter DJ, Jeuken JW. Multiplex ligation-dependent probe amplification for the detection of chromosomal gains and losses in formalin-fixed tissue. Diagn Mol Pathol. 2005;14:9–16. doi: 10.1097/01.pas.0000146701.98954.47. [DOI] [PubMed] [Google Scholar]
- Van Gils W, Lodder EM, Mensink HW, Kilic E, Naus NC, Bruggenwirth HT, Van Ijcken W, Paridaens D, Luyten GP, De Klein A. Gene expression profiling in uveal melanoma: two regions on 3p related to prognosis. Invest Ophthalmol Vis Sci. 2008;49:4254–4262. doi: 10.1167/iovs.08-2033. [DOI] [PubMed] [Google Scholar]
- Van Hees CL, De Boer A, Jager MJ, Bleeker JC, Kakebeeke HM, Crijns MB, Vandenbroucke JP, Bergman W. Are atypical nevi a risk factor for uveal melanoma? A case–control study. J Invest Dermatol. 1994;103:202–205. doi: 10.1111/1523-1747.ep12392754. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Fitch KR, Fuchs H, De Angelis MH, Barsh GS. Effects of G-protein mutations on skin color. Nat Genet. 2004;36:961–968. doi: 10.1038/ng1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, Wilkinson KD. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008;68:6953–6962. doi: 10.1158/0008-5472.CAN-08-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Hengge UR, Urbanik D, Markwart A, Mirmohammadsaegh A, Reichel MB, Wittekind C, Wiedemann P, Tannapfel A. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab Invest. 2003;83:1771–1776. doi: 10.1097/01.lab.0000101732.89463.29. [DOI] [PubMed] [Google Scholar]
- White VA, Chambers JD, Courtright PD, Chang WY, Horsman DE. Correlation of cytogenetic abnormalities with the outcome of patients with uveal melanoma. Cancer. 1998;83:354–359. [PubMed] [Google Scholar]
- Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltshire RN, Elner VM, Dennis T, Vine AK, Trent JM. Cytogenetic analysis of posterior uveal melanoma. Cancer Genet Cytogenet. 1993;66:47–53. doi: 10.1016/0165-4608(93)90148-f. [DOI] [PubMed] [Google Scholar]
- Worley LA, Onken MD, Person E, Robirds D, Branson J, Char DH, Perry A, Harbour JW. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007;13:1466–1471. doi: 10.1158/1078-0432.CCR-06-2401. [DOI] [PubMed] [Google Scholar]
- Worley LA, Long MD, Onken MD, Harbour JW. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res. 2008;18:184–190. doi: 10.1097/CMR.0b013e3282feeac6. [DOI] [PubMed] [Google Scholar]
- Young LH, Egan KM, Walsh SM, Gragoudas ES. Familial uveal melanoma. Am J Ophthalmol. 1994;117:516–520. doi: 10.1016/s0002-9394(14)70014-5. [DOI] [PubMed] [Google Scholar]
- Yucel YH, Johnston MG, Ly T, et al. Identification of lymphatics in the ciliary body of the human eye: a novel “uveolymphatic” outflow pathway. Exp Eye Res. 2009;89:810–819. doi: 10.1016/j.exer.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Zeschnigk M, Tschentscher F, Lich C, Brandt B, Horsthemke B, Lohmann DR. Methylation Analysis of Several Tumour Suppressor Genes Shows a Low Frequency of Methylation of CDKN2A and RARB in Uveal Melanomas. Comp Funct Genomics. 2003;4:329–336. doi: 10.1002/cfg.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuidervaart W, Van Der Velden PA, Hurks MH, Van Nieuwpoort FA, Out-Luiting CJ, Singh AD, Frants RR, Jager MJ, Gruis NA. Gene expression profiling identifies tumour markers potentially playing a role in uveal melanoma development. Br J Cancer. 2003;89:1914–1919. doi: 10.1038/sj.bjc.6601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuidervaart W, Van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92:2032–2038. doi: 10.1038/sj.bjc.6602598. [DOI] [PMC free article] [PubMed] [Google Scholar]