

Adipose tissue fibrosis occurs during obesity and is associated with metabolic dysfunction. Iwayama et al. identify perivascular cells as fibro/adipogenic progenitors in white adipose tissue and show that PDGFRα targets progenitor cell plasticity as a profibrotic mechanism.
Keywords: platelet-derived growth factor, fibrosis, adipogenesis, Nestin, pericyte, imprinting
Abstract
Fibrosis is a common disease process in which profibrotic cells disturb organ function by secreting disorganized extracellular matrix (ECM). Adipose tissue fibrosis occurs during obesity and is associated with metabolic dysfunction, but how profibrotic cells originate is still being elucidated. Here, we use a developmental model to investigate perivascular cells in white adipose tissue (WAT) and their potential to cause organ fibrosis. We show that a Nestin-Cre transgene targets perivascular cells (adventitial cells and pericyte-like cells) in WAT, and Nestin-GFP specifically labels pericyte-like cells. Activation of PDGFRα signaling in perivascular cells causes them to transition into ECM-synthesizing profibrotic cells. Before this transition occurs, PDGFRα signaling up-regulates mTOR signaling and ribosome biogenesis pathways and perturbs the expression of a network of epigenetically imprinted genes that have been implicated in cell growth and tissue homeostasis. Isolated Nestin-GFP+ cells differentiate into adipocytes ex vivo and form WAT when transplanted into recipient mice. However, PDGFRα signaling opposes adipogenesis and generates profibrotic cells instead, which leads to fibrotic WAT in transplant experiments. These results identify perivascular cells as fibro/adipogenic progenitors in WAT and show that PDGFRα targets progenitor cell plasticity as a profibrotic mechanism.
Vascular smooth muscle cells and pericytes are classified as mural cells. While vascular smooth muscle cells occur in layers around arteries and veins, pericytes reside directly on the abluminal surface of capillaries (Armulik et al. 2011). Perivascular adventitial cells form the outermost layer around larger vessels (Majesky et al. 2011). Pericytes and adventitial cells have been implicated as an adult reservoir of progenitor cells with the potential to generate adipocytes, osteoblasts, and other mesenchymal cell types (Crisan et al. 2008; Hoshino et al. 2008; Passman et al. 2008; Mendez-Ferrer et al. 2010; Corselli et al. 2012). Lineage tracing and cell ablation studies strongly suggest that tissue injury can induce pericytes or pericyte-like cells in diverse organs to leave their perivascular niche and differentiate into profibrotic cells (Humphreys et al. 2010; Goritz et al. 2011; Rock et al. 2011; Dulauroy et al. 2012; Soderblom et al. 2013; Kramann et al. 2015). Sometimes called myofibroblasts, these profibrotic cells secrete extracellular matrix (ECM) and are the main effector cells of organ fibrosis (Tomasek et al. 2002; Hinz et al. 2007).
Fibrosis is a disease process involving the destruction of normal tissue by deposition of collagen-rich ECM. This is a serious medical problem because every organ can be damaged by fibrosis, and, in severe cases, it may lead to organ failure. Unfortunately, in the majority of fibrotic diseases, there is no specific treatment to mitigate or reverse the destructive tissue remodeling. The generally accepted model of fibrosis posits that sustained injury to the organ parenchyma causes inflammation and production of cytokines and growth factors that recruit immune cells and profibrotic cells to the injured site. In fibrotic disease, the normal repair processes fail to resolve, and ECM-producing cells persist, creating scar tissue and impairing organ function (Wynn and Ramalingam 2012; Friedman et al. 2013). Transforming growth factor β (TGF-β) induces profibrotic cells with contractile properties (i.e., myofibroblasts) and is often considered the predominant profibrotic signal. However, platelet-derived growth factor (PDGF) is another important profibrotic signal that binds the receptor tyrosine kinases PDGFRα and PDGFRβ (Bonner 2004; Andrae et al. 2008). Multiple studies have shown that PDGF signaling is needed for fibrosis: Chemical inhibitors of PDGFRα/β tyrosine kinase activity can reduce fibrosis in animal models of skin and lung fibrosis, blocking antibodies against PDGFRα/β reduce kidney and atrial fibrosis in mice, and PDGFRα heterozygous mice are resistant to hepatic fibrosis (Abdollahi et al. 2005; Akhmetshina et al. 2009; Liao et al. 2010; Chen et al. 2011; Hayes et al. 2014). Furthermore, increased PDGF signaling via transgenic expression of PDGF ligands (Ponten et al. 2003) or conditional activation of a PDGFRα gain-of-function mutation (Olson and Soriano 2009) is sufficient to cause fibrosis. However, the cells targeted by profibrotic PDGF signaling have not been characterized.
Previously, we explored the role of PDGFRα signaling in organ fibrosis using R26-CreER; PDGFRα+/D842V mice. In this model, a Cre/lox-inducible gain-of-function knock-in mutation (D842V) in PDGFRα increased receptor tyrosine kinase activity. This knock-in was targeted to the endogenous Pdgfrα gene, and therefore Cre-induced expression of the activated receptor recapitulated the natural Pdgfrα expression pattern. Activation of PDGFRαD842V with R26-CreER caused progressive fibrosis in many organs, including the skin (affecting dermal adipose tissue), gastrointestinal tract, skeletal muscle, heart, kidneys, and lungs (Olson and Soriano 2009). Because this occurred after a single pulse of tamoxifen given in late embryogenesis or adulthood, activation of the PDGFRα pathway in some adult cells must be sufficient to generate significant profibrotic activity. Interestingly, activation of a PDGFRβD849V gain-of-function mutation did not cause fibrosis (Olson and Soriano 2011; T Iwayama and LE Olson, unpubl.), which suggests that PDGFRα may be uniquely poised to promote the emergence of profibrotic cells. However, because the R26-driven Cre exhibits global activity, more specific Cre drivers were needed to develop detailed information about the cellular targets of profibrotic PDGFRα signaling.
White adipose tissue (WAT) fibrosis is significant because of its intimate relationship with WAT dysfunction, chronic inflammation, and insulin resistance in humans and mice (Khan et al. 2009; Pasarica et al. 2009; Divoux et al. 2010; Sun et al. 2013; Vila et al. 2014). To investigate pericytes and adventitial cells in WAT and their potential to become profibrotic cells, we evaluated the effects of conditional activation of PDGFRαD842V in perivascular cells, whether the resulting ECM-producing cells arose from perivascular cells, and the fibro/adipogenic cell fate potential of WAT pericyte-like cells that are marked by the Nestin-GFP reporter. Our lineage tracing experiments showed that Nestin-Cre1Kln targets pericyte-like cells and adventitial cells in WAT, while the Nestin-GFP reporter specifically labels pericyte-like cells. Activation of PDGFRαD842V with Nestin-Cre results in fibrosis by causing perivascular cells to transition into ECM-producing cells. Whole-genome RNA sequencing (RNA-seq) of Nestin-GFP+ cells identified gene signatures suggestive of progenitor cell status and increased mTOR signaling and ribosome biogenesis functions in response to PDGFRαD842V. Isolated Nestin-GFP+ cells have adipocyte progenitor cell surface markers and differentiate into adipocytes ex vivo. However, PDGFRα signaling opposes adipogenesis and generates profibrotic cells ex vivo or when PDGFRαD842V cells are transplanted into recipient mice. Therefore, Nestin-GFP+ PDGFRα+ cells are progenitor cells with the potential to become adipocytes or profibrotic cells, and PDGFRα activation causes fibrosis cell-autonomously by perturbation of progenitor function.
Results
An approach for activating PDGFRα in perivascular cells
We sought Cre drivers predicted to be active in perivascular cells to test whether these cells give rise to profibrotic cells in response to PDGFRα signaling. Chondroitin sulfate proteoglycan 4 (Cspg4/NG2) and PDGFRβ are typical pericyte markers. However, mature adipocytes express Cspg4 (Chang et al. 2012), and PDGFRβ-Cre was shown to have broad activity in WAT (Tang et al. 2008). Therefore, these drivers are not useful for precise lineage tracing in WAT. Indeed, when we tested Cspg4-Cre (Zhu et al. 2008) and PDGFRβ-Cre (Foo et al. 2006) BAC transgenic mice by crossing with the R26-Tomato reporter line, we found abundant labeling outside the perivascular compartment (Supplemental Fig. 1).
Nestin (Nes) has been used as a pericyte marker, and Nes-GFP has served as a useful transgenic reporter to label subsets of pericyte-like cells that display somatic stem/progenitor cell properties (Dore-Duffy et al. 2006; Mendez-Ferrer et al. 2010), but expression in WAT has not been examined. Therefore, we crossed Nes-GFP (Mignone et al. 2004) and Nes-Cre1Kln (Tronche et al. 1999) transgenic mice to generate double-transgenic mice with Cre and GFP driven by the promoter and first intron of the rat nestin gene (Zimmerman et al. 1994). We also introduced the R26-Tomato Cre-dependent reporter, resulting in triple-transgenic Nes-GFP; Nes-Cre; R26-Tomato mice (Fig. 1A). There were two distinct types of Tomato+ cells in the WAT of 3-wk-old mice. First, there were individual Tomato+ cells closely associated with capillaries with a pericyte-like morphology (Fig. 1B,C,E). Second, there were clustered Tomato+ cells around arterioles and venules but separated from the endothelium by a layer of vascular smooth muscle cells, indicating that they were adventitial cells (Fig. 1D, arrow). Interestingly, Tomato+ adventitial cells were not colabeled by Nes-GFP, but the pericyte-like cells were consistently colabeled with both reporters (Fig. 1D–I). The Tomato+GFP+ pericyte-like cells expressed PDGFRβ and Cspg4 (Fig. 1F,G) and were embedded in the capillary basement membrane (Fig. 1H), further suggestive of a pericyte identity. Fluorescent Tomato+GFP+ pericyte-like cells were seen on the abluminal surface of capillaries isolated from WAT by anti-CD31-coated magnetic beads (Supplemental Fig. 2). Tomato+GFP+ pericyte-like cells and Tomato+ adventitial cells also expressed PDGFRα (Fig. 1I). We conclude that the Nes-GFP reporter is active in PDGFRα+ pericytes or pericyte-like cells, while the cells targeted by Nes-Cre include pericyte-like cells and adventitial cells (together called perivascular cells). This difference is explained by the fact that Nes-GFP is restricted to cells where the transgenic nestin promoter is active, while Nes-Cre/Tomato is a lineage reporter that indelibly labels a larger population, including nestin-expressing cells and their progeny. These results show that Nes-driven transgenes are aligned with our primary goal of targeting perivascular cells in WAT.
Figure 1.

Nestin-Cre and Nestin-GFP identify perivascular cells in WAT. (A) Schematic of the genetic tools in Nes-GFP; Nes-Cre; R26-Tomato dual-reporter mice used in this figure. GFP and Cre are expressed from distinct nestin-driven transgenes. Cre acts on a Cre/lox-dependent R26 knock-in fluorescent Tomato reporter, which serves as a lineage trace. (B) Epifluorescence of Nes-Cre/Tomato lineage tracing in visceral WAT, imaged by whole-mount with isolectin-IB4 staining for capillary endothelial cells. (C) Measurement of the distance between DAPI+ nuclei of individual Nes-GFP+ cells (n = 167) and the nearest IB4+ capillary membrane, as shown in the example at the right. A distance <10 µm means the cell is on the abluminal surface of the capillary. (D) Epifluorescence of Nes-GFP and Nes-Cre/Tomato plus immunofluorescence staining of Myh11 in vascular smooth muscle cells. The GFP/Tomato reporters are coexpressed in pericyte-like cells (arrowhead). Tomato also identifies adventitial cells in which Nes-GFP is not expressed (arrow). (E–I) Epifluorescence of Nes-GFP and Nes-Cre/Tomato plus IB4 labeling for endothelial cells (E), immunofluorescence staining of Cspg4 or PDGFRβ for pericytes (F,G), immunofluorescence staining of collagen IV for basement membrane (H), and immunofluorescence staining of PDGFRα (I). (J) Epifluorescence of Nes-GFP and Nes-Cre/Tomato plus immunofluorescence staining of Perilipin (PLIN1) for adipocytes. Tomato identifies rare adipocytes (arrow) that do not express Nes-GFP. A Tomato+GFP+ pericyte-like cell is also shown (arrowhead). Bars: B, 100 µm; D, 30 µm; E–I, 20 µm; J, 30 µm.
We also used flow cytometry as an independent approach to characterize and quantify the specificity of the Nes-Cre/Tomato and Nes-GFP reporters in the stromal vascular fraction (SVF) of WAT. In the subcutaneous and visceral WAT, between 20% and 35% of the whole SVF was labeled with either reporter. Between 20% and 45% of the PDGFRα+ population was Tomato/GFP+, but a very low percentage (1%–5%) of the CD31+ and CD45+ populations was labeled (Supplemental Fig. 3). These data show that Nes-driven transgenes are specific for a subset of mesenchymal cells in the SVF and have minimal expression in endothelial and hematopoietic cells. We also found that a large fraction of the adipocyte precursor population in the SVF was Tomato/GFP+ (Rodeheffer et al. 2008). However, by fluorescent microscopy, <1% of the mature adipocytes were Tomato+ in the WAT of 3-wk-old mice, and none was GFP+ (Fig. 1J). This may indicate that Nes-Cre/Tomato labels a subset of the adipocyte precursors that do not contribute to adipogenesis during the juvenile period. Interestingly, the number of Tomato+ adipocytes in visceral WAT increased significantly when mice were fed a high-fat diet (HFD), as described later (Fig. 5, below).
Analysis of heart muscle, intestine, and spleen from Nes-GFP; Nes-Cre; R26-Tomato mice showed that, as in WAT, only perivascular cells were Tomato+ (data not shown). Thus, Nes-Cre should be useful for precise lineage tracing in these organs. In the kidneys, lungs, and skeletal muscle, perivascular cells as well as many parenchymal cells were Tomato+.
PDGFRα activation in perivascular cells is sufficient for fibrosis
We generated Nes-Cre; PDGFRα+/D842V mutant mice to test whether perivascular expression of activated PDGFRα would cause fibrosis (Fig. 2A). Histological analysis identified fibrosis in mutant WAT, beginning as perivascular lesions at ∼12 wk and progressing to interstitial fibrosis at later times (Fig. 2B,C). The tissue area containing extracellular collagen fibers was significantly increased in mutants at 12- and 24-wk of age (Fig. 2C,D). Col1a1 and Col3a1 expression were higher in 24-wk-old fibrotic WAT compared with age-matched control WAT (Fig. 2E). Cell proliferation was increased in mutant WAT at 12-wk of age (Fig. 2F,G). These results demonstrate that PDGFRα activation in perivascular cells, including pericyte-like cells and/or adventitial cells, is sufficient to cause WAT fibrosis. Nes-Cre; PDGFRα+/D842V mutants also developed severe fibrosis of the intestinal submucosa and skeletal muscle as well as perivascular-restricted fibrosis in the heart, lung, spleen, and kidney (Supplemental Fig. 4). Collectively, these phenotypes demonstrate the high fibrogenic potential of perivascular cells in response to PDGFRα signaling.
Figure 2.

PDGFRα activation in perivascular cells is sufficient for fibrosis. (A) Schematic of the genetic tools in Nes-Cre; PDGFRα+/[S]D842V mutant mice used in this figure. Cre acts on the PDGFRαD842V knock-in allele to induce expression of an activated mutant PDGFRα. (B) Masson's trichrome staining for collagen (blue) in dermal and subcutaneous WAT. (C) Picosirius red staining for collagen (orange) in subcutaneous WAT. (D) Quantification of a picosirius red-stained area as a measure of fibrosis. n = 3–6 mice per data point; (*) P < 0.01. (E) Quantitative PCR (qPCR) analysis of collagen transcripts in subcutaneous WAT at 24 wk of age. n = 3; mean ± SEM; (*) P < 0.05. (F) DAPI staining of cell nuclei and EdU labeling for proliferating cells in WAT at 12-wk of age. Arrows indicate EdU/DAPI double-positive nuclei. (G) Flow cytometric quantification of EdU+ cells as a percentage of total cells sorted from subcutaneous WAT at 12 wk of age. n = 6; mean ± SEM; (*) P < 0.05. Bars: B, 120 µm; C, 120 µm.
Perivascular cells generate profibrotic cells cell-autonomously
Perivascular cells might generate profibrotic cells directly, or fibrosis might occur non-cell-autonomously; for instance, by paracrine signaling to stimulate nonperivascular cells to become profibrotic. To test for a cell-autonomous mechanism, we crossed Nes-Cre; PDGFRα+/D842V mutants with R26-Tomato reporter mice to lineage trace perivascular cells during fibrosis. We also used a Collagen1a1-GFP reporter (Magness et al. 2004) to identify profibrotic cells (Fig. 3A). In control mice without fibrosis, Tomato+ perivascular cells did not express Col-GFP (Fig. 3B). In mutant mice at 28-wk of age, there was a dramatic increase in Col-GFP+ cells, which was mirrored by an expansion of Tomato+ cells (Fig. 3B). Within fibrotic lesions, close to 100% of Tomato+ cells were Col-GFP+ (Fig. 3C,D). Furthermore, Col-GFP+ cells were almost exclusively Tomato+, indicating that profibrotic cells were derived from the perivascular lineage, including pericyte-like cells and adventitial cells. Profibrotic cells were also PDGFRα+ (Fig. 3E). Importantly, Nes-GFP was not expressed in fibrotic lesions of Nes-GFP; Nes-Cre; PDGFRα+/D842V mutants (Supplemental Fig. 5). This suggests that pericytes lose Nes-GFP expression during the transition to profibrotic cells. We took advantage of the selective Nes-GFP marker to isolate pericyte-like cells for whole-transcriptome sequencing and functional studies in vitro.
Figure 3.

Profibrotic cells originate from perivascular cells. (A) Schematic of the genetic tools in Nes-Cre; PDGFRα+/[S]D842V; R26-Tomato; Col-GFP mutant double-reporter mice used in this figure. Cre acts on the PDGFRαD842V knock-in allele and an R26-Tomato reporter to induce expression of an activated mutant PDGFRα and lineage trace perivascular cells. GFP is expressed from a distinct Col1a1-driven transgene. (B, left panels) Epifluorescence of Nes-Cre/Tomato and Col-GFP plus isolectin-IB4 staining of endothelial cells in nonfibrotic subcutaneous or visceral WAT. (Middle panels) Epifluorescence of Nes-Cre/Tomato plus isolectin-IB4 in fibrotic WAT. (Right panels) Epifluorescence of Col-GFP for identification of ECM-producing cells in the same tissue. The large increase in Tomato+ area in Nestin-D842V samples is mirrored by Col-GFP area. (C) Epifluorescence of Nestin-Cre/Tomato colocalized with Col-GFP in a fibrotic lesion of subcutaneous WAT. (D) Quantification of the proportion of Col-GFP-labeled cells coexpressing Nestin-Cre/Tomato and the proportion of Nestin-Cre/Tomato-labeled cells coexpressing Col-GFP. n = 3; mean ± SEM. (E) Immunofluorescence staining of PDGFRα colocalized with epifluorescence of Nes-Cre/Tomato. Bars: B, 100 µm; C,E, 50 µm.
RNA-seq analysis of gene expression in Nes-GFP+ cells
We performed genome-wide RNA-seq to characterize Nes-GFP+ cells at a transcriptional level and elucidate gene expression changes in PDGFRαD842V cells. We flow-sorted Nes-GFP+ cells from Nes-GFP; Nes-Cre; PDGFRα+/D842V mutant mice and Nes-GFP; Nes-Cre control mice. Because our goal was to identify genomic signatures preceding fibrosis, we chose dermal adipose tissue from 3-d-old mice as a tissue source. We confirmed the enrichment of pericyte and mesenchymal markers on sorted Nes-GFP+ cells by quantitative RT–PCR (qRT–PCR) for Pdgfrα, Pdgfrβ, Rgs5, and Adam12 (Supplemental Fig. 6). We then prepared cDNA libraries and performed RNA-seq to generate a list of the top 50 most highly expressed genes in Nes-GFP+ cells (Supplemental Table 1). This list included many genes encoding ECM proteins (e.g., Col1a1, Col1a2, Col3a1, Fn1, and Eln) and pericyte-enriched genes (e.g., Col4a1, Col4a2, Fstl1, Ahnak, and Bgn) (Bondjers et al. 2006; Zhang et al. 2014). Also on the list were five genes that are regulated by epigenetic imprinting mechanisms in mice: Dlk1, H19, Igf2, Gnas, and Peg3. Imprinted genes are rare, estimated at <1% of mouse genes (Morison et al. 2005). Therefore, five imprinted genes among the top 50 genes in Nes-GFP+ cells was an unusually high proportion.
By global comparison of gene expression between control and PDGFRαD842V cells, we identified 1356 differentially expressed genes (DEGs; P < 0.01) out of 37,992 genes scored. In general, the ECM genes were not DEGs, which is consistent with analysis of cells in a prefibrotic state. However, by checking a full list of ∼70 known imprinted genes (Morison et al. 2005) against the DEGs, we discovered that there were 15 up-regulated imprinted genes and two down-regulated imprinted genes (Fig. 4A). Among 1356 DEGs, the occurrence of 17 imprinted genes is significantly more than would be expected by chance (P < 0.0001, χ2 test). Interestingly, Dlk1, H19, Igf2, and Peg3 are part of the “imprinted gene network” (IGN), which includes 11 monoallelically expressed genes that are down-regulated in most tissues postnatally but remain highly expressed in many somatic stem and progenitor cells (Varrault et al. 2006; Lui et al. 2008; Berg et al. 2011; Besson et al. 2011). In our experiment, seven IGN genes were overexpressed in PDGFRαD842V cells: Dlk1, H19, Igf2, Plagl1, Mest, Cdkn1c, and Slc38a4 (Fig. 4A). By in situ hybridization (ISH) with antisense RNA probes, increased expression of Dlk1, H19, and Igf2 was apparent in adventitial cells of Nes-Cre; PDGFRαD842V subcutaneous WAT (Fig. 4B, arrowheads). Scattered cells with a high ISH signal were also seen in both control and mutant samples, consistent with expression of Dlk1, H19, and Igf2 in pericytes or pericyte-like cells (arrows). Together, these results suggest that IGN gene expression is perivascular cell-specific.
Figure 4.

PDGFRα signaling perturbs imprinted gene expression and mTOR/mRNA translation pathways. (A) RNA-seq analysis of differentially expressed imprinted genes. P-values are indicated. Bold genes are part of the IGN. (B) ISH for H19, Dlk1, and Igf2 mRNA in subcutaneous WAT of 4-d-old mice. The blue stain identifies intense gene expression in adventitial cells (arrowheads) in PDGFRαD842V tissue. Scattered pericyte-like cells also show expression in both samples (arrows). (C) Differentially expressed pathways in Nes-GFP+ cells of wild-type or PDGFRαD842V mice, identified by Ingenuity Pathway Analysis of the RNA-seq data. “Canonical pathways” are defined by a cluster of related signature genes, which constitutes the ratio's denominator. The numerator is the number of signature genes that were changed with P < 0.01 in the RNA-seq data set. P-value is the probability that the ratio occurred by chance. (D,E) Heat map of six mTOR pathway signature genes that were differentially expressed and a model representing the PI3K/Akt/mTOR pathway with functional association of the six DEGs. (F) Heat map of 61 genes encoding ribosomal proteins that were differentially expressed and represented as signature genes in the EIF2, mTOR, and EIF4/p70S6K canonical pathways.
We used Ingenuity Pathway Analysis to categorize the DEGs according to canonical pathways (Fig. 4C). The three most significantly changed pathways were related to mRNA translation: IEF2 signaling, mTOR signaling, and regulation of EIF2 and p70S6K. Six of the DEGs from the mTOR signaling pathway were regulators of PI3K/Akt/mTOR signaling, which is one of the major effector pathways downstream from PDGFRα (Fig. 4D,E). Increased ribosome biogenesis was also predicted by up-regulation of 61 genes for ribosomal proteins in association with the IEF2, mTOR, and p70S6K canonical pathways (Fig. 4F). An attractive possibility is that increased mRNA translation contributes to the emergence of a profibrotic phenotype in part by increasing ECM protein synthesis, but this mechanism requires further testing.
TGF-β signaling is frequently implicated in fibrosis, but TGF-β target genes Ctgf, Acta2, Trpc6, and Adam12 were not up-regulated in PDGFRαD842V cells. The canonical pathway for hepatic fibrosis was highly scored (Fig. 4C), but DEGs in this pathway were not TGF-β-related. Instead, they consisted mainly of growth factors and receptors (e.g., Pdgfa, Igf2, Fgfr1, and Kdr), myosins (Myh8, Myl1, and Myl9), and miscellaneous collagens (e.g., Col25a1, Col15a1, and Col28a1). As our analysis was performed on Nes-GFP+ cells in a prefibrotic state, it is possible that increased TGF-β signaling would be seen later in the fibrosis process.
Nes-GFP+ cells are adipocyte precursors
PDGFRα is a marker for adipocyte precursors but is not expressed on mature adipocytes (Lee et al. 2012; Berry and Rodeheffer 2013). Analysis of sorted cells from dermal adipose tissue of 3-d-old mice indicated that most Nes-GFP+ cells coexpressed markers for adipocyte precursors, including CD29, CD34, and CD24 (Supplemental Fig. 7; Rodeheffer et al. 2008; Church et al. 2014). As a functional test, we cultured Nes-GFP+ cells and GFP-negative cells ex vivo with a standard adipogenic cocktail. GFP+ cells were far more adipogenic than GFP-negative cells, based on lipid droplet formation (Fig. 5A) and qRT–PCR to measure adipocyte markers (Fig. 5B).
Figure 5.
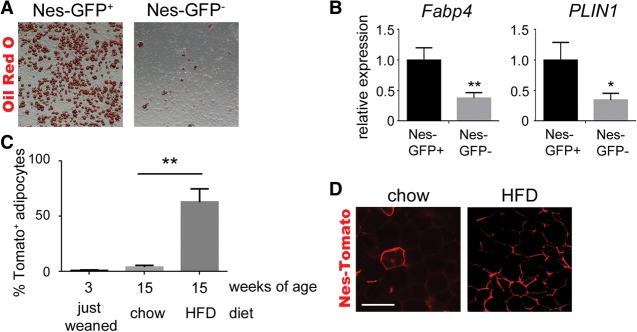
Nes-GFP+ cells are adipocyte precursors. (A) Representative images of Oil Red O-stained adipocytes differentiated from Nes-GFP− or Nes-GFP+ cells. (B) qPCR analysis of mature adipocyte markers after differentiation. Expression in the noninduced group was normalized to 1. n = 3; mean ± SEM; (*) P < 0.05; (**) P < 0.01. (C) Quantification of the percentage of adipocytes in visceral WAT expressing the Nes-Cre/R26-Tomato lineage reporter after 12 wk of chow diet or HFD. n = 3; mean ± SEM; (**) P < 0.0001. (D) Epifluorescence of Nes-Cre/R26-Tomato lineage reporter in visceral WAT after 12 wk of chow diet or HFD. Bar, 100 µm.
As shown previously, few adipocytes were Tomato+ in Nes-Cre; R26-Tomato lineage tracing mice (Fig. 1J), which seemed to conflict with the high adipogenic potential of Nes-GFP+ cells ex vivo. To further investigate adipogenesis in vivo, we fed Nes-Cre; R26-Tomato mice a HFD or normal chow diet starting at 3 wk of age. Remarkably, after 12 wk of HFD there was Tomato labeling in >50% of adipocytes in the visceral WAT (Fig. 5C,D). Feeding chow diet for 12 wk caused a small increase in Tomato+ adipocytes; although not statistically significant, the trend suggested that longer aging would eventually lead to a significant increase. These results suggest that Nes-GFP+ cells are part of the adipocyte precursor population and show, by lineage tracing, that Nes-Cre/Tomato+ perivascular cells generate new adipocytes in adult visceral WAT (Fig. 5C,D).
PDGFRα activation increases proliferation and ECM production while inhibiting adipogenesis
We next investigated how PDGFRα signaling influences proliferation and fibro/adipogenic differentiation. In these experiments, we cultured Nes-GFP+ cells isolated from dermal adipose tissue of 3-d-old Nes-GFP; Nes-Cre control mice or Nes-GFP; Nes-Cre; PDGFRα+/D842V mutant mice. Treating cells with EdU revealed a greater than twofold increase in the percentage of proliferating mutant cells compared with controls (Fig. 6A). This is in agreement with the increased proliferation seen in vivo in adult tissue undergoing fibrotic remodeling (Fig. 2F,G). Collagen expression was similar in the two populations of freshly sorted Nes-GFP+ cells (Fig. 6B). However, after growing Nes-GFP+ cells for 1 wk, we found that collagen expression was significantly increased in mutant cells compared with controls (Fig. 6B). We extended our ex vivo analysis by culturing the cells for 3 wk on transwell inserts to allow the formation of three-dimensional cell–ECM complexes (Karamichos et al. 2010). Cell–ECM complexes were then transferred to glass slides and stained for collagen III. Weak staining was present in controls, but the collagen III area was expanded in complexes formed from PDGFRαD842V mutant cells (Fig. 6C,D). Adipocyte differentiation was strongly inhibited in PDGFRαD842V mutant cells treated with adipogenic cocktail (Fig. 6E,F). PDGFRαD842V mutant cells showed weak induction of the transcription factor Pparg2 following treatment with adipogenic cocktail (Fig. 6F). PPARγ2 marks fate-determined preadipocytes and is the master regulator of adipocyte differentiation. Therefore, PDGFRα signaling may inhibit adipogenesis at the level of fate determination.
Figure 6.

PDGFRα signaling increases proliferation and ECM production while inhibiting adipogenesis. (A) FACS analysis of cultured Nes-GFP+ cells from control or mutant (Nes-GFP; Nes-Cre; PDGFRα+/[S]D842V) mice after 20 min of incubation with EdU. n = 3; mean ± SD; (*) P < 0.05. (B) qPCR analysis of collagen expression in control or mutant Nes-GFP+ cells freshly isolated or after 1 wk of culture with stabilized ascorbic acid. n = 3; mean ± SEM; (*) P < 0.05. (C) Whole-mount immunofluorescence staining of collagen III protein in fibrotic complexes generated by Nes-GFP+ cells after 3 wk of culture. (D) Quantification of the collagen III-stained area. n = 3; mean ± SD; (*) P < 0.05. (E) Oil Red O-stained adipocytes differentiated from control or mutant Nes-GFP+ cells. (F) qPCR analysis of mature adipocyte markers before and after differentiation. (N.D.) Not detectable. Expression in the wild-type group was normalized to 1. n = 3; mean ± SD; (**) P < 0.01; (***) P < 0.001.
Transplanted Nes-GFP+ cells differentiate into adipocytes and generate fibrosis
If WAT perivascular cells are adipocyte precursors and PDGFRαD842V alters progenitor cell plasticity to cause a profibrotic transition, then it should be possible to generate new WAT and fibrosis by transplantation. We used a Matrigel plug assay to transplant Nes-GFP+ cells into syngeneic recipient mice (Kawaguchi et al. 1998). Freshly sorted cells from dermal WAT of 3-d-old Nes-GFP; Nes-Cre; R26-Tomato mice were transplanted as Matrigel/cell mixtures into wild-type hosts. After 3 wk, WAT was formed in the Matrigel, and Nes-GFP+ cells persisted as mural cells within the newly generated WAT (Fig. 7A). There were also Tomato+ adipocytes, demonstrating that transplanted cells had differentiated into adipocytes in situ. Next, we sorted Nes-GFP+ cells from Nes-GFP; Nes-Cre; PDGFRα+/D842V; R26-Tomato mutants or control mice and transplanted them into wild-type hosts. Gross analysis at 12 wk after transplantation demonstrated that the PDGFRαD842V plugs had developed tissue with a paucity of adipocytes compared with plugs containing control cells (Fig. 7B). By histological analysis, the tissue in PDGFRαD842V plugs was fibrotic with prominent perivascular collagen deposition (Fig. 7C). Cells within the fibrotic lesions no longer expressed Nes-GFP but were Tomato+, demonstrating that profibrotic cells had originated from the transplanted progenitor cells (Fig. 7D).
Figure 7.

Transplanted Nes-GFP+ cells generate adipocytes and profibrotic cells. (A) Experimental scheme to transplant Nes-GFP+ cells with Nes-Cre/Tomato lineage tracing in Matrigel plugs into wild-type mice, with analysis at 3 wk after transplantation. (Mural cells) Epifluorescence of Nes-GFP with IB4 staining of endothelial cells identified persistence of GFP+ cells. (Adipocytes) Epifluorescence of Nes-Cre/Tomato identified donor-derived adipocytes. (B) Experimental scheme to transplant Nes-GFP+ cells with Nes-Cre/Tomato and either PDGFRαD842V or wild-type PDGFRα. Analysis at 12 wk after transplantation: phase contrast image of newly generated WAT, with abundant adipocytes in the control but sparse adipocytes in the mutant. (C) Trichrome staining (top) or picosirius red stain (bottom) of newly generated WAT 12 wk after transplantation, with perivascular fibrosis in the mutant. (D) Epifluorescence of Nes-Cre/Tomato in perivascular fibrotic areas demonstrating profibrotic cells of donor origin. Bars: A, 50 µm; C, 100 µm; D, 50 µm.
Discussion
PDGFRα signaling is sufficient to cause spontaneous fibrosis in diverse organs (Olson and Soriano 2009). However, the cellular origin of profibrotic cells remains controversial for most forms of organ fibrosis. Therefore, the first goal of this study was to determine whether pericytes or other perivascular cells could give rise to profibrotic cells in response to PDGFRα signaling. By using Nestin-Cre to lineage trace pericytes and adventitial cells in WAT and Col1a1-GFP to identify ECM-producing cells in fibrotic lesions, we found that nearly all of the ECM-producing cells in PDGFRαD842V mutant mice originated from perivascular cells. The intrinsic profibrotic potential of isolated Nes-GFP+ PDGFRαD842V pericyte-like cells was also shown by ex vivo differentiation assays and transplantation. Of interest, Cspg4-Cre; PDGFRα+/D842V and PDGFRβ-Cre; PDGFRα+/D842V mutant mice also developed fibrosis (data not shown), but precise lineage tracing was not possible in WAT because of significant Cre activity in the adipocyte compartment.
Our conclusion that profibrotic cells have a perivascular origin is complementary to other lineage tracing studies performed in the context of specific tissue injury (Humphreys et al. 2010; Goritz et al. 2011; Rock et al. 2011; Dulauroy et al. 2012; Soderblom et al. 2013; Kramann et al. 2015). However, unlike these previous studies, ours was not designed to simulate an organ-specific fibrotic response to acute injury. Instead, the spontaneous nature of our model provided a unique opportunity to explore the earliest fibrosis mechanisms under simplified, noninflammatory conditions. Under physiological conditions, WAT fibrosis is likely to be driven by local hypoxia, inflammation, increased adipocyte turnover, and macrophage infiltration. An important question for the future is how these stresses alter the expression of PDGF ligands or PDGFRα signaling in a manner that would cause progenitors to transition into profibrotic cells. Intriguingly, a recent study suggested that profibrotic cells in the skin can transition from adiponectin-expressing cells or mature adipocytes in the context of bleomycin-induced fibrosis (Marangoni et al. 2015). Although mature adipocytes do not express PDGFRα under normal conditions, its re-expression in disease states could be important.
We considered the possibility that Nes-GFP+ cells from WAT might have properties of mesenchymal stem cells (MSCs) because the same reporter was previously shown to mark such cells in the bone marrow (Mendez-Ferrer et al. 2010). We avoided the MSC label here because Nes-GFP+ cells exhibited poor differentiation into Alizarin red-stained osteogenic cells compared with bone-derived PDGFRα+ Sca1+ MSCs (data not shown). Skeletal muscle contains PDGFRα+ fibro/adipogenic progenitors (Joe et al. 2010; Uezumi et al. 2010); whether these cells express Nes-GFP is unknown. It has been reported that Nes-GFP+ pericytes do not produce collagen or accumulate in fibrotic scars of the injured skeletal muscle, lung, kidney, heart, or nervous system (Birbrair et al. 2013a,b, 2014). However, these studies with Nes-GFP did not include a lineage tracing component. We also did not see Nes-GFP+ cells accumulating in fibrotic scars in Nes-GFP; Nes-Cre; PDGFRα+/D842V mice (Supplemental Fig. 5). With lineage tracing, we found that the progeny of nestin-labeled cells did accumulate in fibrotic scars and transitioned into collagen-producing cells (Fig. 3). Furthermore, Nes-GFP+ cells from Nes-Cre; R26-Tomato; PDGFRα+/D842V mice caused fibrotic fat pads in transplantation experiments (Fig. 7), and lineage tracing confirmed that profibrotic cells originated from the transplanted cells. Together, these results suggest that the Nes-GFP reporter becomes silenced when pericytes transition to profibrotic cells.
The cellular origin of adipocytes has recently become a subject of intense investigation (Gesta et al. 2007; Berry et al. 2014; Sanchez-Gurmaches and Guertin 2014). Our study contributes new information to this question by showing how Nes-Cre/Tomato+ perivascular cells are a functional part of the adipocyte precursor population in the adult. Lineage tracing showed very little contribution of Nes-Cre/Tomato+ cells to the juvenile WAT. However, after a HFD challenge, there was significant recruitment of Tomato+ precursors into the adult adipocyte population of visceral WAT. In support of there being distinct sources for juvenile and adult adipocytes, a recent study found that adult adipocytes are generated from perivascular cells, while juvenile adipocytes develop from a different origin (Jiang et al. 2014). Dermal and subcutaneous WAT develop as independent depots, but adipocytes in both depots are derived from Dlk1+ PDGFRα+ precursors (Berry and Rodeheffer 2013; Driskell et al. 2013, 2014; Wojciechowicz et al. 2013; Hudak et al. 2014). Our results are consistent with the existence of a sublineage of Dlk1+ PDGFRα+ cells that seeds juvenile WAT with progenitors for adipogenesis and fibrosis in the adult. The importance of PDGFRα signaling in adipose tissue development remains to be tested.
Our study highlights perturbations in progenitor cell plasticity in the formation of profibrotic cells and eventual organ fibrosis. Altered cell fate was apparent in isolated Nes-GFP+ PDGFRαD842V cells that resisted the effects of adipogenic inducers and transitioned to a profibrotic phenotype. Among the most highly expressed genes in Nes-GFP+ cells were Dlk1, H19, and Igf2, which have roles in cell fate and differentiation of fat, muscle, and bone (Moon et al. 2002; Hardouin et al. 2011; Dey et al. 2014). Individually, these three genes could have mechanistic roles in altered fibro/adipogenic fate. However, the coordinate up-regulation of 15 imprinted genes and down-regulation of two more, together constituting >20% of the known imprinted genes in mice, suggests that PDGFRα signaling engages core mechanisms governing progenitor cell plasticity. How PDGFRα signaling perturbs imprinted gene expression remains to be explored. Genetic imprinting involves DNA methylation, histone modifications, and lncRNAs to cause monoallelic gene expression in a parent of origin-specific manner (Bartolomei 2009; Adalsteinsson and Ferguson-Smith 2014). PDGFRα signaling might relax the epigenetic mechanisms that maintain monoallelic expression. Alternatively, the network of coregulated imprinted genes might share common promoter elements that are sensitive to PDGFRα signaling pathways. Differentiating between these possibilities will be the subject of future studies.
Materials and methods
Mice
The mouse strains PDGFRα+/D842V (018433), R26-Tomato (007909), Nestin-Cre (003771), and Cspg4-Cre (008533) are available at Jackson Laboratories. Col1a1-GFP mice were a gift from Dr. David Brenner, Nestin-GFP mice were a gift from Dr. Grigori Enikolopov, and PDGFRβ-Cre mice were a gift from Dr. Ralf Adams. Mice were used in a mixed C57BL6/129S4 background. The Institutional Animal Care and Use Committee of the Oklahoma Medical Research Foundation approved all procedures described in this study. All analyses were based on a minimum of three PDGFRα+/842V mutants and littermate controls (expressing Cre without a PDGFRα842V allele). In this study, dermal WAT refers to adipose tissue of the skin hypodermis (Driskell et al. 2014). Visceral WAT refers to perigonadal adipose tissue. Subcutaneous WAT refers to inguinal subcutaneous adipose tissue. For in vivo EdU experiments, 2 mM EdU (Jena Bioscience) in 200 µL of 0.9% saline was injected intraperitoneally twice at 24 and 8 h before analysis. To study Nestin-Cre/Tomato labeling of adipocytes, some mice were fed a HFD (60% calories from fat) (Harlan Teklad, #TD06414) for up to 12 wk.
Tissue and histology
Organs were fixed in either 4% paraformaldehyde (PFA)/PBS diluted from 16% stock (Electron Microscopy Sciences) or Bouin's fixative (Sigma). For histochemical staining, Bouin's-fixed tissue was embedded in paraffin and sectioned at 7 µm before staining with Masson's trichrome (Sigma) or picosirius red staining (Electron Microscopy Sciences). For immunohistochemistry, 4% PFA-fixed samples were soaked in 20% sucrose/PBS overnight, embedded in O.C.T. compound (Sakura Tek), and stained with antibodies for perilipin (1:500; Cell Signaling Technology, #9349), GFP (1:1000; Abcam, ab6673), Cspg4 (1:100; Millipore, AB5320), PDGFRβ (1:100; R&D Systems, BAF1042), PDGFRα (1:100; Upstate Biotechnology, 07-276), or Myh11 (1:100; Biomedical Technologies, BT-562) followed by Alexa fluor 488-conjugated, Cy3-conjugated, and Alexa fluor 647-conjugated secondary antibodies (1:100; Jackson Immunoresearch) and DAPI (Sigma) as a nuclear stain. For vessel labeling, fluorescence-conjugated isolectin-IB4 (Invitrogen) was included during incubation with secondary antibody. EdU+ cells were detected by Click chemistry according to the manufacturer's protocol (Life Technologies). All images were taken with Nikon Eclipse 80i microscopy or Olympus FV1000 confocal microscopy. For Z-stack images, 60-µm sections were stained, and 30–60 slices of 0.45 µm each were obtained for the maximum intensity projection. To determine the percentage of Col1a1-GFP+ cells derived from the Nestin-Cre/Tomato+ lineage, each GFP+ DAPI+ cell was scored as Tomato+ or Tomato-negative. To measure the distance between individual Nes-GFP+ cells and the capillary endothelium, ImageJ was used to measure from the center of the nucleus (DAPI+) of each Nes-GFP+ cell to the nearest IB4+ endothelial membrane. For data collection, three to six images at 400× magnification were analyzed for three different Nes-GFP mice, with a total of 167 Nes-GFP+ cells. Data were transferred to GraphPad Prism to produce the graph of relative frequency distribution. To quantify Tomato+ adipocytes, cryosections were stained with perilipin antibody. After imaging at 200×, the number of Tomato+ adipocytes in each section was divided by the total number of perilipin+ adipocytes to determine the percent of Tomato+ adipocytes.
qRT–PCR
Total RNA was isolated from cells (with TRIzol reagent) or tissue (with RNeasy minikit, Qiagen), and cDNA was synthesized with SuperScript III reverse transcriptase according to the manufacturer's protocol (Life Technologies). PCR was then performed with a Bio-Rad CFX96 real-time system (Bio-Rad) and SYBR Green ReadyMix (Sigma). The expression levels were determined by normalization to the average values of two housekeeping genes (Timm17b and Gapdh) with a ΔΔCt method, and the expression levels relative to experimental control are shown. For primer sequences, please refer to Supplemental Table 2.
Isolation of Nestin-GFP+ cells
For all ex vivo analysis, transplantation, and RNA-seq studies, Nestin-GFP+ cells were isolated by the following procedure. Cell suspensions were prepared from the dermal WAT of 3-d-old mice as described (Lichti et al. 2008). Briefly, the whole skin was floated on 0.25% cold trypsin overnight with the epidermis up. The next day, the epidermis was separated from dermal tissue and discarded. Dermis was then digested with 0.35% collagenase type I (GIBCO) in DMEM for 60 min at 37°C. Hair follicles were removed by low-speed centrifugation. Subsequently, Nes-GFP+ single cells were sorted from cell suspensions with a MoFlo XDP (Beckman Coulter) cell sorter, and the purity of sorted cells was verified as >95% by rerunning the sorted population.
cDNA library construction and sequencing
Total RNA was purified from 2 million to 3 million freshly sorted Nes-GFP+ cells using RNeasy minikit (Qiagen). cDNA libraries were prepared with NEBNext Ultra Directional RNA library preparation kit for Illumina (New England Biolabs) according to the manufacturer's protocol. In short, mRNA was isolated from 1 µg of purified total RNA with oligo dT beads and fragmented. Next, first and second strand cDNA were synthesized, followed by purification using Agencourt AMPure XP beads (Beckman Coulter). The second strand cDNAs were end-repaired, A-tailed, and adaptor-ligated. Size-selected DNA with Agencourt AMPure XP beads was enriched by 13-cycle PCR with each index primer and again purified using the beads. Each indexed library was analyzed by the Agilent 2200 TapeStation system, and RNA integrity number equivalents (RINe) ranged from 9.2 to 9.6. Prior to sequencing, the library pool was absolutely quantified via qPCR using the Illumina library quantification kit by KAPA Biosystems on a Roche LightCycler 480 qPCR instrument. The pool was then diluted to 2 nM followed by NaOH denaturation and further dilution to 12 pM. The final dilution product was then loaded onto a single Illumina HiSeq 2500 high-output lane and sequenced with paired-end, 100-base-pair (bp) reads, which produced 20 million to 40 million read pairs per sample.
RNA-seq data processing and accession numbers
The quality of the reads was assessed at any processing step using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Adapters were removed using Trimmomatic tool. Sequencing data were processed according to guidelines for RNA-seq data analysis (Trapnell et al. 2012). Briefly, reads were aligned to a mouse genome indexed with Bowtie 2 using TopHat version 2.0.4 (Trapnell et al. 2012). The alignment settings were adjusted to set “mate inner distance” to 170 bp and “mate standard deviation” to 100 bp and “microexon search” and “prefilter multihits” were turned on. The Bowtie 2 alignment setting was set to “very sensitive,” and the “library type” was set to “fr-firststrand.” For differential gene expression analysis, expression level was obtained using the htseq-count tool by counting reads in Mus musculus gene annotations provided by Ensembl release 67. Quality control of the expression data was performed using the arrayQualityMetrics R package. DEGs were identified using the DeSeq2 R package, with a controlling false discovery rate of <10%. Functional analysis was performed using Ingenuity Pathway Analysis (Ingenuity Systems, http:// www.ingenuity.com) and the topGO R package. Visualization was performed within the R/Bioconductor environment.
ISH
Template sequences from 292 to 1250 of Dlk1 (NM_010052), from 474 to 1416 of H19 (NR_001592), and from 702 to 1646 Igf2 (NM_001122736) were used to generate antisense RNA probes for ISH. The dissected tissue was fixed with 10% neutral formalin for 7 d, soaked in 20% sucrose/PBS overnight, embedded in OCT compound (Sakura Tek), and sectioned at 14 µm. Dig-labeled RNA probes were synthesized by DIG RNA labeling mix (Roche) from DNA template sequences. Hybridized probes were detected with AP-conjugated anti-Dig Fab fragments (Roche) and developed with NBT/BCIP.
Cell culture
Sorted Nes-GFP+ cells were expanded under hypoxic conditions (5% O2, 10% CO2) with MesenCult medium according to the manufacturer's protocol (Stem Cell Technologies), and cells at passages 1–3 were used for differentiation and proliferation experiments under normoxia conditions. Adipogenic differentiation was induced with two cycles of adipogenic cocktail treatment (one cycle of treatment: DMEM + 10% FBS containing 10 µg/mL insulin, 0.5 mM 3-isobutyl-1-methylxanthine, and 0.25 µg/mL dexamethasone for 2 d followed by 2 d of DMEM + 10% FBS containing 10 µg/mL insulin) and maintained with DMEM + 10% FBS containing 10 µg/mL insulin until day 11, after which the cells were fixed and stained with Oil Red O. For in vitro fibrosis assays, cells were cultured with DMEM + 10% FBS containing 0.5 mM ascorbic acid derivative (2-o-α-D-glucopyranosyl-L-ascorbic acid). For qRT–PCR analysis, after 7 d, cells were grown on cell culture plastic. For long-term culture and formation of cell–ECM complexes, the cells were cultured on polycarbonate transwell inserts for 3 wk. The resulting complexes were fixed with 4% PFA, separated from the transwell, and mounted on glass slides. The resulting complex was then stained with anti-Col3a1 antibody (1:500; Abcam, ab7778), followed by Alexa fluor 647-conjugated secondary antibody (1:100; Jackson Laboratory). Col3a1-positive area was calculated for each complex (n = 3) using ImageJ. For in vitro proliferation assays, the cells were incubated with 20 µM EdU for 20 min, and EdU-positive cells were detected using the LSR II after performing a Click chemistry reaction according to the manufacturer's protocol (Life Technologies).
Transplantation
One million freshly isolated Nes-GFP+ cells were suspended in 50 µL of PBS and mixed with 150 µL of Matrigel (BD, #356234). Next, 50 µL was injected subcutaneously into the foreheads of recipient mice. After 3–4 wk or 3 mo, the newly formed fat pads were dissected and analyzed.
Statistical analysis (excluding RNA-seq)
All calculations were performed using GraphPad Prism 6.0 software (GraphPad Software, Inc.). The differences between two groups were assessed by unpaired Student's t-test, and comparisons of multiple groups involved the use of ANOVA. P < 0.05 was considered to be significant. The significance of imprinted DEGs was assessed by a χ2 test with Yates correction using these values: 17 imprinted DEGs, 1339 nonimprinted DEGs, 53 imprinted genes that were not DEGs, and 37,922 nonimprinted genes that were not DEGs.
Accession numbers
RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) public database under accession number GSE64510.
Supplementary Material
Acknowledgments
We thank Michael Upchurch, Andrew Bachman, and Eli Rhoades for expert technical assistance; the Oklahoma Medical Research Foundation (OMRF) Centers of Biomedical Research Excellence (COBRE) Flow Cytometry Core for cell sorting analysis; the OMRF COBRE Imaging Core facility for tissue processing; the OMRF Genomics facility for RNA-seq; Weidong Wang for assistance with confocal microscopy; and David Brenner, Ralf Adams, and Grigori Enikolopov for sharing mouse strains. We thank James Tomasek, Linda Thompson, David Jones, Philippe Soriano, and our laboratory colleagues for critical comments on the manuscript. This study was supported by grants from the National Institutes of Health (5P20GM103636-02) and the Oklahoma Center for Adult Stem Cell Research (OCASCR). L.E.O. is a Pew Scholar in Biomedical Research, and this work was supported in part by the Pew Charitable Trusts.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.260554.115.
References
- Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, Lee LB, McMahon G, Grone HJ, Lipson KE, et al. 2005. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med 201: 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adalsteinsson BT, Ferguson-Smith AC. 2014. Epigenetic control of the genome-lessons from genomic imprinting. Genes 5: 635–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, Distler O, Distler JH. 2009. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum 60: 219–224. [DOI] [PubMed] [Google Scholar]
- Andrae J, Gallini R, Betsholtz C. 2008. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22: 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. 2011. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: 193–215. [DOI] [PubMed] [Google Scholar]
- Bartolomei MS. 2009. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev 23: 2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Lin KK, Sonnet C, Boles NC, Weksberg DC, Nguyen H, Holt LJ, Rickwood D, Daly RJ, Goodell MA. 2011. Imprinted genes that regulate early mammalian growth are coexpressed in somatic stem cells. PLoS One 6: e26410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R, Rodeheffer MS. 2013. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol 15: 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R, Jeffery E, Rodeheffer MS. 2014. Weighing in on adipocyte precursors. Cell Metab 19: 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson V, Smeriglio P, Wegener A, Relaix F, Nait Oumesmar B, Sassoon DA, Marazzi G. 2011. PW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proc Natl Acad Sci 108: 11470–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O. 2013a. Role of pericytes in skeletal muscle regeneration and fat accumulation. Stem Cells Dev 22: 2298–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. 2013b. Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol 305: C1098–C1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Files DC, Mannava S, Smith T, Wang ZM, Messi ML, Mintz A, Delbono O. 2014. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res Ther 5: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondjers C, He L, Takemoto M, Norlin J, Asker N, Hellstrom M, Lindahl P, Betsholtz C. 2006. Microarray analysis of blood microvessels from PDGF-B and PDGF-Rβ mutant mice identifies novel markers for brain pericytes. FASEB J 20: 1703–1705. [DOI] [PubMed] [Google Scholar]
- Bonner JC. 2004. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15: 255–273. [DOI] [PubMed] [Google Scholar]
- Chang Y, She ZG, Sakimura K, Roberts A, Kucharova K, Rowitch DH, Stallcup WB. 2012. Ablation of NG2 proteoglycan leads to deficits in brown fat function and to adult onset obesity. PLoS One 7: e30637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ, et al. 2011. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 80: 1170–1181. [DOI] [PubMed] [Google Scholar]
- Church CD, Berry R, Rodeheffer MS. 2014. Isolation and study of adipocyte precursors. Methods Enzymol 537: 31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corselli M, Chen CW, Sun B, Yap S, Rubin JP, Peault B. 2012. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev 21: 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. 2008. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3: 301–313. [DOI] [PubMed] [Google Scholar]
- Dey BK, Pfeifer K, Dutta A. 2014. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C, Zucker JD, et al. 2010. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 59: 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore-Duffy P, Katychev A, Wang X, Van Buren E. 2006. CNS microvascular pericytes exhibit multipotential stem cell activity. J Cereb Blood Flow Metab 26: 613–624. [DOI] [PubMed] [Google Scholar]
- Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson-Smith AC, et al. 2013. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504: 277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Jahoda CA, Chuong CM, Watt FM, Horsley V. 2014. Defining dermal adipose tissue. Exp Dermatol 23: 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. 2012. Lineage tracing and genetic ablation of ADAM12+ perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med 18: 1262–1270. [DOI] [PubMed] [Google Scholar]
- Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. 2006. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 124: 161–173. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Sheppard D, Duffield JS, Violette S. 2013. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med 5: 167sr1. [DOI] [PubMed] [Google Scholar]
- Gesta S, Tseng YH, Kahn CR. 2007. Developmental origin of fat: tracking obesity to its source. Cell 131: 242–256. [DOI] [PubMed] [Google Scholar]
- Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. 2011. A pericyte origin of spinal cord scar tissue. Science 333: 238–242. [DOI] [PubMed] [Google Scholar]
- Hardouin SN, Guo R, Romeo PH, Nagy A, Aubin JE. 2011. Impaired mesenchymal stem cell differentiation and osteoclastogenesis in mice deficient for Igf2-P2 transcripts. Development 138: 203–213. [DOI] [PubMed] [Google Scholar]
- Hayes BJ, Riehle KJ, Shimizu-Albergine M, Bauer RL, Hudkins KL, Johansson F, Yeh MM, Mahoney WM Jr, Yeung RS, Campbell JS. 2014. Activation of platelet-derived growth factor receptor α contributes to liver fibrosis. PLoS One 9: e92925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. 2007. The myofibroblast: one function, multiple origins. Am J Pathol 170: 1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Chiba H, Nagai K, Ishii G, Ochiai A. 2008. Human vascular adventitial fibroblasts contain mesenchymal stem/progenitor cells. Biochem Biophys Res Commun 368: 305–310. [DOI] [PubMed] [Google Scholar]
- Hudak CS, Gulyaeva O, Wang Y, Park SM, Lee L, Kang C, Sul HS. 2014. Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Rep 8: 678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. 2010. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Berry DC, Tang W, Graff JM. 2014. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell Rep 9: 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM. 2010. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamichos D, Guo XQ, Hutcheon AE, Zieske JD. 2010. Human corneal fibrosis: an in vitro model. Invest Ophthalmol Vis Sci 51: 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi N, Toriyama K, Nicodemou-Lena E, Inou K, Torii S, Kitagawa Y. 1998. De novo adipogenesis in mice at the site of injection of basement membrane and basic fibroblast growth factor. Proc Natl Acad Sci 95: 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. 2009. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29: 1575–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD. 2015. Perivascular gli1(+) progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16: 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Mottillo EP, Granneman JG. 2012. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab 15: 480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CH, Akazawa H, Tamagawa M, Ito K, Yasuda N, Kudo Y, Yamamoto R, Ozasa Y, Fujimoto M, Wang P, et al. 2010. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J Clin Invest 120: 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichti U, Anders J, Yuspa SH. 2008. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc 3: 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JC, Finkielstain GP, Barnes KM, Baron J. 2008. An imprinted gene network that controls mammalian somatic growth is down-regulated during postnatal growth deceleration in multiple organs. Am J Physiol Regul Integr Comp Physiol 295: R189–R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Bataller R, Yang L, Brenner DA. 2004. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology 40: 1151–1159. [DOI] [PubMed] [Google Scholar]
- Majesky MW, Dong XR, Hoglund V, Mahoney WM Jr, Daum G. 2011. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol 31: 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni RG, Korman B, Wei J, Wood TA, Graham L, Whitfield ML, Scherer PE, Tourtellotte WG, Varga J. 2015. Myofibroblasts in cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol 67: 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS. 2010. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466: 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. 2004. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol 469: 311–324. [DOI] [PubMed] [Google Scholar]
- Moon YS, Smas CM, Lee K, Villena JA, Kim KH, Yun EJ, Sul HS. 2002. Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol Cell Biol 22: 5585–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morison IM, Ramsay JP, Spencer HG. 2005. A census of mammalian imprinting. Trends Genet 21: 457–465. [DOI] [PubMed] [Google Scholar]
- Olson LE, Soriano P. 2009. Increased PDGFRα activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 16: 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, Soriano P. 2011. PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev Cell 20: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, Ravussin E, Bray GA, Smith SR. 2009. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab 94: 5155–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passman JN, Dong XR, Wu SP, Maguire CT, Hogan KA, Bautch VL, Majesky MW. 2008. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci 105: 9349–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponten A, Li X, Thoren P, Aase K, Sjoblom T, Ostman A, Eriksson U. 2003. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. Am J Pathol 163: 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. 2011. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci 108: E1475–E1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, Friedman JM. 2008. Identification of white adipocyte progenitor cells in vivo. Cell 135: 240–249. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Guertin DA. 2014. Adipocyte lineages: tracing back the origins of fat. Biochim Biophys Acta 1842: 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, Krishnan V, Lai-Hsu C, Park KK, Tsoulfas P, et al. 2013. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci 33: 13882–13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clement K, Scherer PE. 2013. Fibrosis and adipose tissue dysfunction. Cell Metab 18: 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, Graff JM. 2008. White fat progenitor cells reside in the adipose vasculature. Science 322: 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. 2002. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. 1999. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99–103. [DOI] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. 2010. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 12: 143–152. [DOI] [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, et al. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11: 711–722. [DOI] [PubMed] [Google Scholar]
- Vila IK, Badin PM, Marques MA, Monbrun L, Lefort C, Mir L, Louche K, Bourlier V, Roussel B, Gui P, et al. 2014. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep 7: 1116–1129. [DOI] [PubMed] [Google Scholar]
- Wojciechowicz K, Gledhill K, Ambler CA, Manning CB, Jahoda CA. 2013. Development of the mouse dermal adipose layer occurs independently of subcutaneous adipose tissue and is marked by restricted early expression of FABP4. PLoS One 8: e59811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. 2012. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. 2008. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135: 145–157. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. 1994. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron 12: 11–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.