

DNA repair pathway choice is thought to be determined by cell cycle timing and chromatin context. van Sluis et al. find that targeted introduction of double-strand breaks (DSBs) into rDNA, but not abutting sequences, results in ATM-dependent inhibition of their transcription by RNA polymerase I. Importantly, DSBs within rDNA recruit the homologous recombination machinery throughout the cell cycle.
Keywords: nucleolus, nucleolar organizer region (NOR), ribosomal genes (rDNA), DNA double-strand breaks (DSBs), homologous recombination (HR), ataxia telangiectasia-mutated (ATM)
Abstract
DNA double-strand breaks (DSBs) are repaired by two main pathways: nonhomologous end-joining and homologous recombination (HR). Repair pathway choice is thought to be determined by cell cycle timing and chromatin context. Nucleoli, prominent nuclear subdomains and sites of ribosome biogenesis, form around nucleolar organizer regions (NORs) that contain rDNA arrays located on human acrocentric chromosome p-arms. Actively transcribed rDNA repeats are positioned within the interior of the nucleolus, whereas sequences proximal and distal to NORs are packaged as heterochromatin located at the nucleolar periphery. NORs provide an opportunity to investigate the DSB response at highly transcribed, repetitive, and essential loci. Targeted introduction of DSBs into rDNA, but not abutting sequences, results in ATM-dependent inhibition of their transcription by RNA polymerase I. This is coupled with movement of rDNA from the nucleolar interior to anchoring points at the periphery. Reorganization renders rDNA accessible to repair factors normally excluded from nucleoli. Importantly, DSBs within rDNA recruit the HR machinery throughout the cell cycle. Additionally, unscheduled DNA synthesis, consistent with HR at damaged NORs, can be observed in G1 cells. These results suggest that HR can be templated in cis and suggest a role for chromosomal context in the maintenance of NOR genomic stability.
In order to maintain and propagate genetic information, it is essential that DNA double-strand breaks (DSBs) arising from normal cellular metabolism and extrinsic sources, such as ionizing radiation and xenobiotics, are efficiently repaired (Jackson 2002; Harper and Elledge 2007; Hoeijmakers 2009). The presence of DSBs initiates a complex DNA damage response (DDR) that involves detection, signaling, and ultimately repair. The kinase ATM (ataxia telangiectasia-mutated) is a key player in transducing DSB recognition into activation of cell cycle checkpoints and repair processes (Shiloh and Ziv 2013). Once activated by recruitment to DSBs, ATM phosphorylates the histone variant H2AX on Ser139 (Burma et al. 2001). This phosphorylation, referred to as γH2AX, can spread for up to 1 Mb away from the break site (Rogakou et al. 1999; Iacovoni et al. 2010).
In mammalian cells, two major pathways are used for repair: nonhomologous end-joining (NHEJ) and homologous recombination (HR) (Chapman et al. 2012b). In NHEJ, the broken ends are minimally processed, aligned, and ligated together. Importantly, NHEJ does not require sequence complementarity and is error-prone, often introducing point mutations, small insertions, or deletions. In contrast, HR involves extensive DNA end resection to generate 3′ single-stranded overhangs. These invade double-stranded undamaged homologous DNA copies that template unscheduled DNA synthesis and accurate repair. Usually, the repair template is a sister chromatid. Thus, it is generally considered that accurate repair by HR is restricted to S and G2 phases of the cell cycle (Aylon et al. 2004), whereas error-prone NHEJ is predominant in G0/G1 cells (Lieber et al. 2003).
Key factors play a role in the NHEJ versus HR selection process. In NHEJ, the broken ends are bound by the Ku heterodimer in complex with the kinase DNA-PK (DNA-dependent protein kinase) to protect the ends from resection (Jackson 2002). Initiation of resection is further regulated by the balance of 53BP1 (p53-binding protein 1) and BRCA1 (breast cancer early onset 1) (Panier and Boulton 2014). BRCA1 can recruit nucleases that carry out resection, while 53BP1 inhibits this process. Once resection is initiated, the cell is committed to repair by HR.
Sequence composition and chromatin status can influence repair choice. While evidence suggests that, even in G2, most DSB repair uses NHEJ (Karanam et al. 2012), repair of DSBs in repeat-rich heterochromatin is predominantly by HR and involves movement of DSBs to improve access by repair factors (Chiolo et al. 2011; Murray et al. 2012). A recent study, made possible by introducing sequence-specific endonucleases into cells, examined DSBs introduced at specific genomic locations. It was shown that HR-mediated repair of DSBs within euchromatin is restricted to transcriptionally active chromatin enriched in trimethylated histone H3K36 (Aymard et al. 2014).
Nucleoli, the sites of ribosome biogenesis, are the most prominent subnuclear domains in all human cells. They form around ribosomal gene (rDNA) arrays in nucleolar organizer regions (NORs) (Sirri et al. 2008; Pederson 2010). In humans, the rDNA repeat is 43 kb in length, 13 kb of which encodes precursor ribosomal RNA (pre-rRNA), and the remaining 30 kb being intergenic spacer (IGS). Approximately 300 rDNA repeats are distributed among NORs situated on the poorly characterized and unsequenced short arms of each of the five human acrocentric chromosomes (Stults et al. 2008). During mitosis, active NORs are bookmarked by upstream binding factor (UBF), a nucleolar-specific HMG-box protein that binds extensively across the rDNA array (Roussel et al. 1993; O'Sullivan et al. 2002; Mais et al. 2005; Grob et al. 2014). After reactivation of rDNA transcription by its dedicated RNA polymerase I (Pol I) transcription machinery, the resulting pre-rRNA seeds reformation of nucleoli (Grob et al. 2014).
Nucleoli are subdivided into three distinct components reflecting the stages of ribosome biogenesis (Sirri et al. 2008). Fibrillar centers (FCs) contain unengaged pools of transcription factors and nontranscribed rDNA sequences (Prieto and McStay 2007). rDNA transcription occurs at the interface between FCs and the surrounding dense fibrillar component (DFC) where early processing of resulting pre-rRNA occurs. Late processing of pre-rRNA occurs in the surrounding granular component (GC), yielding mature 18S, 5.8S, and 28S rRNAs that assemble into ribosome subunits. As the cell cycle progresses, nucleoli—initially formed around individual NORs—fuse, forming large nucleoli. These are characterized by the presence of perinucleolar heterochromatin derived from sequences surrounding rDNA on acrocentric short arms (Nemeth and Langst 2011). Nucleolar structure is dependent on ongoing transcription of rDNA. Specific inhibition of Pol I transcription with a low dose of actinomycin D (AMD) causes a rapid nucleolar reorganization, often referred to as nucleolar segregation, in which the FCs and DFCs migrate along with rDNA to the nucleolar periphery, forming so called nucleolar caps (Hadjiolov 1985). Notably, most nucleolar caps contain a single NOR (Floutsakou et al. 2013).
Recently, we provided the first description of the genomic architecture of human NORs (Floutsakou et al. 2013). In particular, we characterized almost 400 kb of DNA sequence immediately distal to rDNA arrays on the short arms of the acrocentric. These sequences, termed the distal junction (DJ), display >99% sequence identity between the acrocentrics and are not found elsewhere in the genome. Although previously believed to be heterochromatic, characterization of the DJ reveals that it has a complex genomic architecture similar to other euchromatic regions of the genome. It is dominated by a very large inverted repeat, is transcriptionally active, and displays a complex chromatin structure. Moreover, the DJ is embedded within perinucleolar heterochromatin where it appears to play a role in nucleolar organization by anchoring ribosomal gene repeats that extend into the nucleolar interior.
In addition to ribosome biogenesis, the nucleolus plays key roles in growth control and human pathology (Pederson 2010). The nucleolus is also emerging as a major sensor of cellular stress (Boulon et al. 2010). These facts highlight the importance of understanding the response to DNA damage within this essential nuclear subdomain. Moreover, the repetitive nature of rDNA, its isolation in a ribonucleoprotein-dense environment, and the dedicated nature of its transcription and RNA processing machinery provide a unique opportunity to characterize the DDR in detail at highly expressed genetic loci.
A previous study has shown that global introduction of DSBs into mouse cells by γ-irradiation or more local introduction into nucleoli by laser microirradiation results in ATM-dependent inhibition of Pol I transcription (Kruhlak et al. 2007). Importantly, this study also showed that Pol I inhibition was restricted to nucleoli containing damaged DNA. It should be pointed out, however, that similar experiments performed in human cells failed to show any inhibition of Pol I transcription following γ-irradiation (Moore et al. 2011). More surprisingly, it has been demonstrated that laser microirradiation-induced DSBs outside of nucleoli lead to an ATM-dependent pan-nuclear silencing of Pol I transcription (Ciccia et al. 2014; Larsen et al. 2014). The difficulty in interpreting such experiments is that we have no knowledge of the number or distribution of DSBs. Furthermore, as rDNA represents only 0.4% of the genome and is at a low concentration within nucleoli, damage within it represents a minority of the total damage present in irradiation experiments.
The homing endonuclease I-PpoI from Physarum can cleave within the 28S rRNA-coding region of rDNA in human cells, offering an opportunity to study in detail the response of nucleoli to DSBs introduced into rDNA (Stoddard 2005; Berkovich et al. 2007). Here we report that DSBs introduced into rDNA using I-PpoI or the recently developed CRISPR/Cas9 system (Cong et al. 2013; Mali et al. 2013) induce inhibition of Pol I transcription and nucleolar segregation. This results in the formation of γH2AX-positive caps (containing rDNA and the Pol I transcription machinery) at the nucleolar periphery. This response is strictly ATM-dependent. Using the FUCCI (fluorescence ubiquitination cell cycle indicator) system (Sakaue-Sawano et al. 2008), we demonstrated that rDNA DSBs recruit components of the HR pathway and exhibit evidence of repair irrespective of cell cycle stage. Our data suggest that HR-mediated repair of DSBs within rDNA can be templated by rDNA repeats in cis. We reveal that this nucleolar response is restricted to DSBs within rDNA. Finally, we argue that the chromosomal context of NORs is important for maintaining the genomic integrity of rDNA arrays.
Results
DSBs in rDNA induce nucleolar reorganization and inhibition of Pol I transcription
In order to introduce DNA DSBs into rDNA repeats in human cells, we exploited I-PpoI, a homing endonuclease from Physarum polycephalum. I-PpoI has a 15-base-pair (bp) recognition sequence within each of the ∼300 copies of 28S rRNA-coding sequence and up to 13 other sites in the human genome (Muscarella et al. 1990; Pankotai et al. 2012). While non-rDNA sites on chromosomes 1 and 11 have been the focus of a number of earlier studies (Berkovich et al. 2007; Pankotai et al. 2012), here we focus on the consequence of DSBs within rDNA. We adopted an mRNA transfection approach for introduction of I-PpoI (Fig. 1A). The advantages of this are threefold. First, any cell type, including primary cells, can be efficiently transfected. Second, in contrast to plasmid-based transfections, the process of mRNA transfection does not itself initiate a DDR (data not shown). Finally, expression of I-PpoI is rapid; within 6 h, the majority of cells exhibit a response. For most of the experiments described here, we used human telomerase (hTert) immortalized retinal pigmented epithelial cells (referred to here as RPE1) that are karyotypically normal, are p53 wild-type, and have intact DNA damage checkpoints. At 6 h after introduction of V5 epitope-tagged I-PpoI into these cells, ∼20% of rDNA repeats exhibit a DSB (Fig. 1B). This is accompanied by an induction of γH2AX (Fig. 1C). Importantly, introduction of a catalytically dead I-PpoI mutant (H98A) fails to induce either DSBs or a γH2AX response. Analysis of individual cells revealed that transfected cells (V5 staining) have reorganized nucleoli, with UBF relocalizing from the nucleolar interior to perinucleolar caps (Fig. 1D). A strong γH2AX signal is also associated with each of these nucleolar caps. An identical response is also observed upon introduction of I-PpoI into human primary skin fibroblast cell lines 1BR3, BJ, and CCD-1079Sk (Supplemental Fig. S1A). Nucleolar caps also contain the Pol I subunit Paf49, transcription factor RRN3, and other FC/DFC proteins (Treacle and Fibrillarin) (Supplemental Fig. S1B). Treacle, the product of the TCOF1 gene, is a nucleolar phosphoprotein that, like UBF, remains associated with rDNA chromatin throughout the cell cycle (Valdez et al. 2004). The appearance and composition of these nucleolar caps is similar to those formed when Pol I transcription is inhibited by AMD (Hadjiolov 1985). However, at a dose and time when AMD-induced nucleolar segregation was observed, we detected no γH2AX response (Supplemental Fig. S1C), suggesting that damage-induced nucleolar reorganization is also a consequence of transcriptional inhibition. To directly address the transcriptional status of I-PpoI transfected cells, we first performed BrUTP incorporation assays in permeabilized cells under conditions where transcription by Pol II and III is inhibited. Pol I transcription is absent in cells exhibiting nucleolar segregation (Fig. 1E). In order to also assess effects on Pol II and III transcription, we exploited 5-ethynyl uridine (EU) incorporation in living cells followed by “click chemistry.” In these assays, the majority of EU incorporated is by Pol I in the nucleolus, as revealed by antibody staining and AMD inhibition (Supplemental Fig. S1D). The majority of the nonnucleolar nucleoplasmic signal is due to Pol II, as revealed by inhibition with flavopiridol (Chao and Price 2001). EU incorporation assays performed in I-PpoI transfected cells confirm that transcription by Pol I is inhibited and demonstrate that transcription by Pol II remains unaffected (Fig. 1F).
Figure 1.
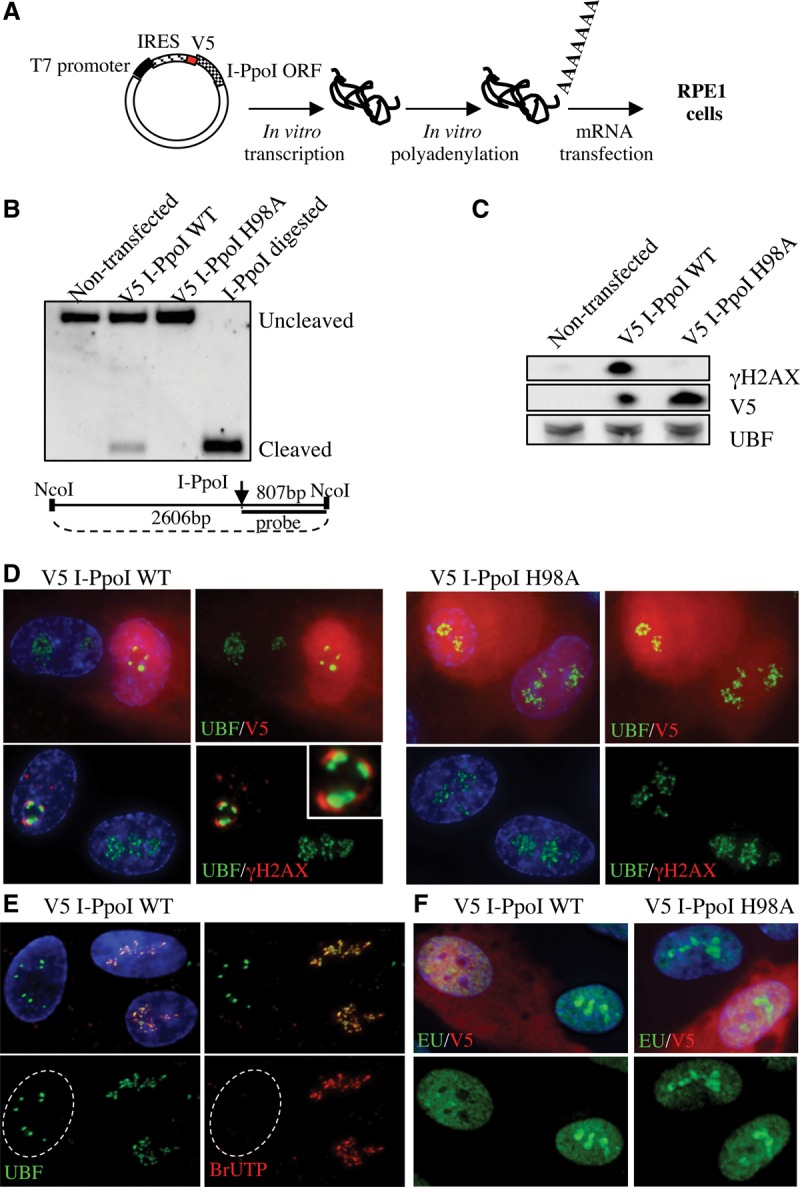
Introduction of DSBs into rDNA with I-PpoI induces nucleolar reorganization and inhibition of transcription. (A) Strategy for expressing I-PpoI. A transcript encoding V5 epitope-tagged I-PpoI preceded by an IRES element was produced in vitro using T7 RNA polymerase. Following polyadenylation, transcripts were transfected into RPE1 cells. (B) Southern blotting reveals that wild-type (WT) I-PpoI introduces DSBs within the 28S rRNA-coding sequence in vivo. Approximately 20% of rDNA repeats contain a DSB. Genomic DNA digested to completion with I-PpoI in vitro is included for comparison. Note the absence of cleavage observed using catalytically dead I-PpoI (H98A). The probing scheme is shown below. (C) Western blotting with V5 antibodies reveals that wild-type and mutant I-PpoI are produced at comparable levels and that only wild-type I-PpoI induces a γH2AX response. (D) Staining of cells 6 h after transfection reveals that wild-type but not mutant V5-tagged I-PpoI induces formation of UBF-containing nucleolar caps that are associated with prominent γH2AX signals. The inset shows an enlargement of a single nucleolus. (E) BrUTP incorporation assays indicate that transcription of rDNA by Pol I is inhibited in wild-type I-PpoI transfected cells. The nucleus of the transfected cell shown is indicated by a white dotted line. (F) EU incorporation assays confirm that wild-type but not mutant V5-tagged I-PpoI induces inhibition of nucleolar (Pol I) transcription and further show that nucleoplasmic (Pol II and III) transcription is unaffected.
We reported previously that NORs are anchored in perinucleolar heterochromatin by DJ sequences located immediately distal to rDNA arrays (Floutsakou et al. 2013). Ectopic DJ sequence arrays integrated into nonacrocentric chromosomes retain the ability to associate with perinucleolar heterochromatin. Furthermore, AMD-induced nucleolar caps contain rDNA from a single NOR and form adjacent to the linked DJ. To determine whether these sequences are similarly organized in damage-induced nucleolar caps, we performed three-dimensional (3D) immunoFISH on I-PpoI transfected RPE1 cells using rDNA and DJ probes (Fig. 2A). We demonstrated that rDNA is not detectable in the nucleolar interior but is highly enriched within damage-induced nucleolar caps where it overlaps with the γH2AX signal (Fig. 2B). Moreover, as with AMD treatment, damage-induced nucleolar caps form immediately adjacent to DJ sequences (Fig. 2C). These results suggest that DJ sequences are involved in the nucleolar reorganization observed in response to rDNA damage.
Figure 2.
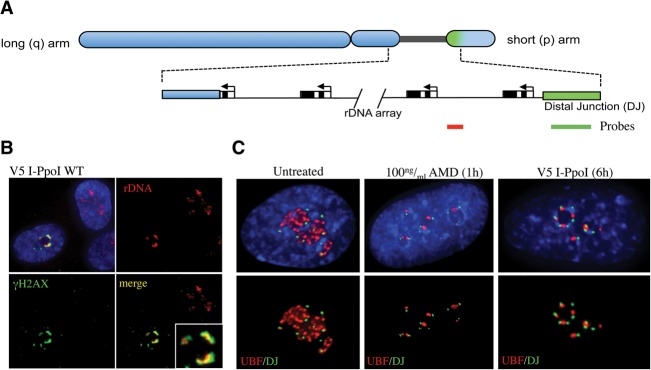
I-PpoI-induced nucleolar caps contain rDNA and form adjacent to DJ sequences. (A) A cartoon representation of a human acrocentric chromosome with enlargement of the rDNA array and surrounding sequences. Sequences immediately distal (DJ) to the rDNA are colored green. The position of probes used in FISH to detect DJs (green) and rDNA (red) are indicated below. (B) 3D immunoFISH performed on I-PpoI transfected cells shows colocalization of rDNA and γH2AX in nucleolar caps. The inset shows an enlarged individual nucleolus. (C) 3D immunoFISH performed on AMD-treated and I-PpoI transfected cells shows that, in both cases, nucleolar caps form immediately adjacent to DJ sequences embedded in perinucleolar heterochromatin.
Specificity of the nucleolar DSB response
Identification of the DJ sequences immediately distal to rDNA provides an opportunity to address spatial restriction of the nucleolar DSB response. To this end, we exploited the ability of CRISPR/Cas9 to introduce DSBs at target sites (Cong et al. 2013; Mali et al. 2013). We designed a series of guide RNAs (gRNAs) that target either rDNA or the DJs present on all five acrocentric short arms (Floutsakou et al. 2013). As Cas9 unbound to gRNA has been reported to target nucleoli, we used modified gRNAs with extended stem–loops (Chen et al. 2013). These gRNAs exhibit tighter Cas9 binding, thus eliminating nonspecific nucleolar localization. rDNA gRNA1 and gRNA2 target the 5′ ETS, ∼120 bp and 1.4 kb, respectively, downstream from the transcriptional start site (Fig. 3A; Supplemental Table S1). To assess the impact of DSBs in nontranscribed sequences, rDNA gRNA3 targets the middle of the IGS. We transfected RPE1 cells with Cas9 and individual rDNA gRNAs. Monitoring Cas9 expression by antibody staining reveals that, despite the presence of a nuclear localization signal at its C terminus, most Cas9 remains cytoplasmic. Nevertheless, sufficient Cas9/gRNA complexes enter the nucleus to induce DSBs. All Cas9-expressing cells exhibit a γH2AX response and are associated with varying degrees of nucleolar reorganization, as adjudged by UBF staining (Fig. 3A) or rDNA FISH (Supplemental Fig. S2). While most transfected cells exhibit complete nucleolar segregation, similar to I-PpoI, a minority exhibits partial segregation (Supplemental Fig. S2). In line with this, rDNA targeted gRNAs also induce silencing of Pol I transcription, ranging from partial to complete (Fig. 3A).
Figure 3.

Introduction of DSBs into rDNA and adjacent sequences using CRISPR/Cas9 reveals a regional specificity of response. (A) Cas9 complexed with gRNAs targeted to rDNA induces a γH2AX response, nucleolar segregation, and inhibition of nucleolar transcription. The locations of gRNA targets within the rDNA repeat are illustrated. In all cases, transfected cells are identified with a Cas9 antibody. (Top three panels) Nucleolar segregation was assayed by UBF staining. Ongoing transcription was assessed by EU incorporation. The middle three panels represent the majority of cells in which nucleolar transcription is absent. The bottom three panels represent the minority of transfected cells in which partial inhibition was observed. (B) Cas9 complexed with DJ targeted gRNAs induces a nucleolar γH2AX response but does not result in nucleolar segregation or inhibition of transcription. The positions of DJ gRNAs are illustrated. Nucleolar segregation and ongoing transcription are shown in the top and bottom panels respectively.
DJ gRNA1 targets Acro138 repeat blocks centered at 138 kb and 290 kb distal to the rDNA. DJ gRNA2 and gRNA3 target sequences 187 kb and 238 kb distal to the rDNA (Fig. 3B; Supplemental Table S1). Note that these gRNAs can target both left and right arms of the large inverted repeat that is present in the DJ (Floutsakou et al. 2013). All Cas9-expressing cells cotransfected with DJ gRNAs exhibit γH2AX signals surrounding nucleoli (Fig. 3B). Combined immuno-FISH using a DJ probe reveals that γH2AX and DJ signals partially overlap (Supplemental Fig. S2). In contrast to results obtained using rDNA gRNAs, in the majority of Cas9/DJ gRNA transfected cells, no nucleolar segregation was observed, and nucleolar transcription was unaffected (Fig. 3B). We conclude that inhibition of Pol I transcription and nucleolar reorganization are specific responses to DSBs within rDNA. We further conclude that DSBs anywhere in the rDNA repeat, including the nontranscribed IGS, induce both transcriptional inhibition and nucleolar segregation.
Inhibition of Pol I transcription is ATM-dependent
In mouse cells treated with ionizing radiation (10 Gy), Pol I transcription is inhibited in an ATM-dependent manner (Kruhlak et al. 2007). In light of similar experiments performed in human cells failing to observe this response (Moore et al. 2011), we sought to determine whether I-PpoI-induced nucleolar segregation and Pol I inhibition are ATM-dependent. Transfected cells treated with either ATM inhibitor KU55933 (Hickson et al. 2004) or DNA-PK inhibitor NU7441 (Leahy et al. 2004) exhibit levels of DSBs similar to those observed in control or DMSO-treated cells (Fig. 4A). A γH2AX response is missing only in ATM-inhibited/I-PpoI transfected cells (Fig. 4B). Pol I transcription is inhibited in only 1% of I-PpoI transfected cells in the presence of the ATM inhibitor but in 98% of cells treated with DMSO (Fig. 4C). Nucleolar segregation resulting from I-PpoI-induced DSBs is also ATM-dependent (Supplemental Fig. S3A). In line with the proposed involvement of ATM, we observed enrichment of total ATM and exclusive localization of activated ATM (pS1981) at nucleolar caps in I-PpoI transfected cells (Fig. 4D,E). Interestingly, while DNA-PK and Ku are both present in nucleoli prior to damage, neither is found at nucleolar caps induced by DSBs or AMD (Supplemental Fig. S3B; data not shown). Thus, we demonstrated that the γH2AX response, Pol I transcription inhibition, and nucleolar segregation in response to DSBs are all ATM-dependent. The nucleolar proteome has been shown to contain a pool of ATM (Andersen et al. 2002), raising the possibility that the responses that we observed are due to pre-existing pools of nucleolar ATM. In support of this view, pre-extraction of cells allows us to visualize a subpopulation of ATM in the nucleoli of undamaged cells (Supplemental Fig. S3C).
Figure 4.
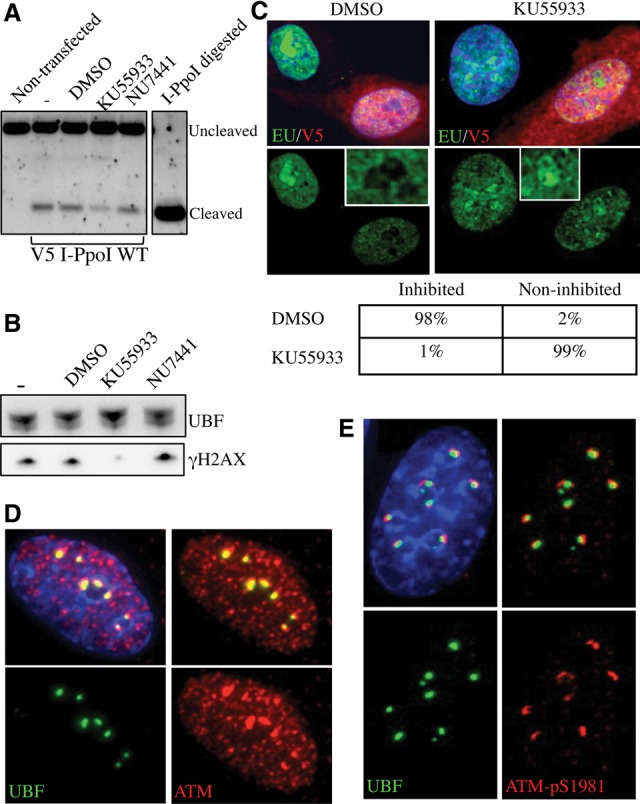
Transcriptional inhibition is ATM-dependent. (A) Southern blotting reveals that ATM and DNA-PK inhibitors (10 μM KU55933 and 10 μM NU7441, respectively) have little effect on the cleavage efficiency of I-PpoI in vivo. (B) Western blotting indicates that ATM inhibitor (KU55933) selectively inhibits the γH2AX response induced by I-PpoI transfection. (C) Nucleolar transcription continues in V5 I-PpoI transfected cells in the presence of ATM inhibitor (KU55933). Nucleolar transcription is inhibited in control (DMSO-treated) cells. Quantification of results from analyzing >100 transfected cells for each treatment are shown below. The insets show enlarged single nucleoli. (D) Staining of I-PpoI transfected cells with an ATM antibody shows enrichment of ATM at nucleolar caps. (E) Staining of I-PpoI transfected cells with antibodies that detect ATM-pS1981 show that activated ATM is restricted to nucleolar caps.
Damage-induced nucleolar caps recruit HR repair machinery
We next sought to determine whether inhibition of transcription and repositioning of damaged rDNA on the nucleolar surface facilitates recruitment of DNA repair proteins. Initially, we focused our attention on BRCA1 and 53BP1, factors involved in the HR versus NHEJ decision process (Panier and Boulton 2014). Staining of I-PpoI transfected cells revealed that both BRCA1 and 53BP1 can be recruited to γH2AX-positive nucleolar caps (Fig. 5A). Costaining revealed that all damage-induced nucleolar caps are positive for 53BP1, and the majority of these (58%) is also positive for BRCA1 (Supplemental Table S2; Supplemental Fig. S4A). The presence of BRCA1 is indicative of repair by HR. As BRCA1 and 53BP1 show differing temporal recruitment to DSBs, the repair pathway used in cells with nucleolar caps that are only 53BP1-positive cannot be unambiguously assigned (Chapman et al. 2012a). Nevertheless, we can conclude that, in all cases where damaged rDNA has moved to the nucleolar periphery, it is recognized by mediators of the DDR.
Figure 5.

I-PpoI-induced DSBs selectively recruit HR repair factors to nucleolar caps. (A) BRCA1 and 53BP1 were individually observed at γH2AX-positive nucleolar caps in I-PpoI transfected cells. (B) Antibody staining of I-PpoI transfected cells shows that HR factors RPA2 (total and phospho-S4/S8), Rad51, and Rad52 are highly enriched at nucleolar caps. (C) 3D immunoFISH was performed on I-PpoI transfected RPE1 cells using a DJ probe, Treacle, and RPA2 antibodies. The insets show enlarged individual caps containing a single NOR.
To determine the repair pathway selected, we sought to determine which repair factors are present at damage-induced nucleolar caps. All cells that exhibit UBF-positive caps are also positive for γH2AX. Of these, 57%, 62%, and 44% exhibit significant enrichment of HR proteins RPA2, Rad51, and Rad52, respectively (Fig. 5B; Supplemental Table S2). NHEJ proteins Ku80 and XRCC4 are distributed throughout the nucleoplasm with no evidence of enrichment at nucleolar caps (Supplemental Table S2; Supplemental Fig. S4B). The lack of recruitment of factors involved in the NHEJ pathway and recruitment of factors required for end resection suggests that repair is homology-directed. In contrast to ATM, evidence suggests that HR factors recruited to DSBs within nucleolar caps are from nucleoplasmic pools rather than nucleolar pools. For example, endogenous RPA2 and GFP-tagged RPA2 are excluded from nucleoli in cells without damage (Supplemental Fig. S4C).
Damage-induced nucleolar caps can contain single or multiple NORs, as revealed by 3D immunoFISH using a DJ probe that enables identification of individual NORs (Fig. 2C). To demonstrate that nucleolar caps comprising a single NOR can still recruit the HR machinery, we combined the DJ FISH probe with antibodies against Treacle and RPA2 (Fig. 5C). RPA2-positive nucleolar caps with a single associated DJ signal can be readily observed (Fig. 5C, see insets).
DSBs in rDNA are repaired by HR independent of cell cycle stage
The experiments described thus far were performed in asynchronous RPE1 cells in which 40%–50% of the population are in G1 and do not have a sister chromatid as a template for homology-directed repair. This prompted closer investigation of the influence of the cell cycle on choice of repair pathway. To this end, we exploited the FUCCI system that can easily discriminate cell cycle stages (Sakaue-Sawano et al. 2008). We constructed stable cell lines in RPE1 that contained either mCherryFP fused to a domain of Cdt1 or Azami-GreenFP (mAG) fused to Geminin. In order to characterize these clones, DNA replication was monitored using 5′-ethynyl-2′-deoxyuridine (EdU) incorporation followed by click chemistry (Supplemental Fig. S5A). mCherry-Cdt1 is present at high levels in G1 cells, decreases in early S, and is absent in the rest of the cell cycle. Fifty-nine percent of the cells in an asynchronous population exhibit red fluorescence and hence are in G1, early S, or mid-S. Importantly, we can distinguish between these stages based on the strength of the signal. Also, mCherry-Cdt1 can be used as a marker for the nucleolus, where it accumulates in G1. mAG-Geminin begins to accumulate in mid-S, peaking in G2 cells. In this stable line, 43% of cells exhibit green fluorescence and hence are in mid-S-, late S-, G2-, and M-phase cells.
In mCherry-Cdt1 RPE1 cells analyzed 6 h after transfection with I-PpoI, we observed that 46% and 44% of cells with Rad51- and RPA2-positive foci, respectively, are in red cells (Fig. 6A). Moreover, in the majority of these cells, Rad51 and RPA2 foci surround bright-red nucleoli, characteristic of G1 cells. In mAG-Geminin RPE1 cells transfected with I-PpoI, we observed 50% and 52% of cells with Rad51- and RPA2-positive foci, respectively, that lack any green fluorescence, again characteristic of G1 cells (Fig. 6B). This result is further underscored by the presence of nucleolar caps that are positive for both BRCA1 and 53BP1 in mCherry-Cdt1-positive (G1) I-PpoI transfected RPE1 cells (Supplemental Fig. S5B). These results indicate that HR repair factors are recruited to damaged rDNA in I-PpoI-induced nucleolar caps throughout the cell cycle, including G1.
Figure 6.

HR factors are recruited, and damage-induced DNA synthesis can be observed at I-PpoI-induced nucleolar caps in G1 cells. (A) I-PpoI transfection of RPE1 cells stably expressing FUCCI component mCherry-Cdt1 reveals that HR factors Rad51 and RPA2 are recruited comparably to nucleolar caps in G1 (red) and late S/G2 (nonred) cells. Quantification is shown below. (B) I-PpoI transfection of RPE1 cells stably expressing FUCCI component mAG-Geminin reveals that HR factors Rad51 and RPA2 are recruited comparably to nucleolar caps in G1 (nongreen) and late S/G2 (green) cells. Quantification is shown below. (C) RPE1 cells stably expressing FUCCI component mAG-Geminin or mCherry-Cdt1 were transfected with I-PpoI mRNA; 4 h after transfection, fresh medium containing EdU was added to cells. Following a 2-h pulse of EdU labeling, cells were fixed. Treacle antibodies detected damage-induced nucleolar caps. Damage-induced DNA synthesis was visualized using biotin-azide and fluorophore-conjugated streptavidin (see the Materials and Methods for details). In the top right panel, EdU incorporation can be observed at damage-induced nucleolar caps in G1 (nongreen) mAG-Geminin cells. In the middle panels, EdU incorporation is observed in nucleolar caps in both G1 (red) and G2 (nonred) mCherry-Cdt1 cells. A DIC image is shown at the right. In the bottom panels, a single enlarged G1 cell from the same experiment is shown.
As homology-directed repair of DSBs involves unscheduled DNA synthesis, we used EdU incorporation and click chemistry to visualize repair in G1 nucleolar caps. To demonstrate the feasibility of this approach, we transfected RPE1 cells with either wild-type or mutant V5-tagged I-PpoI followed by an EdU pulse (Supplemental Fig. S6). In ∼5% of cells containing wild-type I-PpoI, unscheduled DNA synthesis in either G1 or G2 cells can be observed at nucleolar caps. These signals are distinct from EdU incorporation observed during S phase in two respects. They are restricted to nucleolar caps and are far less intense. In no case was unscheduled DNA synthesis observed in nucleoli of cells containing mutant I-PpoI.
To determine whether repair can occur in G1 cells, we performed EdU incorporation assays in RPE1 cells expressing FUCCI components and transfected with I-PpoI (Fig. 6C). In a small proportion of mAG-Geminin-negative (G1) and mCherry-Cdt1-positive (G1) cells, we can observe EdU incorporation in I-PpoI-induced nucleolar caps. For technical reasons, it is difficult to be precise about the number of G1 cells exhibiting unscheduled DNA synthesis in nucleolar caps. Nevertheless, this experiment provides further evidence for homology-directed repair in G1 cells.
Discussion
The ability to introduce targeted breaks in vivo allows us to analyze the nucleolar response to DSBs within rDNA arrays against a low background of DSBs elsewhere in the genome. In brief, we observed that DSBs within rDNA induce ATM-dependent inhibition of Pol I transcription followed by nucleolar reorganization such that damaged rDNA is relocated to the nucleolar periphery. We hypothesize that this reorganization enhances access to rDNA DSBs by the repair machinery. Importantly, recruitment of the HR machinery occurs independently of cell cycle stage. Furthermore, we provided evidence for unscheduled DNA synthesis in nucleolar caps, consistent with HR taking place in G1 cells. By combining these results with our recent work on the genomic architecture of NORs, we can now present a model for how integrity of rDNA arrays is maintained against an onslaught of DNA damage (Fig. 7).
Figure 7.
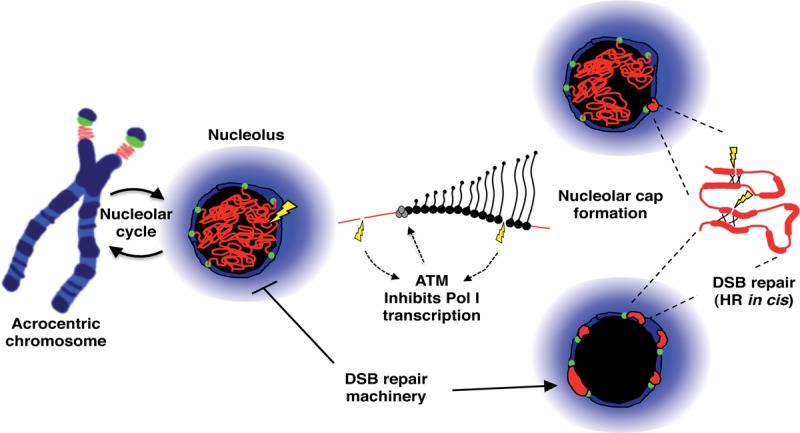
A model for chromosomal context influencing genomic stability of rDNA arrays. rDNA arrays on acrocentric short arms are surrounded by sequences that form perinucleolar heterochromatin during interphase. DJ sequences (green) are localized within this heterochromatin, providing an anchor for the linked rDNA array (red). Introduction of DSBs (yellow) into the rDNA induce ATM-dependent inhibition of rDNA transcription by Pol I followed by subsequent nucleolar reorganization. Positioning of rDNA within caps on the nucleolar surface is a consequence of anchoring by DJ sequences. We hypothesize that this nucleolar reorganization renders damaged rDNA accessible to DSB repair factors normally excluded from nucleoli. Concentration of damaged and intact rDNA repeats within nucleolar caps promotes repair by the HR pathway. As this occurs in G1 cells, HR repair may be templated by repeats in cis within the same NOR. See the Discussion for further details.
While the use of γ-irradiation and laser microirradiation to induce DSBs has yielded conflicting results (Kruhlak et al. 2007; Moore et al. 2011; Larsen et al. 2014), here we unequivocally demonstrated that DSBs within rDNA are sufficient to induce ATM-dependent inhibition of Pol I transcription. The introduction of the CRISPR/Cas9 allowed us to demonstrate that DSBs in both transcribed and nontranscribed regions of the rDNA repeat induce this response. DSBs introduced into DJ sequences closely linked to rDNA arrays but spatially separated fail to inhibit Pol I transcription. These results begin to reveal the regional specificity of the nucleolar response to DSBs.
Cell staining and proteomics revealed that ATM is present within nucleoli prior to damage, suggesting that a pre-existing nucleolar pool of ATM mediates this response. While the mechanism of transcriptional inhibition has yet to be elucidated, phospho-proteomics has revealed that key Pol I transcription factors are targets for ATM (Matsuoka et al. 2007; Stokes et al. 2007). UBF can be phosphorylated on S23 and TAFI110, a component of SL1 on S848 and S858. Interestingly, the nearby T852 in TAFI110 is a target for mitotic phosphorylation that silences Pol I transcription during mitosis (Heix et al. 1998). Other identified nucleolar targets include the Pol I transcription termination factor TTF1 and a component of the early pre-rRNA processing machinery, UTP14A. Finally, Treacle—a factor implicated in transcription and rRNA modification and an interaction partner of Nijmegen breakage syndrome protein 1 (NBS1)—is also an ATM target (Ciccia et al. 2014; Larsen et al. 2014).
Our next major finding is that ATM-dependent inhibition of transcription induces nucleolar reorganization (Fig. 7). The Pol I transcription machinery and rDNA move to the nucleolar periphery, forming nucleolar caps that contain γH2AX and are highly enriched in activated ATM. Interestingly, a DSB introduced into the rDNA array in yeast results in similar rDNA movement to the nucleolar periphery to facilitate repair (Torres-Rosell et al. 2007). Nucleolar caps, albeit without γH2AX and ATM, form in response to transcriptional inhibition by AMD. Both AMD- and DSB-induced nucleolar caps are located adjacent to DJ sequences anchored in perinucleolar heterochromatin. ATM inhibitors prevent both transcriptional inhibition and nucleolar reorganization in response to DSBs. Taken together, these findings argue that the directed movement of rDNA in response to DSBs is a function of transcriptional inhibition, not the induction of DSBs per se. While it has been reported that chromatin mobility is increased at DSBs across the genome as a whole, we believe that sequestration of rDNA in nucleolar caps could act to constrain the mobility of broken ends.
Confining damaged and intact rDNA repeats together within nucleolar caps has a number of predicted consequences. First, it could enhance access by the repair machinery normally excluded from nucleoli, as shown here for RPA2 (Supplemental Fig. S4). Second, the availability of undamaged rDNA in close proximity could facilitate repair by HR. The finding that these DSB-induced nucleolar caps contain both 53BP1 and BRCA1 could suggest that both NHEJ and HR repair pathways are being used. However, analysis of downstream factors suggests that repair is predominantly by the HR pathway even in G1 cells. As we demonstrated the existence of damage-induced nucleolar caps containing a single NOR (Fig. 5C), it is reasonable to propose that homology-directed repair can be templated by intact rDNA repeats within the same NOR (Fig. 7).
NHEJ appears to be the dominant repair pathway across the genome as a whole, irrespective of cell cycle stage (Karanam et al. 2012). Therefore, mechanisms may exist within nucleoli to suppress NHEJ in favor of HR. Possible players include DNA-PK and 53BP1 (Jackson 2002; Panier and Boulton 2014). DNA-PK and Ku are not detected at nucleolar caps and presumably are unable to suppress end resection. Another proposed mechanism for suppressing NHEJ is by preventing 53BP1 binding and promoting BRCA1 binding at or close to ends. This model involves acetylation of histone H4 on K16, preventing recognition of monomethylated or dimethylated histone H4K20 by the Tudor domain of 53BP1 (Tang et al. 2013). High-resolution microscopy has revealed nonoverlapping 53BP1 and BRCA1 signals within γ-irradiation-induced foci (Chapman et al. 2012a). Similarly, we observed nonoverlapping 53BP1 and BRCA1 signals within DSB-induced nucleolar caps. A candidate histone acetyltransferase responsible for H4K16 acetylation is Tip60 (KAT5), a known interaction partner for both ATM and UBF (Halkidou et al. 2004; Sun et al. 2005). We expect that the local chromatin environment plays a role in promoting repair of DSBs within rDNA by the HR pathway.
In our experimental approach, all rDNA repeats within the nucleus are potential DSB targets, resulting in high levels of DNA damage. Consequently, the response that we observed, particularly using I-PpoI, is likely to represent an extreme version of a nucleolar DDR. Notably, in the CRISPR/Cas9 experiments, we often observed incomplete transcriptional inhibition and nucleolar reorganization, possibly reflecting silencing and repositioning of some but not all NORs (Fig. 7). Future experiments with NORs engineered to contain rDNA repeats with novel DSB targets will facilitate examination of responses to lower levels of damage.
Repair of DSBs by the HR pathway in G1 cells (i.e., if templated by repeats in cis) may involve crossover mechanisms that would lead to instability in the rDNA array. This could be prevented by using the Bloom's helicase (BLM)-dependent branch migration pathway, which does not result in crossover (Karow et al. 2000; Renkawitz et al. 2014). Interestingly in human cells lacking functional BLM, 10-fold elevated rates of sister chromatid exchange within rDNA arrays are observed (Killen et al. 2009). The synthesis-dependent strand annealing (SDSA) may provide an alternative noncrossover pathway (Renkawitz et al. 2014).
Targeting DSBs to the DJ sequences immediately adjacent to rDNA arrays is beginning to reveal a regional specificity to the nucleolar DDR. All Cas9/DJ gRNA transfected cells exhibit a γH2AX response, yet most show little or no transcriptional inhibition. As our knowledge of the genomic architecture of acrocentric chromosome short arms improves, we can more fully investigate the spatial restriction of the nucleolar DDR.
A major feature of the nucleolar DDR that we uncovered is the directed movement of damaged rDNA to the nucleolar periphery (Fig. 7). Recently, we demonstrated that DJ sequences embedded in perinucleolar heterochromatin facilitate a similar directed movement in AMD-treated cells (Floutsakou et al. 2013). A corollary of this is that chromosomal context, in particular the presence of adjacent heterochromatin and DJ sequences, plays a role in the long-term genomic stability of rDNA arrays. We predict that in cases where rDNA arrays are out of their normal chromosomal context, damaged repeats may remain within the nucleolar interior and be repaired less efficiently or by the error-prone NHEJ pathway. Interestingly, juxtaposition of rDNA with either centromeric or telomeric heterochromatin appears to be an evolutionarily conserved feature of higher eukaryotes. Moreover, disruption of heterochromatin has been shown to impact on the stability of rDNA arrays in Drosophila (Peng and Karpen 2007). Finally, it remains to be seen whether the response to DSBs in rDNA arrays observed here can be extended to other transcriptionally active repeat arrays.
Materials and methods
Cell lines
hTERT-RPE1 cells (obtained from American Type Culture Collection) were maintained in DMEM/nutrient mixture F-12 Ham containing 2 mM L-glutamine, 10% (v/v) FBS, and 0.25% (v/v) sodium bicarbonate. 1BR3, CCD-1079Sk, and BJ primary cells were grown in DMEM/nutrient mixture F-12 Ham supplemented with 2 mM L-glutamine, 10% FBS, and nonessential amino acids.
RPE1 cells were transfected with FUCCI expression plasmids pcDNA3-mCherry-Cdt1 (30–120) or pcDNA3-mAG-Geminin (1–110) (Dr. A. Miyawaki, RIKEN Institute; modified by Chelly van Vuuren), and stable integrants were selected with 1 mg/mL G418 (Melford). A stable cell line expressing RPA2-GFP was generated by transfecting RPE1 cells with eGFP-RPA2-N1 (Dr. M Carty, National University of Ireland, Galway).
For ATM and DNA-PK inhibition, kinase inhibitors KU55933 (SelleckChem) and NU7441 (Axon Medchem) were added to a final concentration of 10 μM at the point of I-PpoI transfection.
I-PpoI mRNA production and transfection
In order to produce the in vitro transcribed I-PpoI message, we constructed pIRES I-PpoI, which comprises an IRES element (EcoRI–NcoI) fused to the I-PpoI ORF (NcoI–XbaI) and cloned into the vector pBluescriptSK+ (Stratagene). To facilitate visualization of I-PpoI in transfected cells, a sequence encoding a V5 epitope tag was inserted into the NcoI site to generate pIRES V5 I-PpoI. To construct a catalytically dead version, histidine 98 of I-PpoI was converted to an alanine by site-directed mutagenesis to generate pIRES V5 I-PpoI H98A (Eklund et al. 2007). Plasmids were linearized at a NotI site positioned in the polylinker downstream from the I-PpoI ORF and transcribed using a MEGAscript T7 kit (Ambion) according to the manufacturer's instructions. I-PpoI mRNA was subsequently polyadenylated using a Poly(A) tailing kit (Ambion) according to the manufacturer's instructions.
Cells were seeded on glass coverslips in a six-well plate at least 36 h prior to transfection with in vitro transcribed mRNA using the TransMessenger transfection reagent (Qiagen). One microgram of I-PpoI mRNA and 2 μL of Enhancer R were diluted in buffer EC-R to a final volume of 100 μL and incubated for 5 min at room temperature. Two microliters of TransMessenger transfection reagent was added and further incubated for 10 min at room temperature. After addition of 900 μL of serum-free medium, the transfection cocktail was added to cells. Following 4 h of incubation, the transfection medium was replaced by full medium, and cells were grown for an additional 2 h.
CRISPR/Cas9
We used a CRISPR/Cas9 system described by Mali et al. (2013). hCas9 (a human codon-optimized Cas9 expression plasmid with a C-terminal NLS) and a gRNA empty vector were obtained from Addgene. The gRNA empty vector was modified by site-directed mutagenesis to incorporate the A–U flip (F) and stem extension (E) mutations that stabilize Cas9/gRNA complexes (Chen et al. 2013). gRNA target sequences were integrated into AflII linearized gRNA-FE vector by Gibson assembly (New England Biolabs). Optimized CRISPR Design (http://crispr.mit.edu) aided gRNA target selection. The gRNA target sequences are provided in Supplemental Table S1. To introduce targeted DSBs, hCas9 and gRNA plasmid were cotransfected in a 1:4 weight ratio into RPE1 cells using a standard calcium phosphate protocol.
Antibodies
The commercial antibodies used were as follows: ATM (clone MAT3-4G10/8; Sigma-Aldrich, A1106), Rad52 (Cell Signaling Technology, #3425), Ku80 (Cell Signaling Technology, #2180), BRCA1 (Millipore, #07-434), γH2AX (Millipore, #05-636; Cell Signaling Technology, #9718), 53BP1(Bethyl Laboratories, A300-272A), XRCC4 (Novus, NBP1-30878), Rad51 (Thermo Scientific, MA1-23271), DNA-PK (Thermo Scientific, MA5-15813), phospho-ATM S1981 (Thermo Scientific, MA1-2020), V5 (Serotec, MCA1360), BrdU (Roche, 11170376001), Cas9 mAb (Diagenode, C15200203), RPA2 (Millipore, #MABE285), and phospho-RPA2-S4/S8 (Bethyl Laboratories, A300-245A). Fibrillarin antibody (mAb clone72B9) was a gift from U. Scheer (Wurzburg). Antibodies against UBF, Treacle, Nop52, Rrn3 and Paf49 were raised in sheep against recombinant proteins. For double-labeling experiments, 53BP1 and BRCA1 antibodies were directly labeled using Zenon tricolor rabbit IgG-labeling kit #2 (Life Technologies) according to the manufacturer's instructions. Secondary antibodies for immunofluorescence were purchased from Jackson ImmunoResearch.
Cell staining and imaging
For immunofluorescent staining, cells were fixed in 4% (w/v) paraformaldehyde (PFA) in PBS for 10 min and permeabilized with 0.5% (w/v) saponin and 0.5% (v/v) Triton X-100/PBS for 10 min. Primary antibodies were diluted in 1% (w/v) bovine serum albumin (BSA)/PBS and incubated in a humidity box for 1 h at 37°C followed by incubation with secondary antibodies for 40 min. Coverslips were mounted in VectorShield (Vector Laboratories) with or without DAPI, as appropriate. For pre-extraction, coverslips were incubated for 10 min on ice in 0.5% (v/v) Triton X-100/PBS before fixation.
For 3D immunoFISH, cells grown on Superfrost Plus microscopic slides (VWR) were fixed, denatured, probed, and antibody-stained as described previously (Mais et al. 2005; Prieto and McStay 2007), with the exception that cells were stained with γH2AX or RPA2 primary and secondary antibodies and fixed with 2% PFA prior to FISH. All DNA probes used have been described previously (Floutsakou et al. 2013).
In all imaging experiments, 30–40 Z-stacks of fluorescent images were captured using a Photometric Coolsnap HQ camera and Volocity 6 imaging software (PerkinElmer) with a 63× Plan Apochromat Zeiss objective mounted on a Zeiss Axioplan2 imaging microscope. Images were deconvolved by iterative restoration in Volocity 6 with a confidence limit of 95% and a maximum of 50 iterations. In some cases, extended focus projections of deconvolved Z-stacks are presented, while, in other cases, individual focal planes are shown.
Transcription and replication assays
BrUTP incorporation assays were performed as previously described (Mais et al. 2005). For click chemistry-based transcription and replication assays, cells were incubated with 1 mM EU or 10 μM EdU (Berry & Associates, Inc.) for 1 h or 30 min, respectively. Coverslips were fixed with 4% (w/v) PFA/PBS, and cells were permeabilized with 0.5% (w/v) saponin and 0.5% (v/v) Triton X-100/PBS. Sites of synthesis were visualized with the 6-carboxyfluorescein-TEG azide (Berry & Associates, Inc.), Click-IT Alexa 488, or Click-IT Alexa 594 (Life Technologies) according to the manufacturers’ instructions.
Visualization of damage-induced DNA synthesis
RPE1, mAG-Geminin RPE1, or mCherry-Cdt1 RPE1 cells grown on coverslips were transfected with I-PpoI mRNA as described above. Four hours after transfection, fresh medium containing 10 μM EdU was added, and cells were incubated for a further 2 h. Coverslips were then fixed with 4% (w/v) PFA/PBS, and cells were permeabilized with 0.5% (w/v) saponin and 0.5% (v/v) Triton X-100/PBS. EdU incorporation in RPE1 and mAG-Geminin RPE1 cells was visualized using biotin-TEG azide (Berry & Associates, Inc.) followed by staining with Cy3-conjugated streptavidin (Rockland, S000-04). In mCherry-Cdt1 RPE1 cells, biotin-TEG azide was followed by sequential staining with Alexa488-conjugated streptavidin (Life Technologies, S32354), biotin-conjugated anti-streptavidin (Rockland, 200–4695), and Alexa488-conjugated streptavidin.
Supplementary Material
Acknowledgments
We thank Chelly van Vuuren and Eleanor Glancy for help with CRISPR/Cas9 and providing reagents, Atsushi Miyawaki for the FUCCI system, and Michael Carty for RPA2 reagents. We also thank Carol Duffy and Noel Lowndes for comments on the manuscript. M.v.S. acknowledges support from the Beckman Fund. B.M. acknowledges Science Foundation Ireland (PI grant 07/IN.1/B924) and the Science Foundation Ireland-Health Research Board-Wellcome Trust Biomedical Research Partnership (Investigator Award 106199/Z/14/Z) for funding work in his laboratory.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.260703.115.
Freely available online through the Genes & Development Open Access option.
References
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI. 2002. Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1–11. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Kupiec M. 2004. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23: 4868–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. 2014. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21: 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJ Jr, Kastan MB. 2007. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 9: 683–690. [DOI] [PubMed] [Google Scholar]
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. 2010. The nucleolus under stress. Mol Cell 40: 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276: 42462–42467. [DOI] [PubMed] [Google Scholar]
- Chao SH, Price DH. 2001. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276: 31793–31799. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. 2012a. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 125: 3529–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, Boulton SJ. 2012b. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47: 497–510. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. 2013. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155: 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. 2011. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Huang JW, Izhar L, Sowa ME, Harper JW, Elledge SJ. 2014. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc Natl Acad Sci 111: 18631–18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund JL, Ulge UY, Eastberg J, Monnat RJ Jr. 2007. Altered target site specificity variants of the I-PpoI His-Cys box homing endonuclease. Nucleic Acids Res 35: 5839–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floutsakou I, Agrawal S, Nguyen TT, Seoighe C, Ganley AR, McStay B. 2013. The shared genomic architecture of human nucleolar organizer regions. Genome Res 23: 2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob A, Colleran C, McStay B. 2014. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev 28: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjiolov AA. 1985. The nucleolus and ribosome biogenesis. Springer-Verlag, Wien, NY. [Google Scholar]
- Halkidou K, Logan IR, Cook S, Neal DE, Robson CN. 2004. Putative involvement of the histone acetyltransferase Tip60 in ribosomal gene transcription. Nucleic Acids Res 32: 1654–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. 2007. The DNA damage response: ten years after. Mol Cell 28: 739–745. [DOI] [PubMed] [Google Scholar]
- Heix J, Vente A, Voit R, Budde A, Michaelidis TM, Grummt I. 1998. Mitotic silencing of human rRNA synthesis: inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. Embo J 17: 7373–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. 2009. DNA damage, aging, and cancer. N Engl J Med 361: 1475–1485. [DOI] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G. 2010. High-resolution profiling of γH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29: 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP. 2002. Sensing and repairing DNA double-strand breaks. Carcinogenesis 23: 687–696. [DOI] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A, Lahav G. 2012. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 47: 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow JK, Constantinou A, Li JL, West SC, Hickson ID. 2000. The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci 97: 6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killen MW, Stults DM, Adachi N, Hanakahi L, Pierce AJ. 2009. Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum Mol Genet 18: 3417–3428. [DOI] [PubMed] [Google Scholar]
- Kruhlak M, Crouch EE, Orlov M, Montano C, Gorski SA, Nussenzweig A, Misteli T, Phair RD, Casellas R. 2007. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447: 730–734. [DOI] [PubMed] [Google Scholar]
- Larsen DH, Hari F, Clapperton JA, Gwerder M, Gutsche K, Altmeyer M, Jungmichel S, Toledo LI, Fink D, Rask MB, et al. 2014. The NBS1–Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol 16: 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC. 2004. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett 14: 6083–6087. [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K. 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4: 712–720. [DOI] [PubMed] [Google Scholar]
- Mais C, Wright JE, Prieto JL, Raggett SL, McStay B. 2005. UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev 19: 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166. [DOI] [PubMed] [Google Scholar]
- Moore HM, Bai B, Boisvert FM, Latonen L, Rantanen V, Simpson JC, Pepperkok R, Lamond AI, Laiho M. 2011. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol Cell Proteomics 10: M111 009241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JM, Stiff T, Jeggo PA. 2012. DNA double-strand break repair within heterochromatic regions. Biochem Soc Trans 40: 173–178. [DOI] [PubMed] [Google Scholar]
- Muscarella DE, Ellison EL, Ruoff BM, Vogt VM. 1990. Characterization of I-Ppo, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Mol Cell Biol 10: 3386–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth A, Langst G. 2011. Genome organization in and around the nucleolus. Trends Genet 27: 149–156. [DOI] [PubMed] [Google Scholar]
- O'Sullivan AC, Sullivan GJ, McStay B. 2002. UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat. Mol Cell Biol 22: 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Boulton SJ. 2014. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7–18. [DOI] [PubMed] [Google Scholar]
- Pankotai T, Bonhomme C, Chen D, Soutoglou E. 2012. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat Struct Mol Biol 19: 276–282. [DOI] [PubMed] [Google Scholar]
- Pederson T. 2010. The nucleolus. Cold Spring Harb Perspect Biol 3: a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, Karpen GH. 2007. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol 9: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto JL, McStay B. 2007. Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev 21: 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawitz J, Lademann CA, Jentsch S. 2014. Mechanisms and principles of homology search during recombination. Nat Rev Mol Cell Biol 15: 369–383. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146: 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel P, Andre C, Masson C, Geraud G, Hernandez VD. 1993. Localization of the RNA polymerase I transcription factor hUBF during the cell cycle. J Cell Sci 104: 327–337. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. 2008. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132: 487–498. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210. [PubMed] [Google Scholar]
- Sirri V, Urcuqui-Inchima S, Roussel P, Hernandez-Verdun D. 2008. Nucleolus: the fascinating nuclear body. Histochem Cell Biol 129: 13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard BL. 2005. Homing endonuclease structure and function. Q Rev Biophys 38: 49–95. [DOI] [PubMed] [Google Scholar]
- Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, Yang V, Beausoleil SA, Gygi SP, Livingstone M, et al. 2007. Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci 104: 19855–19860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Pierce HH, Pierce AJ. 2008. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res 18: 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. 2005. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci 102: 13182–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. 2013. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragon L, Lisby M. 2007. The Smc5–Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9: 923–931. [DOI] [PubMed] [Google Scholar]
- Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. 2004. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci 101: 10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.