

In this study, Kang et al. investigate how PcG complexes form repressive chromatin domains. The findings show that Scm, a transcriptional repressor, is an important regulator of PRC1, PRC2, and transcriptional silencing and suggest that Scm coordinates PcG complexes and polymerizes, resulting in PcG silencing.
Keywords: Polycomb group (PcG), Drosophila, Scm, SAM domain, BioTAP-XL
Abstract
The Polycomb group (PcG) proteins are key regulators of development in Drosophila and are strongly implicated in human health and disease. How PcG complexes form repressive chromatin domains remains unclear. Using cross-linked affinity purifications of BioTAP-Polycomb (Pc) or BioTAP-Enhancer of zeste [E(z)], we captured all PcG-repressive complex 1 (PRC1) or PRC2 core components and Sex comb on midleg (Scm) as the only protein strongly enriched with both complexes. Although previously not linked to PRC2, we confirmed direct binding of Scm and PRC2 using recombinant protein expression and colocalization of Scm with PRC1, PRC2, and H3K27me3 in embryos and cultured cells using ChIP-seq (chromatin immunoprecipitation [ChIP] combined with deep sequencing). Furthermore, we found that RNAi knockdown of Scm and overexpression of the dominant-negative Scm-SAM (sterile α motif) domain both affected the binding pattern of E(z) on polytene chromosomes. Aberrant localization of the Scm-SAM domain in long contiguous regions on polytene chromosomes revealed its independent ability to spread on chromatin, consistent with its previously described ability to oligomerize in vitro. Pull-downs of BioTAP-Scm captured PRC1 and PRC2 and additional repressive complexes, including PhoRC, LINT, and CtBP. We propose that Scm is a key mediator connecting PRC1, PRC2, and transcriptional silencing. Combined with previous structural and genetic analyses, our results strongly suggest that Scm coordinates PcG complexes and polymerizes to produce broad domains of PcG silencing.
The Polycomb group (PcG) genes were discovered in Drosophila based on their essential roles in pattern formation. Although expressed ubiquitously, they maintain repression of developmental regulators in precise spatial patterns delineated by earlier pattern formation regulatory decisions (Lewis 1978; Struhl 1981; Simon et al. 1992). PcG proteins are now known to repress many other target genes in Drosophila (Negre et al. 2006; Schwartz et al. 2006; Tolhuis et al. 2006). Furthermore, PcG proteins function in diverse regulatory pathways such as cell type specificity and X inactivation in mammals, and both PcG loss-of-function and gain-of-function mutations have been strongly implicated in cancer (Sparmann and van Lohuizen 2006; Gieni and Hendzel 2009).
Many of the initially characterized PcG proteins can be classified into two principal complexes: PcG-repressive complex 1 (PRC1), implicated in chromatin compaction, and PRC2, which mediates H3K27 histone methylation. Polycomb (Pc), Psc, Su(z)2, Polyhomeotic (Ph), and Sce (dRING) are core components of Drosophila PRC1, while Enhancer of zeste [E(z)], Su(z)12, Esc, and Nurf55 are core components of PRC2. These soluble PRC1 and PRC2 complexes purify separately and do not share components (Shao et al. 1999; Saurin et al. 2001; Czermin et al. 2002; Muller et al. 2002). However, in flies as well as in mammalian cells, there are additional PcG complexes and subcomplexes (such as PhoRC, dRAF, and PR-DUB) (Klymenko et al. 2006; Lagarou et al. 2008; Scheuermann et al. 2010; Schwartz and Pirrotta 2013) and proteins classified as substoichiometric components of PRC1 (e.g., Sex comb on midleg [Scm]) or PRC2 (e.g., Jarid2 and Pcl) (O'Connell et al. 2001; Saurin et al. 2001; Tie et al. 2003; Li et al. 2010; Herz et al. 2012). Furthermore, several PcG factors are still not clearly linked to any complex (e.g., super sex combs [sxc] and multi sex combs [mxc]) (Ingham 1984; Santamaria and Randsholt 1995).
How PcG complexes find Pc response elements (PREs) and spread to create repressive domains is not known on a mechanistic level. Here we focus on a central role for Scm, a PcG protein whose genetic function in silencing is as strong as any core component of PRC1 and PRC2 (Breen and Duncan 1986; Bornemann et al. 1998), but whose biochemical relationships and mechanistic role in silencing are less clear. Scm protein is recovered as a substoichiometric component of soluble PRC1 and can directly bind the Ph subunit of PRC1 in reconstitution experiments (Saurin et al. 2001; Peterson et al. 2004). The Scm protein contains several chromatin interaction motifs, including two MBT (malignant brain tumor) histone interaction domains and a zinc finger domain (Fig. 6A, below; Bornemann et al. 1996; Wang et al. 2010). It also contains a C-terminal SAM (sterile α motif)/SPM (Scm, Ph, and MBT) domain, which has been implicated in homopolymerization and heteropolymerization in both genetic and structural studies (Peterson et al. 1997, 2004; Kim et al. 2005). Scm interaction with Ph is through their respective SAM domains, and self-polymerization of the Scm-SAM domain can occur in vitro (Kim et al. 2005). Therefore, it is interesting to speculate that Scm may be involved in the spreading of silencing complexes from PREs along the chromosome.
Figure 6.
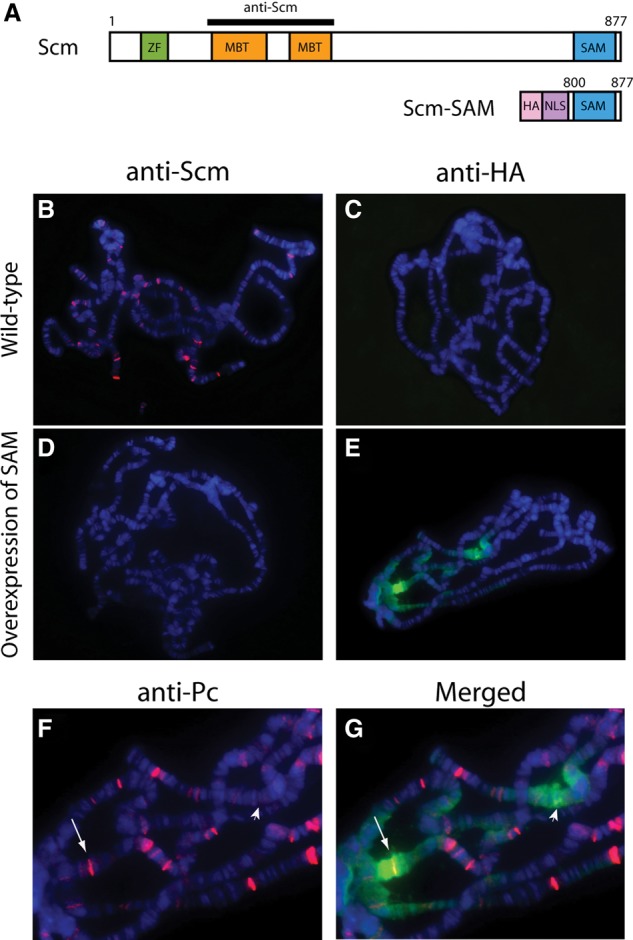
The overexpressed Scm-SAM domain binds sporadically in long contiguous regions on polytene chromosomes. (A) Scm contains a zinc finger domain (ZF; green), two MBT motifs (orange), and a SAM domain (blue). An Scm antibody raised against a peptide antigen encompassing the MBT repeats is represented as a black bar. The overexpressed Scm-SAM transgene contains an HA-epitope tag (pink), nuclear localization signal (NLS; puple), and C-terminal SAM domain (amino acids 800–877). (B–G) The P{GawB}c729 GAL4 line was crossed with Oregon R or the UAS-SAM transgenic line, and polytene chromosomes were immunostained with anti-Scm (B,D, red), anti-HA (C,E, green), and anti-Pc (F, red). DNA was counterstained with Hoechst. (B) Wild-type distribution of the Scm protein. (C) The anti-HA antibody did not detect any signal in wild type. (D) Endogenous Scm was depleted by overexpression of Scm-SAM. (E) Detection of the overexpressed SAM domain by anti-HA shows long contiguous signals on polytene chromosomes. (F) Polytene chromosomes shown in E were also labeled with anti-Pc antibody. Zoomed-in view of two regions showing strong HA signals in E are marked by an arrow and arrowhead. (G) Merged image showing that one of two strong HA signal regions is overlapped with Pc (arrow), but the other region does not contain a strong Pc band (arrowhead), indicating that SAM domain stretches do not appear to require strong PREs as nucleation sites.
To improve our understanding of mechanisms for PcG targeting, assembly, and spreading, we analyzed the composition of PRC1 and PRC2 using cross-linking prior to tandem affinity purification (BioTAP-XL) to identify protein–protein interactions that may be disrupted by removal from chromatin. Previously, BioTAP-XL was successful in our recovery of new interactors of the Drosophila MSL and HP1 proteins as well as of EZH2 in human cells (Alekseyenko et al. 2014a,b). Applying this method to PcG proteins, we found robust recovery of PRC1 subunits using Pc as bait and strong enrichment of PRC2 components using E(z) as bait. Furthermore, we identified new candidate interactors for each complex. Interestingly, Scm was the only protein to be strongly enriched with both PRC1 and PRC2, suggesting a central function that may have been lost in conventional biochemical purifications. We further explored Scm function in PcG silencing, analyzing recombinant protein interactions, ChIP-seq (chromatin immunoprecipitation [ChIP] combined with deep sequencing), mass spectrometry, and in vivo RNAi and SAM domain overexpression. Our results strongly suggest that Scm is a key mediator connecting PRC1 and PRC2. Disruption of PRC2 [E(z)] via loss of Scm or overexpression of the Scm-SAM domain implicates Scm polymerization in the establishment and spreading of silent PcG-dependent chromatin domains.
Results
BioTAP-XL reveals classical and noncanonical binding partners of PRC1
To search for new factors involved in PcG function, we performed BioTAP-XL cross-linking and affinity purifications followed by mass spectrometry. Pc and E(z) were selected to be tagged subunits to capture PRC1 and PRC2 complexes, respectively (Supplemental Fig. S1A). We incorporated a C-terminal BioTAP tag into a genomic Pc transgene, including promoter and flanking upstream regions to preserve endogenous regulation. Previously, Nekrasov et al. (2007) demonstrated that an N-TAP-tagged E(z) cDNA transgene driven by the α-tubulin promoter successfully rescued a transheterozygous E(z)-null mutant. Therefore, we replaced the TAP tag with a BioTAP tag to create a BioTAP-N-E(z) cDNA transgene. Transgenic embryos carrying these constructs expressed BioTAP fusion proteins of the expected sizes (Supplemental Fig. S1B), and the tagged proteins were detectable on larval polytene chromosomes in a wild-type background, indicating that they could compete with their endogenous counterparts (Supplemental Fig. S1C). Importantly, the BioTAP-N-E(z) and Pc-C-BioTAP transgenes successfully rescued E(z) and Pc transheterozygous-null mutants, respectively, indicating that these BioTAP fusion proteins were functional substitutes for their endogenous counterparts (Supplemental Fig. S1D).
Using BioTAP-XL coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Supplemental Fig. S2), we identified proteins in Pc or E(z) embryonic pull-downs that were enriched relative to input and mock pull-downs (Table 1A,B). The core components of the PRC1 complex—Psc, Su(z)2, Sce (dRING), Pc, and Ph-p/Ph-d—were all highly enriched in the Pc pull-down. Scm, previously proposed to be a substoichiometric component of PRC1 (Saurin et al. 2001), was also a high-ranking interactor, strongly validating our approach.
Table 1.
Top enrichment lists of Pc- and E(z)-interacting proteins in Drosophila embryos
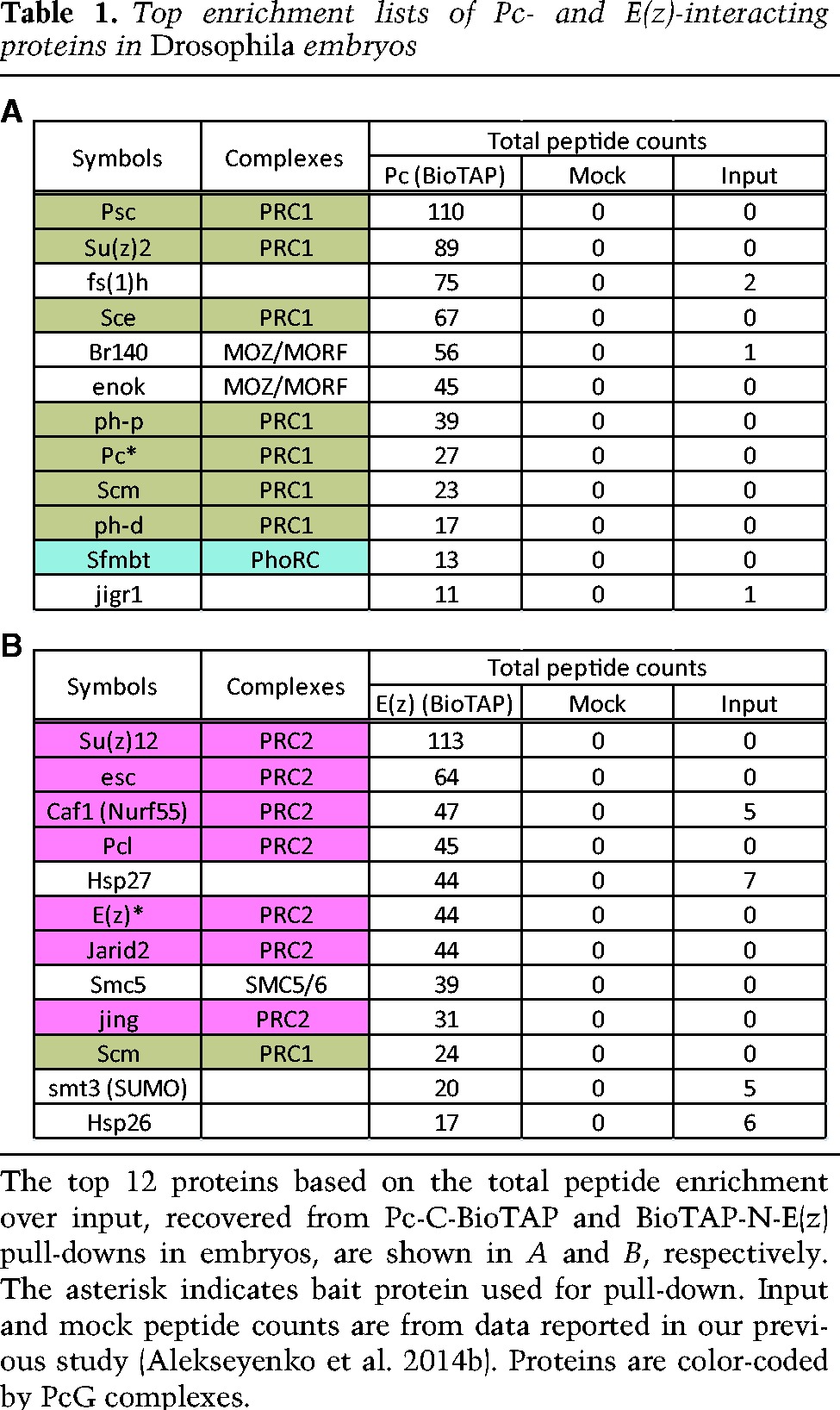
Interestingly, Enoki mushroom (Enok) and Br140, Drosophila orthologs of two subunits of the mammalian MOZ/MORF complex, as well as female sterile homeotic 1 [Fs(1)h], the Drosophila ortholog of mammalian BRD4, were also highly enriched in the Pc pull-down. Considering that BRD4 and the MOZ/MORF complex are involved in transcriptional activation in mammals (Pelletier et al. 2002; Jang et al. 2005; Voss et al. 2009; Kanno et al. 2014), their strong interaction with Pc is surprising. However, these same interactors were previously observed after affinity purification of Pc without cross-linking (Strubbe et al. 2011), supporting their identification as genuine binding partners of Pc. Classical studies of Fs(1)h and recent genetic analyses of Enok have revealed very early developmental functions in oogenesis and pattern formation that are likely to be relevant to our observed PcG interactions (Digan et al. 1986; Shearn 1989; Huang et al. 2014).
BioTAP-XL reveals classical and noncanonical binding partners of PRC2
The results of the BioTAP-N-E(z) pull-down are listed in Table 1B. As expected, the top-ranking proteins are components of the PRC2 complex, including the four core components Su(z)12, Esc, E(z), and Caf1(aka Nurf55) as well as Jarid2 and Pcl. In addition, we identified Jing, whose mammalian ortholog, AEBP2, is an established PRC2 component in mammals (Cao et al. 2002; Kim et al. 2009). AEBP2 contributes to mammalian PRC2 stability and has been incorporated into the PRC2 EM structure (Ciferri et al. 2012). Jarid2 and AEBP2 enhance H3K27 trimethylation (H3K27me3) by PRC2 on H2Aub nucleosomes (Kalb et al. 2014); the ubiquitylation event is achieved by variant PRC1-type complexes such as dRAF in Drosophila and RYBP–PRC1 in mammals (Lagarou et al. 2008; Tavares et al. 2012). The only previously reported evidence for association between Jing and PRC2 in Drosophila was copurification of Jing in an affinity pull-down using Flag-HA-Jarid2 (Herz et al. 2012).
The non-PcG protein that most strongly interacted with E(z) in our assay was SMC5, a structural maintenance of chromosome (SMC) family protein previously implicated in DNA repair through the formation of the SMC5/6 complex (Fujioka et al. 2002; Harvey et al. 2004; De Piccoli et al. 2006; Stephan et al. 2011). Previous reports are consistent with a role for the mammalian PRC2 complex in DNA double-strand break repair (Chou et al. 2010; Campbell et al. 2013). However, we did not identify the SMC6 subunit in our pull-down, suggesting that an alternative SMC5 complex may be associated with E(z). Smt3 (SUMO) was also enriched in our E(z) pull-down, and mammalian PRC2 core components EZH2 and SUZ12 have been shown to be sumoylated in vitro and in vivo (Riising et al. 2008). Scm, a new interactor of PRC2 (see below) is another PcG protein that could be regulated by sumoylation when associated with PRC2 (Smith et al. 2011). Interestingly, in yeast, SMC5 can interact with Nse2/Mms21, a SUMO E3 ligase (Sergeant et al. 2005), suggesting that SMC5 could potentially be involved in sumoylation of PRC2 components.
Scm is a uniquely shared subunit of PRC1 and PRC2
PRC1 and PRC2 show substantial colocalization by genome-wide ChIP analyses (Schwartz et al. 2006; Tolhuis et al. 2006). However, they purify as independent complexes using traditional biochemical methods (Shao et al. 1999; Saurin et al. 2001; Czermin et al. 2002; Muller et al. 2002). Interestingly, even with cross-linking, we found that PRC1 and PRC2 were still largely independent, with nonoverlapping lists of enriched proteins (Fig. 1; Table 1). The notable exception to this was Scm, which was similarly enriched in both Pc and E(z) pull-downs. Enrichment of Scm in the E(z) pull-down was unexpected and led us to investigate whether Scm could directly interact with PRC2 as it does with reconstituted PRC1 through interaction with Ph (Peterson et al. 2004). Using the Sf9/baculovirus system, we coexpressed Scm with the four recombinant PRC2 core components, including a Flag-tagged Esc subunit. As a negative control, we expressed Scm without PRC2 under the same conditions. We performed affinity purification for the Flag epitope (Fig. 2A) and confirmed the formation of purified PRC2 complex after coexpression through silver staining (Fig. 2B). Scm was not recovered after anti-Flag affinity purification in the absence of PRC2, but it was difficult to determine whether Scm assembled with the reconstituted PRC2 complex through silver staining because Scm and Su(z)12 have similar molecular weights. Therefore, we confirmed the copurification of Scm with PRC2 by Western blot (Fig. 2C). The ability of Scm to directly interact with both PRC1 and PRC2 suggests that Scm may play a unique role in communicating between or connecting the two major PcG complexes.
Figure 1.
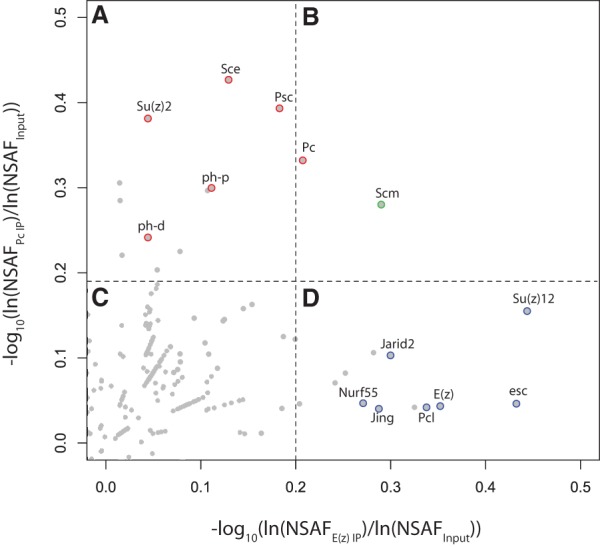
Scatter plot of E(z) and Pc pull-down enrichment over total embryonic chromatin input. Each point represents an individual protein, with coordinates (X and Y) corresponding to its enrichment in E(z) pull-down and Pc pull-down relative to input, as quantified by log10{ln[NSAFE(z) IP]/ln(NSAFInput)} and log10[ln(NSAFPc IP)/ln(NSAFInput)], respectively. Dashed lines represent the 99th percentile of E(z) enrichment (vertical line) and Pc enrichment (horizontal line), where A and B represent the top percentile of proteins enriched in Pc pull-down, and B and D represent the top percentile of proteins enriched in E(z) pull-down. Known PRC1 and PRC2 components are highlighted in red and blue, respectively. Scm is highlighted in green. Enriched proteins in C fell below the top percentile in both pull-downs. See Supplemental Table S2 for the full range of data.
Figure 2.
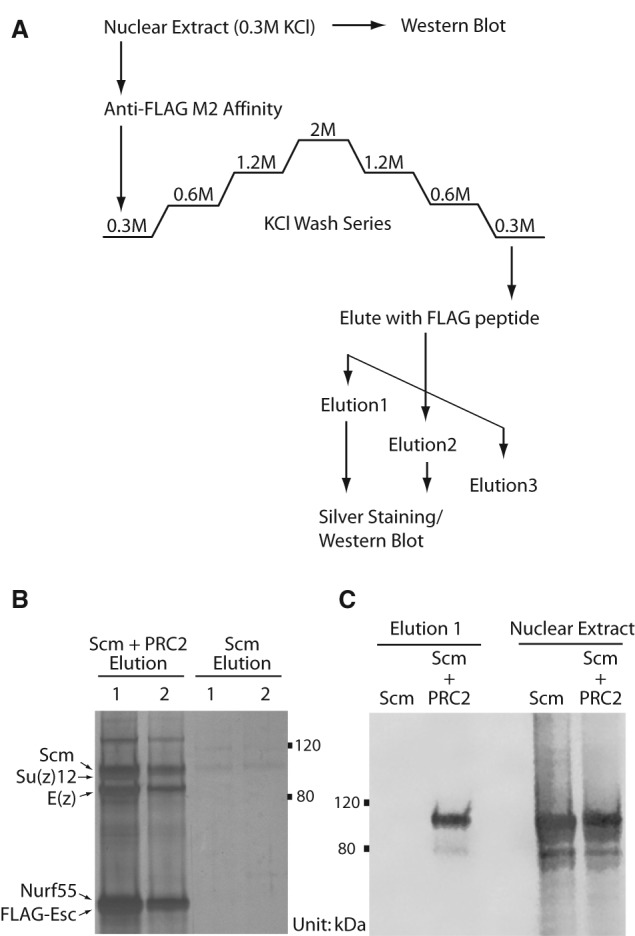
Recombinant Scm interacts with recombinant PRC2 complex. (A) The purification scheme using M2 anti-Flag affinity. Nuclear extracts were generated from Sf9 cells that were infected with baculovirus expressing recombinant Scm protein or coinfected with Scm and the four members of the core PRC2 complex [E(z), Su(z)12, Nurf-55, and Flag-Esc]. Elution fractions were boiled in SDS-PAGE loading buffer and resolved on an 8% Tris-glycine gel. (B) The presence of the intact PRC2 complex was verified by silver staining. (C) The specificity of the Scm–PRC2 interaction was confirmed by Western blotting using anti-Scm antibodies. The Scm–PRC2 interaction was observed in two independently generated coinfected nuclear extracts. Nuclear extract lanes show that Scm expression levels were similar in Scm only and Scm + PRC2 inputs (0.5% input loaded).
Scm colocalizes with the repressive mark H3K27me3 genome-wide
To map the genomic locations of Scm interaction with PRC1 and PRC2, we generated a fly line and a stable S2 cell line that expressed a transgenic BioTAP-Scm fusion protein. The BioTAP tag was fused to the N terminus of Scm and expressed from a genomic copy of Scm (Supplemental Fig. S1A). The fusion protein was fully functional, as confirmed by expected size, polytene staining in a wild-type background, and transgenic rescue of Scm mutant lethality in flies (Supplemental Fig. S1B–D). We performed BioTAP-XL from transgenic embryos and the S2 line followed by DNA isolation, library preparation, and DNA sequencing (Supplemental Fig. S2). We also performed ChIP-seq of Pc-BioTAP and E(z)-BioTAP fusion proteins from embryos and/or S2 cell lines and compared our results with H3K27me3 and/or PcG protein localizations from modENCODE (http://data.modencode.org) analyses. Consistent with our ChIP mass spectrometry results, we found that BioTAP-Scm colocalizes with Pc and E(z) in embryos and that all three proteins displayed a high degree of overlap with the H3K27me3-binding profiles from modENCODE (Fig. 3; Supplemental Fig. S3). We also found similar colocalization in S2 cells (Supplemental Fig. S4A), with selected regions viewed at higher resolution (Supplemental Fig. S5). Comparison of the genome-wide binding profile of N-BioTAP-Scm with the mapping data for all available factors in S2 cells from the modENCODE consortium (http://data.modencode.org) revealed that H3K27me3 displayed the highest Pearson correlation coefficient with Scm (Supplemental Fig. S4B). Taken together, our genome-wide mapping results strongly suggest that Scm protein interactions detected by BioTAP-XL occur in the context of PcG silenced chromatin.
Figure 3.

Genome-wide colocalization of BioTAP-N-Scm, Pc-C-BioTAP, BioTAP-N-E(z), H3K27me3, and H3K36me3 profiles in embryos. (A) Genome browser view of a representative region of chromosome 3R (chr3R) showing that the BioTAP-N-Scm ChIP-seq profile significantly overlaps with Pc-C-BioTAP, BioTAP-N-E(z), and H3K27me3 in embryos [Scm, Pc, and E(z) from the present study; H3K27me3 and H3K36me3 from the modENCODE project (http://data.modencode.org)]. (B) Enrichment patterns of BioTAP-tagged Scm, Pc, E(z), H3K27me3, and H3K36me3 at the top 25% of Scm peaks (n = 457). Each row was centered at the Scm peak and ordered by Scm intensity.
Scm depletion affects the binding patterns of PRC2 and H3K27me3 on polytene chromosomes
Since our BioTAP-XL affinity purifications revealed that Scm was uniquely enriched with both PRC1 and PRC2, we investigated the functional consequences of Scm loss on the global distribution of these silencing complexes. Embryonic genome-wide analyses were not feasible due to the difficulty in removing both maternal and zygotic contributions of Scm in a sufficient number of embryos. Furthermore, RNAi knockdown of Scm mRNA in tissue culture cells was relatively ineffective in our hands at removal of Scm protein from chromatin genome-wide (data not shown). However, we found that expression of shRNA hairpins driven by a salivary gland-specific GAL4 driver resulted in significant decrease or loss of cognate PcG proteins on polytene chromosomes (Fig. 4B; Supplemental Fig. S6D,G), suggesting that we could test their interdependence in this tissue. Therefore, for a broad overview of the potential interdependence of PRC1, PRC2, and Scm, we compared the localization patterns of Pc, E(z), and Scm on polytene chromosomes in wild-type and after salivary gland-specific shRNA knockdowns.
Figure 4.
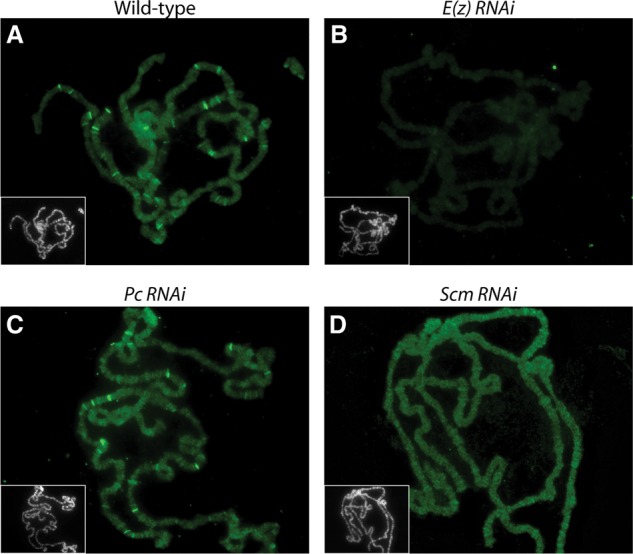
The effects of RNAi knockdown of E(z), Pc, and Scm on E(z) distribution on polytene chromosomes. The P{GawB}c729 salivary gland-specific GAL4 line was crossed with Oregon R (A), UAS-E(z) shRNA (B), UAS-Pc shRNA (C), or UAS-Scm shRNA (D) Transgenic RNAi Project (TRiP) lines. Polytene chromosomes from the resulting third instar larvae were immunostained with anti-E(z) (green). (Grayscale insets) DNA was counterstained with Hoechst. (A) The wild-type distribution of E(z) protein. (B) E(z) knockdown results in a decrease of overall signal level on whole chromosomes as well as loss of E(z) bands. (C) Many E(z) bands still remain after Pc knockdown. (D) Scm knockdown results in loss of strongly staining E(z) bands on polytene chromosomes.
In a seminal study using S2 cells and imaginal discs, Wang et al. (2010) found that binding of Scm to the bxd PRE upstream of the Ultrabithorax (Ubx) Hox gene was not affected by mutation or knockdown of PRC1 or PRC2 components, suggesting that Scm is recruited independently of PRC1 and PRC2. In contrast, depletion of Scm affected recruitment of both PRC1 and PRC2 to the bxd PRE. Consistent with the bxd PRE results, we found that Scm was still present at numerous sites after shRNA knockdown of Pc or E(z) (Supplemental Fig. S6A,C,E). In contrast to the bxd study, however, we observed that Pc also remained on chromatin after E(z) or Scm RNAi (Supplemental Fig. S6B,F,H). Therefore, our results suggest that Scm and Pc can bind target sites independently of each other and PRC2, although increased variability of the immunostaining after knockdown did suggest that wild-type interactions normally increase or stabilize binding.
Wild-type polytene chromosome immunofluorescence using a polyclonal E(z) antiserum detected ∼25 strong E(z) bands, on average, as well as lower-level staining of all chromosome arms (Fig. 4A). E(z) RNAi caused a loss of both the general signal on all chromosomes and the distinct strong E(z) bands (Fig. 4B). Following Pc knockdown, the distinct E(z) bands were still observable, although their numbers were slightly decreased (Fig. 4C). In contrast, Scm knockdown resulted in the loss of the strongly staining E(z) bands (Fig. 4D). This result suggests that PRC2 distribution is more dependent on Scm than on PRC1, at least in this tissue.
Since PRC2 function on chromatin leads to methylation of H3K27, we proceeded to ask whether H3K27me3 was particularly affected in this tissue. As expected, we found that H3K27me3 was undetectable after E(z) shRNA induction (Supplemental Fig. S7B). Compared with the wild-type control (Supplemental Fig. S7A), a noticeable decrease of H3K27me3 levels was not observed after Pc or Scm knockdown (Supplemental Fig. S7C,D). However, a change in H3K27me3 distribution between wild-type and knockdown of Pc or Scm could be detected, with a stronger effect in the Scm knockdown. Following Scm RNAi, H3K27me3 immunostaining was consistently increased in the heterochromatic chromocenter region, while many of the strongly staining H3K27me3 bands seen in wild type were weakened along with an apparent increase in weakly staining sites (Supplemental Fig. S7E,F). These results suggest that, at least in this tissue, both PRC2 and H3K27me3 are redistributed from their normal patterns after depletion of Scm. However, additional validation experiments such as genome-wide ChIP-seq will be required to understand the nature of this apparent targeting defect.
To extend our analyses to diploid tissue, we expressed an Scm-directed shRNA using an engrailed (en)-GAL4 driver. This resulted in a decrease of anti-Scm immunofluorescence in the posterior compartment of the wing imaginal disc, correlating with expression of a GAL4-dependent GFP reporter (Supplemental Fig. S8A–C). However, we did not observe any difference in H3K27me3 levels between the anterior and posterior sides of Scm-depleted wing discs (Supplemental Fig. S8D–F). In contrast, E(z)-depleted wing disc morphology was significantly compromised and displayed a marked loss of H3K27me3 in the GFP-marked region (Supplemental Fig. S8G–I). This result suggests that Scm does not affect PRC2-dependent H3K27me3 levels in imaginal discs. To confirm this result, we generated clones of Scm homozygous mutant cells in imaginal discs and looked for depletion of H3K27me3 by immunofluorescence, as is seen in E(z)-null mutant clones. Consistent with the RNAi knockdown experiments, depletion of E(z) resulted in a clear loss of H3K27me3 (Fig. 5A–C), whereas no visible difference in H3K27me3 levels was detected in Scm mutant clones (Fig. 5D–F; J Müller, pers. comm.). In the absence of genomic ChIP studies, we cannot evaluate whether loss of a wild-type distribution pattern of H3K27me3 occurs in diploid tissues after Scm depletion. However, combined with our studies of polytene chromosome immunostaining, we can speculate that interaction with Scm is required for proper targeting or stable binding of E(z) rather than for PRC2 histone methyltransferase enzymatic activity.
Figure 5.
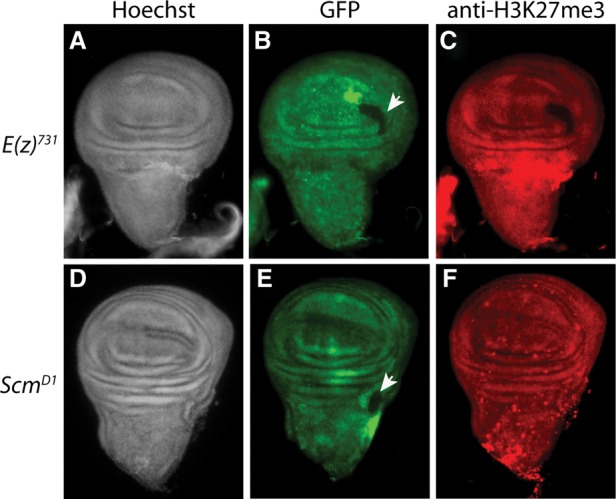
H3K27me3 levels are unaffected in imaginal disc Scm mutant clones. Wing imaginal discs with homozygous clones for E(z)731(A–C) or ScmD1(D–F) were labeled with Hoechst (A,D) and antibody against H3K27me3 (C,F). Homozygous-null mutant clones were distinguished from neighboring wild-type cells by the absence of GFP (arrowheads in B,E). Loss of H3K27me3 was clearly evident in E(z) mutant clones (C), but no change in H3K27me3 immunostaining was detected in Scm mutant clones (F).
Independent Scm-SAM modules bind sporadic and contiguous stretches on polytene chromosomes
Overexpression of the Scm-SAM domain using various GAL4 driver systems causes dominant-negative phenotypes of PcG loss of function (Peterson et al. 2004). In addition, whereas a full-length wild-type transgene rescues Scm mutants, a transgene carrying a point mutation in the SAM domain failed to rescue (Peterson et al. 2004). Furthermore, in a reporter gene assay using transgenic flies, a truncated Scm protein lacking the SAM domain failed to silence the reporter, revealing that the domain is required in Scm for PcG silencing (Roseman et al. 2001). It has been suggested that Scm may participate in the expansion of higher-order chromatin structures resulting from PRC1 function based on the fact that Scm can form a heteropolymer with Ph through the interaction of their SAM domains in vitro (Kim et al. 2005). Thus, one possibility to explain the dominant-negative effect of Scm-SAM overexpression is that the domain interferes with the copolymerization of wild-type Scm and Ph. To test this possibility, we expressed the UAS-SAM transgene specifically in the salivary gland and assessed its consequences by immunostaining of polytene chromosomes for Pc, H3K27me3, E(z), and Scm. Consistent with a competition model, we found that Pc was still retained (Supplemental Fig. S9A,B). H3K27me3 redistribution—including increased chromocenter immunostaining and changes in strong and weak bands (Supplemental Fig. S9C,D)—and the loss of most of the strong E(z) bands (Supplemental Fig. S9E,F) were observed after Scm-SAM overexpression. These results all resemble the effect of Scm knockdown and are consistent with the previously described dominant-negative phenotype. Furthermore, we found that endogenous Scm was no longer detectable using a polyclonal antibody raised against the MBT domain (Fig. 6A,B,D; Grimm et al. 2009). To determine the fate of the overexpressed SAM domain, polytene chromosomes were immunostained with anti-HA antibodies to detect the epitope tag fused to the SAM domain module (Fig. 6A). Based on the competition model, we expected that anti-HA immunostaining would correlate with a Pc pattern, with the SAM domain modules outcompeting endogenous Scm for the interaction with PRC1. Unexpectedly, we found that the HA-SAM domains were frequently localized in sporadic but long contiguous regions on the polytene chromosomes, suggestive of unlimited homopolymerization (Fig. 6C,E). The contiguous stretches were found at apparently random locations, and double staining with anti-Pc showed that SAM domain binding was able to occur in the absence of a strong Pc band (Fig. 6F,G). Our results are consistent with a model in which the Scm-SAM domain is normally involved in homopolymerization and in which other regions of the Scm protein, such as the MBT or zinc finger domains, are required for proper Scm targeting, interaction with PRC2, and regulation of oligomerization (Grimm et al. 2007).
BioTAP-XL links Scm to transcriptional repressors
Because Scm is the only PcG protein to strongly copurify in both Pc- and E(z)- BioTAP-XL mass spectrometry, we tested whether Scm pull-down could reciprocally copurify PRC1 and PRC2 components. Furthermore, we hoped to find novel binding partners that might help explain the potential targeting and spreading roles of Scm in PcG silencing. We performed BioTAP-XL pull-down as before from stable transgenic S2 cell lines and embryos expressing BioTAP-N-Scm. Proteins enriched in Scm pull-downs from both S2 cells and two technical replicates of embryos were identified (Fig. 7). Unlike Pc and E(z) pull-downs, in which the two PcG proteins mainly copurified the components of their respective complexes, the BioTAP-XL analysis of Scm revealed strong copurification of both PRC1 and PRC2 in embryos and S2 cells (Fig. 7C; Supplemental Table S1). The Jarid2 and Jing proteins were notable exceptions. They were strongly purified with E(z) (Table 1B) and known to interact with each other (Herz et al. 2012) but were low or undetectable in the Scm pull-downs, suggesting that they are present in a PRC2 subcomplex separate from Scm. Interestingly, Jigr1 (jing-interacting gene regulatory 1), a protein identified in our Pc pull-down (Table 1A), was identified from genetic screening for enhancers of a jing gain-of-function phenotype in the Drosophila eye (Sun et al. 2006). Therefore, it is intriguing to speculate that a relationship between jigr1 and jing might provide an additional functional connection between PRC1 and PRC2.
Figure 7.

Proteomic analysis of Scm complexes. (A) Volcano plot of proteins identified in the Scm pull-downs and inputs. The vertical dashed line indicates fold change cutoff of the average normalized spectral abundance factors (NSAF) of each protein from input to immunoprecipitation or x = 0.15, while the horizontal dashed line indicates significance cutoff of NSAF difference from input to immunoprecipitation or y = −log10(0.05) = 1.30103. Twenty-three proteins (pink) or <1% of the total protein data set fell within both cutoffs and represent the most significantly enriched proteins in the Scm pull-downs. (B) Zoomed in region of the most enriched proteins from A. (C) Total peptide counts of Scm-specific interactors from S2 tissue culture cells and Drosophila embryos along with their counts in input. Proteins are color-coded by known molecular complex, and the asterisk indicates the bait protein used for pull-down. See Supplemental Table S3 for the full range of data.
In addition to the canonical PRC1 and PRC2 components, we observed high enrichment of the PhoRC complex in the Scm pull-down (e.g., 27 peptides of Sfmbt and 18 peptides of Pho in S2 cells, with similar results in embryos). In contrast, we found much lower and asymmetric recovery of Sfmbt and Pho in the Pc (Sfmbt > Pho) and E(z) (Pho > Sfmbt) experiments (Supplemental Table S1). Pho, the ortholog of mammalian YY1, is a DNA-binding protein that is found at many PREs (Brown et al. 1998, 2003; Busturia et al. 2001; Mishra et al. 2001; Kwong et al. 2008; Oktaba et al. 2008). Therefore, our results may suggest a mechanism for Scm to connect PRC1 and PRC2 to DNA through PhoRC. It should be noted, however, that Pho is also localized to many active genes, so the specific context that defines PREs still remains poorly understood (Schuettengruber et al. 2009).
In addition to Sfmbt, Scm BioTAP-XL copurified another MBT domain protein [dL(3)mbt] and additional LINT complex components (Lint-1 and CoRest) (Meier et al. 2012), suggesting that all three MBT domain proteins encoded in Drosophila may functionally interact. Surprisingly, G9a was one of the top hits in the Scm pull-down. G9a, as a SET domain protein, functions in monomethylation or dimethylation of H3K9 (H3K9me1 or H3K9me2) in euchromatin and is an essential protein for gene silencing in mammals (Tachibana et al. 2002, 2005; Smallwood et al. 2007; Mozzetta et al. 2014). However, in Drosophila, G9a is dispensable for H3K9 methylation, and null mutants are surprisingly viable and fertile (Seum et al. 2007; Figueiredo et al. 2012). Nevertheless, it still remains likely that G9a participates in gene silencing in Drosophila, as G9a acts as a component of the CtBP repressor complex in mammals (Shi et al. 2003), and we noted that the Drosophila orthologs of CtBP repressor complex components [G9a, CoRest, Rpd3, Su(var)3-3, and CtBP] were all identified in the Scm mass spectrometry (Supplemental Table S1). Furthermore, two zinc finger proteins—CG9932, containing a CtBP PXDLS motif (PTDLSQK), and peb (pebbled), the Drosophila ortholog of RREB1—copurified in S2 cells and/or embryos, suggesting that they could play a role analogous to zinc finger protein components of the mammalian CtBP repressor complex, such as ZNF217 and RREB1 (Shi et al. 2003; Quinlan et al. 2006; Flajollet et al. 2009; Ray et al. 2014). Taken together, Scm may function as an important mediator to coordinate not only PRC1, PRC2, and PhoRC but also additional repressor complexes.
Discussion
The role of Scm in the spreading of PcG silencing
One of the most interesting properties of chromatin modification is the ability, under certain circumstances, to propagate in cis independent of sequence (Gelbart and Kuroda 2009; Moazed 2011). This ability to “spread” may be important for the inheritance of chromatin states initially established through interactions at nucleation sites such as PREs. SAM domain-mediated polymerization is therefore an attractive model to explain the propagation of PcG silencing (Kim et al. 2002, 2005; Peterson et al. 2004). From that perspective, Ph, one of the core components of PRC1, may be responsible for the spreading of PRC1 through Ph-SAM polymerization (Robinson et al. 2012; Isono et al. 2013). The compact chromatin environment formed by PRC1 spreading may improve the enzymatic activity of PRC2 (Yuan et al. 2012), and the capacity of the Pc chromodomain to interact with H3K27me3 may also contribute to the synergistic spreading of PcG silencing. However, consistent with the identification of PRC1 and PRC2 as distinct complexes that purify independently, E(z) RNAi does not significantly affect binding patterns of Pc on polytene chromosomes (Supplemental Fig. S6F), and E(z) binding is likewise still detected after Pc RNAi (Fig. 4C). In this study, we found that Scm directly interacts with the PRC2 complex and colocalizes with H3K27me3 in genome-wide analyses. Scm RNAi results in the loss of major sites of E(z) binding and the redistribution of H3K27me3 on polytene chromosomes. In addition, overexpression of the Scm-SAM domain interferes with binding of endogenous Scm to chromosomes and appears to self-polymerize for long distances on polytene chromosomes independently of PRC1 and PRC2. Taken together, we suggest that the interaction of Scm with PRC2 and polymerization by the Scm-SAM domain may be key factors contributing to PRC2 and H3K27me3 spreading (Fig. 8).
Figure 8.
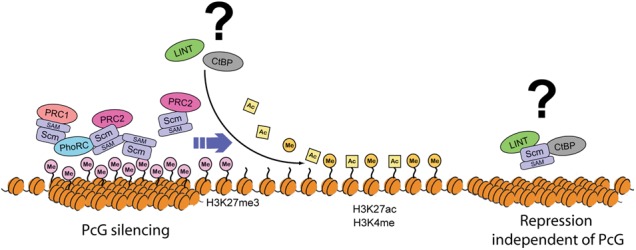
Model for PcG spreading facilitated by Scm. Interaction of Scm with multiple PcG complexes and the polymerization ability of the Scm-SAM domain lead to sequential extension of PcG silencing from initial target sites to neighboring regions. Transient or temporal interaction of LINT or CtBP repressor complexes with Scm may support spreading of PcG silencing through the removal of active histone marks. Alternatively, our results do not exclude the possibility that the interaction of Scm with LINT or CtBP repressor complexes result in separate repression processes independent of PcG silencing.
Despite the loss of strong sites of E(z) binding after Scm RNAi, H3K27me3 is still clearly present at many sites on polytene chromosomes, and the overall level of H3K27me3 in imaginal disc cells is not visibly altered by Scm knockdown or knockout. It is also noteworthy that E(z) RNAi results in not only the loss of strong sites of E(z) binding but also a decrease in overall signal across the chromosomes. These observations imply that PRC2 is still able to catalyze the formation of the H3K27me3-repressive histone modification in the absence of Scm. We speculate that PRC2 propagation through interaction with Scm may locally reinforce the repressive H3K27me3 histone mark to ensure silencing of specific spatiotemporally sensitive target genes.
Although Scm had previously been known as a component of PRC1, core components of PRC1 without Scm are sufficient to inhibit chromatin remodeling and transcription in vitro (Francis et al. 2001; King et al. 2002), suggesting that Scm does not need to be involved directly in the silencing roles of PRC1. Also, we did not observe strong effects on Pc binding after Scm knockdown; rather, our results show that Scm plays an especially important role for PRC2 targeting. Nevertheless, strong silencing of a mini-white reporter gene resulting from tethered Scm is disrupted by mutation of Ph (Roseman et al. 2001), indicating that the silencing achieved by Scm requires the capacity to interact with PRC1.
Overexpression of the Scm-SAM domain on polytene chromosomes revealed its localization in contiguous stretches apparently unlinked from PRC1, suggesting the formation of stable Scm-SAM homopolymers in vivo. This observation also implies that regions of the Scm protein outside of the SAM domain are required for the proper targeting and regulation of SAM-mediated polymerization. These are likely to include the zinc fingers, MBT histone interaction domains, and sequences that govern sumoylation of Scm (Bornemann et al. 1998; Smith et al. 2011).
Linking the PcG to additional repressive complexes
The strong interaction that we discovered between Scm and the G9a SET domain protein, an H3K9 methyltransferase, suggests a new link between H3K27 and H3K9 methylation in Drosophila. These classical histone marks, associated with silent chromatin, were once thought to be largely distinct but are now proposed to have a functional relationship in PcG silencing in mammals, most notably in X inactivation (Rougeulle et al. 2004; Escamilla-Del-Arenal et al. 2013). There was also evidence for colocalization of these two marks in early ChIP analyses at the HOX gene Ubx in imaginal discs (Papp and Muller 2006). G9a may also play a role in regulation of H3K27 methylation (Mozzetta et al. 2014). Alternatively, the abundance of G9a may reflect a key role for the CtBP corepressor complex in PcG function rather than for G9a itself, which is a nonessential gene. Genome-wide binding profile analyses have shown that the components of the CtBP complex such as Su(var)3-3 (LSD1) and Rpd3 (HDAC1) are mainly enriched on active genes rather than repressed genes in human cells (Wang et al. 2009; Zhang et al. 2013), and L(3)mbt, one component of the LINT complex, colocalizes with insulator proteins, including CP190 and mod(mdg4), rather than with PcG proteins in Drosophila (Richter et al. 2011). Therefore, the possibility that the interactions of Scm with CtBP or LINT repressor complexes occur as independent complexes irrelevant to PcG silencing cannot be excluded (Fig. 8). However, considering that PcG silencing could require dynamic interactions during development, components of these repressor complexes may not be permanently stationed in PcG silenced domains but rather participate in PcG silencing transiently. Furthermore, some of the CtBP subunits were copurified in our Pc and E(z) affinity purifications (Supplemental Table S1), and previous studies reported that CtBP complex components can contribute to PcG silencing in Drosophila. For example, CtBP mutation causes the loss of Pc recruitment to many PREs (Atchison et al. 2003; Srinivasan and Atchison 2004; Basu and Atchison 2010). Furthermore, Rpd3 deacetylates H3K27ac, which is mutually exclusive with H3K27me3 (Tie et al. 2009), and Su(var)3-3 demethylates H3K4me1 and H3K4me2 (Rudolph et al. 2007), which are active marks linked to Trithorax (Trx) activity and H3K27ac (Tie et al. 2014).
How PcG complexes find PREs and spread to create repressive domains is not known on a mechanistic level. Perhaps repressor complexes such as CtBP help remove active chromatin marks to attract the PcG initially or enable cycles of spreading to maintain those domains (Fig. 8). Future analyses will entail dissecting the direct interactions of Scm, including nucleosomes and their post-translational modifications. Furthermore, through iterative use of BioTAP-XL, the wealth of additional candidates in the Pc, E(z), and Scm pull-downs featured in this study will be invaluable in extending our understanding of chromatin-based PcG repression.
Materials and methods
BioTAP-tagged transgene constructs and transgenesis
The pFly vector was used as the backbone to construct all transgenes (Wang et al. 2013). A 2.4-kb genomic fragment containing the promoter, coding, and intron regions of the Pc gene was amplified from BAC RP98-2C22 and inserted between NotI and AscI sites of pFly followed by insertion of C-BioTAP from pCAndy-Bio-2xProteinA (Alekseyenko et al. 2014b) between AscI and NheI sites to create pFly-Pc-C-BioTAP. The α-tubulin-NTAP-E(z) CaSpeR plasmid was kindly provided by J. Müller (Nekrasov et al. 2007). The α-tubulin 1 promoter fragment and E(z) cDNA fragment were transferred separately into pFly, and the BioTAP region (N-BioTAP) from pCAndy-1xProteinA-Bio was placed between the α-tubulin 1 promoter and E(z) cDNA fragments [pFly-BioTAP-N-E(z)]. In the case of Scm, the N-terminal BioTAP tag was first introduced into BAC RP98-19N7 using the Counter-Selection BAC modification kit (Gene Bridges). The 7.8-kb genomic fragment containing the BioTAP-N-Scm gene and its upstream and downstream regions was then recombineered into the pFly vector (pFly-BioTAP-N-Scm). Maps of all BioTAP-fused constructs and BAC counterselection details are available on request. The transgenes were injected into PhiC31 integrase-containing attP-docking site embryos (Bloomington Drosophila Stock Center [BDSC] no. 9736; cytogenetic map: 53B2). The pFly-Pc-C-BioTAP and pFly-BioTAP-N-Scm constructs were also cotransfected with a hygromycin-resistant vector at a 10:1 ratio into S2 cells using a calcium phosphate transfection kit (Invitrogen), and stable S2 cell lines were selected by adding hygromycin B (Invitrogen) to a final concentration of 300 µg/mL into the Schneider's Drosophila medium.
BioTAP-XL purification from embryos and S2 cells
We followed the BioTAP-XL protocol as described in Alekseyenko et al. (2015). In brief, embryos of BioTAP transgenic fly lines [Pc-C-BioTAP, BioTAP-N-E(z) and BioTAP-N-Scm] were collected between 12 and 24 h after fertilization and stored for up to 3 d at 4°C. Every 3 d, embryonic nuclei were cross-linked, and extracts were prepared as described, snap-frozen with liquid nitrogen, and stored at −80°C. These steps were repeated until extracts from ∼50 g of embryos were pooled. Stable S2 cell lines expressing BioTAP transgenes (Pc-C-BioTAP and BioTAP-N-Scm) were incubated in four 2.8-L Fernbach glass flasks at 90 rpm and 26.5°C. In each flask, cells were grown in 500 mL of HyClone CCM3 serum-free medium to a density of ∼1 × 107 cells per milliliter. Cross-linked nuclear extracts from S2 cell lines were prepared from ∼2 × 1010 cells grown in four flasks. After sonication, TAP was performed to isolate the BioTAP-tagged bait along with its protein interaction partners and associated genomic DNA. Interacting proteins were identified by on-bead trypsinization of bound complexes followed by LC-MS/MS of the resulting peptides. Genomic localization was determined by high-throughput sequencing of libraries generated from the tandem affinity-purified material using the NEBNext ChIP-seq library preparation master mix set for Illumina (New England Biolabs, catalog no. E6240S) and TruSeq adaptors (Illumina).
Fly genetics (rescue tests, RNAi knockdowns, null mutant clones, and overexpression of the SAM domain)
The crossing schemes for viability rescue tests of BioTAP fusion transgenes are described in Supplemental Figure S1D. The rescue test for temperature-sensitive E(z)12 and E(z)61 transheterozygotic mutants was performed at 29°C; all other rescue tests were performed at 25°C. Rescued adult flies were assessed by the absence of balancer markers and the expression of the mini-white marker incorporated into the transgenic construct, and all transheterozygotic mutants were lethal in the absence of BioTAP-tagged transgenes. Pc and E(z) alleles were kindly provided by Dr. W. Bender. For RNAi knockdown in imaginal discs, the en-GAL4; UAS-GFP line, kindly provided by Dr. N. Perrimon, was crossed with Scm (BDSC no. 35389) and E(z) (BDSC no. 33659) Transgenic RNAi Project (TRiP) lines. For salivary gland-specific RNAi knockdown, the P{GawB}c729 line (BDSC no. 6983, kindly shared with us by Dr. J. Kassis), which expresses Gal4 in the salivary glands, was crossed with TRiP lines [Pc: BDSC no. 36070; E(z): BDSC no. 33659; Scm: BDSC no. 35389]. For generation of ScmD1 and E(z)731clones, w; FRT82B ScmD1/TM6C and w; E(z)731 FRT2A/TM6C fly lines were crossed with the yw hs-flp; FRT82B hs-nGFP line and w hs-flp; hs-nGFP FRT2A line, respectively. The subsequent procedure of heat-shock treatment was performed as described in Beuchle et al. (2001). These fly strains were kindly provided by Dr. J. Müller. Fly stocks bearing UAS-SAM domain constructs on both the second and third chromosomes, kindly provided by Dr. J. Simon (Peterson et al. 2004), were crossed with the P{GawB}c729 line for salivary gland-specific overexpression of the SAM domain.
Immunostaining of imaginal disc tissues and polytene chromosomes
The imaginal disc tissues dissected from third instar larvae were fixed with 1 mL of 4% formaldehyde in PBST and labeled with rabbit antibodies against H3K27me3 (1:1000 dilution; Cell Signaling) or Scm (1:1000 dilution; a gift from J. Müller). Donkey anti-rabbit Alexa fluor 594 (1:500 dilution) was used as secondary antibody, and imaginal discs were mounted in Prolong Gold (Invitrogen) prior to imaging. Salivary glands from third instar larvae were fixed for 1 min in fixation solution #1 (1% Triton X-100, 4% formaldehyde in PBS) followed by fixation for 2 min in fixation solution #2 (50% acetic acid, 4% formaldehyde) prior to squashing to spread the polytene chromosomes. Primary antibodies used for immunostaining included rabbit antibodies against Pc (1:100 dilution; Santa Cruz Biotechnology), H3K27me3 (1:100 dilution; Cell Signaling), Scm (1:100 dilution), and E(z) (1:200 dilution; both gifts from J Müller,); rabbit peroxidase anti-peroxidase (PAP) antibody (1:100 dilution; Sigma) for detection of BioTAP fusion proteins [Pc-C-BioTAP, BioTAP-N-E(z), and BioTAP-N-Scm]; and mouse antibody against HA (1:100 dilution; Abcam) for detection of the HA-tagged Scm-SAM domain module. Chicken anti-rabbit Alexa fluor 488, donkey anti-rabbit Alexa fluor 594, and donkey anti-mouse Alexa fluor 488 (1:500 dilution) were used as secondary antibodies. Samples were mounted in Prolong Gold (Invitrogen) prior to imaging.
Western blots
Embryos of BioTAP transgenic flies were collected and dechorionated by immersion for 3 min in 50% bleach. The embryos were rinsed with distilled water and blot-dried. Embryos (0.2 g) were suspended in 150 µL of nuclear extraction buffer (10% sucrose, 20 mM HEPES at pH 7.6, 10 mM NaCl, 3 mM MgCl2, 0.1% Triton X-100) with 0.1 mM PMSF in a 1.5-mL tube and homogenized on ice with a motorized pestle. Nuclear extraction buffer was added to the homogenate up to 1 mL and centrifuged at 2000g for 5 min at 4°C. Supernatant was removed, and the pellet was resuspended in 150 µL of nuclear extraction buffer. Homogenization and centrifugation steps were repeated. The final crude nuclear pellet was resuspended in 600 µL of Novex Tris-glycine SDS sample buffer (Invitrogen), including NuPAGE sample-reducing agent (Invitrogen), and then boiled for 10 min. Twenty microliters was used for each Western blot. PAP antibody (Sigma) against the Protein A epitope (1:1000 dilution) and Clarity Western ECL substrate (Bio-Rad) were used for the detection of BioTAP fusion proteins.
Preparation of baculovirus-infected nuclear extracts and Flag purification
Baculovirus constructs and viruses were the generous gifts of R. Kingston (Muller et al. 2002). The Scm baculovirus was constructed by insertion of a full-length cDNA from Drosophila Genomics Resource Center stock RE16782 into pFastBac1. Viral stocks were individually reamplified in T-225 flasks for 5–7 d in Insect-XPRESS medium (Lonza) supplemented with 10% FBS (Sigma) prior to use in protein expression experiments. For protein coexpression infections, baculoviruses were mixed at a ratio of 1:1:1:0.5:0.4 for Scm:E(z):Suz12:Nurf55:Flag-Esc and incubated for 72 h at 26.7°C in 500-mL total volume (starting cell density = 2 × 106 Sf9 cells per milliliter). Extracts were made as previously described (Ito et al. 1999) with the following modification. Instead of a half-pellet volume during salt extraction, a 5× pellet volume of low-salt buffer was used, followed by the addition of high-salt buffer to a final concentration of 300 mM KCl. Flag purification was performed as previously described (Francis et al. 2001; Muller et al. 2002). For each Flag purification, 50–60 µL of M2 anti-Flag agarose resin or magnetic beads (Sigma) was washed with BC300N (20 mM HEPES at pH 7.9, 0.2 mM EDTA, 20% glycerol, 300 mM KCl, 0.05% NP40) supplemented with protease inhibitors and PMSF. After washing, beads were incubated overnight with 1 mL of the appropriate extract. Beads and bound complexes were captured on a magnetic stand for 2 min or by centrifugation at 1000g for 2 min at 4°C. Bound complexes were subjected to a series of wash steps of BC-N buffer with increasing KCl concentrations, with a maximum of 2 M KCl. Washes were for 5 min each with end-over-end rotation at 4°C followed by recapture as above. Bound complexes were eluted with 100 µL of 0.4 mg/mL Flag peptide in BC300N for 1–1.5 h at 4°C. Typically, three elutions were taken. Protein content of elution fractions was analyzed by silver staining of SDS-PAGE gels, and the presence of Scm was further probed by Western blotting using anti-Scm (a gift from J. Simon).
Volcano and scatter plot analysis
To identify the most significantly enriched proteins by the BioTAP-tagged fusion proteins, the natural logarithm of the normalized spectral abundance factor (NSAF) was determined to calculate enrichment ratios (pull-down/input) for each identified protein (Zybailov et al. 2006). To allow the calculation for nonabundant proteins not recovered in the input, a peptide pseudocount of 0.5 was introduced for proteins not identified in any given experiment and thus containing zero peptide counts in order to avoid taking the logarithm of 0. For volcano plot, an unpaired two-sample two-tailed t-test was used to compare the ln(NSAF) values of a given protein averaged across the three Scm immunoprecipitations and averaged across the three inputs (one S2 cell input and two biological replicate inputs of embryos).
BioTAP-XL ChIP-seq analyses
Sequencing reads were aligned to the fly genome fb5_22 using Bowtie version 0.12.7 with the unique option enabled (Langmead et al. 2009). For paired-end reads, 1000 base pairs (bp) of insertion was used. Consistency of replicates was checked by genome-wide correlation analysis and visual inspection. The smoothed fold enrichment profiles at log2 scale were calculated using the get.smoothed.enrichment.mle function (window size of 150 bp for the Gaussian kernel function, and step size of 50 bp) in the SPP R package (Kharchenko et al. 2008). The fly genome dm3 annotation was used for all analyses. For H3K27me3 and H3K36me3 data in embryos, aligned ChIP-seq reads were downloaded from modENCODE (http://data.modencode.org/cgi-bin/findFiles.cgi?download=3955; ID 3955) and processed as described above. For the genome-wide correlation analysis of BioTAP-tagged proteins, H3K27me3, and H3K36me3 in embryos (Supplemental Fig. S3), the log2 fold enrichment values were averaged into 1-kb bins. For heat map generation (Fig. 3B), Scm peaks were identified in the BioTAP-N-Scm data using MACS 1.4 with default parameters (Zhang et al. 2008). Peak summit locations were centered in 4-kb windows, and each row was ordered by Scm intensity. For comparison of binding profiles between H3K27me3 and BioTAP-tagged proteins in S2 cells (Supplemental Fig. S4A), H3K27me3 M-values and peak calling results were downloaded from modENCODE (http://data.modencode.org/cgi-bin/findFiles.cgi?download=298; ID 298). For correlation analysis between BioTAP-N-Scm and other factors in S2 cells (Supplemental Fig. S4B), log2 M-values for other factors (ChIP–chip) in S2 were downloaded from modENCODE (http://data.modencode.org). To calculate correlations, Scm enrichment values (sequencing) were interpolated for the array probe positions.
Accession numbers
ChIP-seq data have been submitted to the NCBI Gene Expression Omnibus public repository under the accession number GSE66183.
Supplementary Material
Acknowledgments
We are grateful to R. Tomaino (Taplin Mass Spectrometry Facility, Harvard Medical School) for helpful advice, and J. Kassis, R. Kingston, J. Müller, J. Simon, and W. Bender for generous sharing of advice, fly stocks, baculoviruses, and antibodies. We thank the TRiP at Harvard Medical School (National Institutes of Health/National Institute of General Medical Sciences R01-GM084947) and the BDSC (National Institutes of Health P40OD018537) for providing fly stocks and/or DNA constructs used in this study. We are grateful to J. Al Haj-Abed for valuable help with data analyses and, along with J. Haswell and H. Wallace, critical reading of the manuscript. This work was supported by the National Institutes of Health (GM101958 to M.I.K.) and a post-doctoral fellowship from the Jane Coffin Child Memorial Fund to B.M.Z.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.260562.115.
References
- Alekseyenko AA, Gorchakov AA, Kharchenko PV, Kuroda MI. 2014a. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci 111: 2488–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Gorchakov AA, Zee BM, Fuchs SM, Kharchenko PV, Kuroda MI. 2014b. Heterochromatin-associated interactions of Drosophila HP1a with dADD1, HIPP1, and repetitive RNAs. Genes Dev 28: 1445–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, McElroy KA, Kang H, Zee BM, Kharchenko PV, Kuroda MI. 2015. BioTAP-XL: cross-linking/tandem affinity purification to study DNA targets, RNA, and protein components of chromatin-associated complexes. Curr Protoc Mol Biol 109: 21.30.1–21.30.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchison L, Ghias A, Wilkinson F, Bonini N, Atchison ML. 2003. Transcription factor YY1 functions as a PcG protein in vivo. EMBO J 22: 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Atchison ML. 2010. CtBP levels control intergenic transcripts, PHO/YY1 DNA binding, and PcG recruitment to DNA. J Cell Biochem 110: 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuchle D, Struhl G, Muller J. 2001. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development 128: 993–1004. [DOI] [PubMed] [Google Scholar]
- Bornemann D, Miller E, Simon J. 1996. The Drosophila Polycomb group gene sex comb on midleg (Scm) encodes a zinc finger protein with similarity to polyhomeotic protein. Development 122: 1621–1630. [DOI] [PubMed] [Google Scholar]
- Bornemann D, Miller E, Simon J. 1998. Expression and properties of wild-type and mutant forms of the Drosophila sex comb on midleg (SCM) repressor protein. Genetics 150: 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen TR, Duncan IM. 1986. Maternal expression of genes that regulate the bithorax complex of Drosophila melanogaster. Dev Biol 118: 442–456. [DOI] [PubMed] [Google Scholar]
- Brown JL, Mucci D, Whiteley M, Dirksen ML, Kassis JA. 1998. The Drosophila Polycomb group gene pleiohomeotic encodes a DNA binding protein with homology to the transcription factor YY1. Mol Cell 1: 1057–1064. [DOI] [PubMed] [Google Scholar]
- Brown JL, Fritsch C, Mueller J, Kassis JA. 2003. The Drosophila pho-like gene encodes a YY1-related DNA binding protein that is redundant with pleiohomeotic in homeotic gene silencing. Development 130: 285–294. [DOI] [PubMed] [Google Scholar]
- Busturia A, Lloyd A, Bejarano F, Zavortink M, Xin H, Sakonju S. 2001. The MCP silencer of the Drosophila Abd-B gene requires both Pleiohomeotic and GAGA factor for the maintenance of repression. Development 128: 2163–2173. [DOI] [PubMed] [Google Scholar]
- Campbell S, Ismail IH, Young LC, Poirier GG, Hendzel MJ. 2013. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell cycle 12: 2675–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. [DOI] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. 2010. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci 107: 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. 2012. Molecular architecture of human polycomb repressive complex 2. Elife 1: e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. 2002. Drosophila Enhancer of zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185–196. [DOI] [PubMed] [Google Scholar]
- De Piccoli G, Cortes-Ledesma F, Ira G, Torres-Rosell J, Uhle S, Farmer S, Hwang JY, Machin F, Ceschia A, McAleenan A, et al. 2006. Smc5–Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat Cell Biol 8: 1032–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digan ME, Haynes SR, Mozer BA, Dawid IB, Forquignon F, Gans M. 1986. Genetic and molecular analysis of fs(1)h, a maternal effect homeotic gene in Drosophila. Dev Biol 114: 161–169. [DOI] [PubMed] [Google Scholar]
- Escamilla-Del-Arenal M, da Rocha ST, Spruijt CG, Masui O, Renaud O, Smits AH, Margueron R, Vermeulen M, Heard E. 2013. Cdyl, a new partner of the inactive X chromosome and potential reader of H3K27me3 and H3K9me2. Mol Cell Biol 33: 5005–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo ML, Philip P, Stenberg P, Larsson J. 2012. HP1a recruitment to promoters is independent of H3K9 methylation in Drosophila melanogaster. PLoS Genet 8: e1003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flajollet S, Poras I, Carosella ED, Moreau P. 2009. RREB-1 is a transcriptional repressor of HLA-G. J Immunol 183: 6948–6959. [DOI] [PubMed] [Google Scholar]
- Francis NJ, Saurin AJ, Shao Z, Kingston RE. 2001. Reconstitution of a functional core polycomb repressive complex. Mol Cell 8: 545–556. [DOI] [PubMed] [Google Scholar]
- Fujioka Y, Kimata Y, Nomaguchi K, Watanabe K, Kohno K. 2002. Identification of a novel non-structural maintenance of chromosomes (SMC) component of the SMC5–SMC6 complex involved in DNA repair. J Biol Chem 277: 21585–21591. [DOI] [PubMed] [Google Scholar]
- Gelbart ME, Kuroda MI. 2009. Drosophila dosage compensation: a complex voyage to the X chromosome. Development 136: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieni RS, Hendzel MJ. 2009. Polycomb group protein gene silencing, non-coding RNA, stem cells, and cancer. Biochem Cell Biol 87: 711–746. [DOI] [PubMed] [Google Scholar]
- Grimm C, de Ayala Alonso AG, Rybin V, Steuerwald U, Ly-Hartig N, Fischle W, Muller J, Muller CW. 2007. Structural and functional analyses of methyl-lysine binding by the malignant brain tumour repeat protein Sex comb on midleg. EMBO Rep 8: 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Matos R, Ly-Hartig N, Steuerwald U, Lindner D, Rybin V, Muller J, Muller CW. 2009. Molecular recognition of histone lysine methylation by the Polycomb group repressor dSfmbt. EMBO J 28: 1965–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SH, Sheedy DM, Cuddihy AR, O'Connell MJ. 2004. Coordination of DNA damage responses via the Smc5/Smc6 complex. Mol Cell Biol 24: 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garrett AS, Miller C, Casto D, Zhang Y, Seidel C, Haug JS, Florens L, Washburn MP, et al. 2012. Polycomb repressive complex 2-dependent and -independent functions of Jarid2 in transcriptional regulation in Drosophila. Mol Cell Biol 32: 1683–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Paulson A, Dutta A, Venkatesh S, Smolle M, Abmayr SM, Workman JL. 2014. Histone acetyltransferase Enok regulates oocyte polarization by promoting expression of the actin nucleation factor spire. Genes Dev 28: 2750–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW. 1984. A gene that regulates the bithorax complex differentially in larval and adult cells of Drosophila. Cell 37: 815–823. [DOI] [PubMed] [Google Scholar]
- Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, Ishikura T, Toyoda T, Bernstein BE, Koseki H. 2013. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell 26: 565–577. [DOI] [PubMed] [Google Scholar]
- Ito T, Levenstein ME, Fyodorov DV, Kutach AK, Kobayashi R, Kadonaga JT. 1999. ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev 13: 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19: 523–534. [DOI] [PubMed] [Google Scholar]
- Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M, Muller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21: 569–571. [DOI] [PubMed] [Google Scholar]
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun HW, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D, et al. 2014. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 21: 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ. 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CA, Gingery M, Pilpa RM, Bowie JU. 2002. The SAM domain of polyhomeotic forms a helical polymer. Nat Struct Biol 9: 453–457. [DOI] [PubMed] [Google Scholar]
- Kim CA, Sawaya MR, Cascio D, Kim W, Bowie JU. 2005. Structural organization of a Sex-comb-on-midleg/polyhomeotic copolymer. J Biol Chem 280: 27769–27775. [DOI] [PubMed] [Google Scholar]
- Kim H, Kang K, Kim J. 2009. AEBP2 as a potential targeting protein for Polycomb Repression Complex PRC2. Nucleic Acids Res 37: 2940–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Francis NJ, Kingston RE. 2002. Native and recombinant polycomb group complexes establish a selective block to template accessibility to repress transcription in vitro. Mol Cell Biol 22: 7919–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymenko T, Papp B, Fischle W, Kocher T, Schelder M, Fritsch C, Wild B, Wilm M, Muller J. 2006. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes Dev 20: 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong C, Adryan B, Bell I, Meadows L, Russell S, Manak JR, White R. 2008. Stability and dynamics of polycomb target sites in Drosophila development. PLoS Genet 4: e1000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarou A, Mohd-Sarip A, Moshkin YM, Chalkley GE, Bezstarosti K, Demmers JA, Verrijzer CP. 2008. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev 22: 2799–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276: 565–570. [DOI] [PubMed] [Google Scholar]
- Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. 2010. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev 24: 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier K, Mathieu EL, Finkernagel F, Reuter LM, Scharfe M, Doehlemann G, Jarek M, Brehm A. 2012. LINT, a novel dL(3)mbt-containing complex, represses malignant brain tumour signature genes. PLoS Genet 8: e1002676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra RK, Mihaly J, Barges S, Spierer A, Karch F, Hagstrom K, Schweinsberg SE, Schedl P. 2001. The iab-7 polycomb response element maps to a nucleosome-free region of chromatin and requires both GAGA and pleiohomeotic for silencing activity. Mol Cell Biol 21: 1311–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D. 2011. Mechanisms for the inheritance of chromatin states. Cell 146: 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, Margueron R, Ait-Si-Ali S. 2014. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 53: 277–289. [DOI] [PubMed] [Google Scholar]
- Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197–208. [DOI] [PubMed] [Google Scholar]
- Negre N, Hennetin J, Sun LV, Lavrov S, Bellis M, White KP, Cavalli G. 2006. Chromosomal distribution of PcG proteins during Drosophila development. PLoS Biol 4: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov M, Klymenko T, Fraterman S, Papp B, Oktaba K, Kocher T, Cohen A, Stunnenberg HG, Wilm M, Muller J. 2007. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J 26: 4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell S, Wang L, Robert S, Jones CA, Saint R, Jones RS. 2001. Polycomblike PHD fingers mediate conserved interaction with enhancer of zeste protein. J Biol Chem 276: 43065–43073. [DOI] [PubMed] [Google Scholar]
- Oktaba K, Gutierrez L, Gagneur J, Girardot C, Sengupta AK, Furlong EE, Muller J. 2008. Dynamic regulation by polycomb group protein complexes controls pattern formation and the cell cycle in Drosophila. Dev Cell 15: 877–889. [DOI] [PubMed] [Google Scholar]
- Papp B, Muller J. 2006. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev 20: 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier N, Champagne N, Stifani S, Yang XJ. 2002. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene 21: 2729–2740. [DOI] [PubMed] [Google Scholar]
- Peterson AJ, Kyba M, Bornemann D, Morgan K, Brock HW, Simon J. 1997. A domain shared by the Polycomb group proteins Scm and ph mediates heterotypic and homotypic interactions. Mol Cell Biol 17: 6683–6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson AJ, Mallin DR, Francis NJ, Ketel CS, Stamm J, Voeller RK, Kingston RE, Simon JA. 2004. Requirement for sex comb on midleg protein interactions in Drosophila polycomb group repression. Genetics 167: 1225–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan KG, Nardini M, Verger A, Francescato P, Yaswen P, Corda D, Bolognesi M, Crossley M. 2006. Specific recognition of ZNF217 and other zinc finger proteins at a surface groove of C-terminal binding proteins. Mol Cell Biol 26: 8159–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray SK, Li HJ, Metzger E, Schule R, Leiter AB. 2014. CtBP and associated LSD1 are required for transcriptional activation by NeuroD1 in gastrointestinal endocrine cells. Mol Cell Biol 34: 2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C, Oktaba K, Steinmann J, Muller J, Knoblich JA. 2011. The tumour suppressor L(3)mbt inhibits neuroepithelial proliferation and acts on insulator elements. Nat Cell Biol 13: 1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riising EM, Boggio R, Chiocca S, Helin K, Pasini D. 2008. The polycomb repressive complex 2 is a potential target of SUMO modifications. PLoS One 3: e2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AK, Leal BZ, Chadwell LV, Wang R, Ilangovan U, Kaur Y, Junco SE, Schirf V, Osmulski PA, Gaczynska M, et al. 2012. The growth-suppressive function of the polycomb group protein polyhomeotic is mediated by polymerization of its sterile α motif (SAM) domain. J Biol Chem 287: 8702–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman RR, Morgan K, Mallin DR, Roberson R, Parnell TJ, Bornemann DJ, Simon JA, Geyer PK. 2001. Long-range repression by multiple polycomb group (PcG) proteins targeted by fusion to a defined DNA-binding domain in Drosophila. Genetics 158: 291–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeulle C, Chaumeil J, Sarma K, Allis CD, Reinberg D, Avner P, Heard E. 2004. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol Cell Biol 24: 5475–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph T, Yonezawa M, Lein S, Heidrich K, Kubicek S, Schafer C, Phalke S, Walther M, Schmidt A, Jenuwein T, et al. 2007. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol Cell 26: 103–115. [DOI] [PubMed] [Google Scholar]
- Santamaria P, Randsholt NB. 1995. Characterization of a region of the X chromosome of Drosophila including multi sex combs (mxc), a Polycomb group gene which also functions as a tumour suppressor. Mol Gen Genet 246: 282–290. [DOI] [PubMed] [Google Scholar]
- Saurin AJ, Shao Z, Erdjument-Bromage H, Tempst P, Kingston RE. 2001. A Drosophila Polycomb group complex includes zeste and dTAFII proteins. Nature 412: 655–660. [DOI] [PubMed] [Google Scholar]
- Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J. 2010. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465: 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Ganapathi M, Leblanc B, Portoso M, Jaschek R, Tolhuis B, van Lohuizen M, Tanay A, Cavalli G. 2009. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol 7: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. 2013. A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet 14: 853–864. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, Pirrotta V. 2006. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet 38: 700–705. [DOI] [PubMed] [Google Scholar]
- Sergeant J, Taylor E, Palecek J, Fousteri M, Andrews EA, Sweeney S, Shinagawa H, Watts FZ, Lehmann AR. 2005. Composition and architecture of the Schizosaccharomyces pombe Rad18 (Smc5–6) complex. Mol Cell Biol 25: 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seum C, Bontron S, Reo E, Delattre M, Spierer P. 2007. Drosophila G9a is a nonessential gene. Genetics 177: 1955–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, Kingston RE. 1999. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 98: 37–46. [DOI] [PubMed] [Google Scholar]
- Shearn A. 1989. The ash-1, ash-2 and trithorax genes of Drosophila melanogaster are functionally related. Genetics 121: 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. 2003. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422: 735–738. [DOI] [PubMed] [Google Scholar]
- Simon J, Chiang A, Bender W. 1992. Ten different Polycomb group genes are required for spatial control of the abdA and AbdB homeotic products. Development 114: 493–505. [DOI] [PubMed] [Google Scholar]
- Smallwood A, Esteve PO, Pradhan S, Carey M. 2007. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev 21: 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M, Mallin DR, Simon JA, Courey AJ. 2011. Small ubiquitin-like modifier (SUMO) conjugation impedes transcriptional silencing by the polycomb group repressor sex comb on midleg. J Biol Chem 286: 11391–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. 2006. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 6: 846–856. [DOI] [PubMed] [Google Scholar]
- Srinivasan L, Atchison ML. 2004. YY1 DNA binding and PcG recruitment requires CtBP. Genes Dev 18: 2596–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AK, Kliszczak M, Dodson H, Cooley C, Morrison CG. 2011. Roles of vertebrate Smc5 in sister chromatid cohesion and homologous recombinational repair. Mol Cell Biol 31: 1369–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubbe G, Popp C, Schmidt A, Pauli A, Ringrose L, Beisel C, Paro R. 2011. Polycomb purification by in vivo biotinylation tagging reveals cohesin and Trithorax group proteins as interaction partners. Proc Natl Acad Sci 108: 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G. 1981. A gene product required for correct initiation of segmental determination in Drosophila. Nature 293: 36–41. [DOI] [PubMed] [Google Scholar]
- Sun X, Morozova T, Sonnenfeld M. 2006. Glial and neuronal functions of the Drosophila homolog of the human SWI/SNF gene ATR-X (DATR-X) and the jing zinc-finger gene specify the lateral positioning of longitudinal glia and axons. Genetics 173: 1397–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, et al. 2002. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. 2005. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 19: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, et al. 2012. RYBP–PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148: 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Prasad-Sinha J, Birve A, Rasmuson-Lestander A, Harte PJ. 2003. A 1-megadalton ESC/E(Z) complex from Drosophila that contains polycomblike and RPD3. Mol Cell Biol 23: 3352–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ. 2009. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136: 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Saiakhova AR, Howard B, Monteith KE, Scacheri PC, Cosgrove MS, Harte PJ. 2014. Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development 141: 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, van Lohuizen M. 2006. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat Genet 38: 694–699. [DOI] [PubMed] [Google Scholar]
- Voss AK, Collin C, Dixon MP, Thomas T. 2009. Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev Cell 17: 674–686. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. 2009. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138: 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jahren N, Miller EL, Ketel CS, Mallin DR, Simon JA. 2010. Comparative analysis of chromatin binding by sex comb on midleg (SCM) and other polycomb group repressors at a Drosophila Hox gene. Mol Cell Biol 30: 2584–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, Kuroda MI. 2013. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat Struct Mol Biol 20: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al. 2012. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337: 971–975. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bonasio R, Strino F, Kluger Y, Holloway JK, Modzelewski AJ, Cohen PE, Reinberg D. 2013. SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes Dev 27: 749–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. 2006. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 5: 2339–2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.