

Abstract
Abnormal high mobility group protein B1 (HMGB1) activation is involved in the pathogenesis of pulmonary fibrosis. Pulmonary rehabilitation mixture (PRM), which combines extracts from eight traditional Chinese medicines, has very good lung protection in clinical use. However, it is not known if PRM has anti-fibrotic activity. In this study, we investigated the effects of PRM on transforming growth factor-β1 (TGF-β1)-mediated and bleomycin (BLM)-induced pulmonary fibrosis in vitro and in vivo. The effects of PRM on TGF-β1-mediated epithelial-mesenchymal transition (EMT) in A549 cells, on the proliferation of human lung fibroblasts (HLF-1) in vitro, and on BLM-induced pulmonary fibrosis in vivo were investigated. PRM treatment resulted in a reduction of EMT in A549 cells that was associated with attenuating an increase of vimentin and a decrease of E-cadherin. PRM inhibited the proliferation of HLF-1 at an IC50 of 0.51 µg/mL. PRM ameliorated BLM-induced pulmonary fibrosis in rats, with reduction of histopathological scores and collagen deposition, and a decrease in α-smooth muscle actin (α-SMA) and HMGB1 expression. An increase in receptor for advanced glycation end-product (RAGE) expression was found in BLM-instilled lungs. PRM significantly decreased EMT and prevented pulmonary fibrosis through decreasing HMGB1 and regulating RAGE in vitro and in vivo. PRM inhibited TGF-β1-induced EMT via decreased HMGB1 and vimentin and increased RAGE and E-cadherin levels. In summary, PRM prevented experimental pulmonary fibrosis by modulating the HMGB1/RAGE pathway.
Keywords: High mobility group protein B1, Epithelial-mesenchymal transition, Pulmonary rehabilitation mixture, Receptor for advanced glycation end-product
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive lung disease that is irreversible, usually lethal, and does not have a known cause. Among the different types of IPF, the most common form is idiopathic interstitial pneumonia (1). The characteristic histopathological pattern of IPF includes type II pneumocyte hyperplasia and abnormal mesenchymal cell proliferation. Varying degrees of fibrosis and collagen and extracellular matrix (ECM) deposition can lead to distortion of pulmonary architecture and honeycombing. Fibrotic foci are often observed in the honeycomb lesions, which accumulate fibroblasts and myofibroblasts within the developing ECM (2,3). Although significant advances have been made in understanding the pathogenesis of pulmonary fibrosis, the underlying mechanisms that contribute to IPF remain unclear.
High mobility group protein B1 (HMGB1), also known as high mobility group protein 1 (HMG-1), is a nuclear protein and an architectural chromatin-binding factor that binds DNA and promotes protein assembly on specific DNA targets. Outside the cell, HMGB1 is a potent mediator of inflammation that binds to the receptor for advanced glycation end products (RAGE) with high affinity (4). It causes an acute inflammatory response with accumulation of neutrophils in the interstitial and intra-alveolar areas and production of proinflammatory cytokines in the lung (5). HMGB1 is upregulated in experimental pulmonary fibrosis, particularly during the phase of acute exacerbation, which suggests HMGB1 can be a therapeutic target for IPF (6,7).
Pulmonary rehabilitation mixture (PRM) comprises eight traditional Chinese medicines, including Astragalus membranaceus (Fisch) Bge., Codonopsis pilosula (Franch.) Nannf., Ophiopogon japonicas, Schisandra chinensis, Panax notoginseng (Burk.) F. H. Chen., Bulbus fritillariae thunbergii, Rhizoma anemarrhenae, and Glycyrrhiza uralensis. Pharmacological activity of PRM has not yet been reported in experimental pulmonary fibrosis. We therefore investigated the effects of PRM on experimental pulmonary fibrosis in vitro and in vivo, and propose a mechanism of action.
Material and Methods
PRM extraction
Dried A. membranaceus (Fisch) Bge. 1200 g, C. pilosula (Franch.) Nannf. 700 g, O. japonicas 480 g, S. chinensis 360 g, P. notoginseng (Burk.) F.H. Chen. 240 g, Fritillaria thunbergiiMiq. 240 g, Rhizoma anemarrhenae 240 g, and G. uralensis 120 g were combined and then extracted twice with 20 L 50% EtOH for 24 h each time. The extracts were combined, and the EtOH was collected. Distilled water was added to the crude extract thus obtained to make a total volume of 10 L. The extract was then centrifuged at 3000 g for 15 min at 4°C, and the supernatant was collected.
Animals
Adult male Sprague-Dawley rats (body weight 180-200 g) were housed individually at a constant temperature (22±2°C) and humidity with a 12-h light/dark cycle and free access to chow and water. All experimental procedures in this study were performed in accordance with the Institutional Animal Care and National Institutes of Health Guide for the Care and Use of Laboratory Animals (USA) guidelines. The protocol was approved by the Committee on the Ethics of Animal Experiments of Binzhou Medical University (Permit no. SCXK 2009 0009).
Cell culture
Human lung fibroblasts (HLF-1), human type I alveolar epithelial cells (AT I) and human type II alveolar epithelial cells (A549 cell line) were purchased from the Cell Bank of the Chinese Academy of Sciences. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% newborn calf serum, 100 U/mL penicillin and 100 µg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2 and 95% N2. Cells were subcultured every 3-4 days when reaching a density of 1×105/mL.
Epithelial-to-mesenchymal transition (EMT) of A549 cells in vitro
A549 cells were maintained in DMEM/F12 containing 10% (v/v) fetal bovine serum, 100 kU/L penicillin and 100 mg/L streptomycin at 37°C in a humidified 5% CO2 atmosphere. When cultures reached approximately 70% confluency, cells were starved in serum-free DMEM overnight and treated with PRM at concentrations of 0, 0.1, 0.3, and 1 μg/mL with or without 5 ng/mL of transforming growth factor-β1 (TGF-β1; Sigma, USA) for 48 h. Pictures were then taken of five random fields at 200× magnification using an inverted microscope. To investigate the mechanism of a PRM effect on cell morphology, HMGB1 small interfering RNA (siRNA; Santa Cruz, USA) was added to cultures prior to incubation with or without TGF-β1.
HLF-1 proliferation assay
For in vitro proliferation assays, HLF-1 was inoculated into 96-well (1×105 cells/well) flat bottom plates in triplicate with medium alone (control) or with medium containing different concentrations of PRM (0, 0.1, 0.3, and 1 μg/mL) with or without 5 ng/mL of TGF-β1. Cell proliferation was tested using a methythiazol tetrazolium (MTT) assay. The absorbance was recorded at 490 nm (Spectramax/M5 multi-detection reader, Molecular Devices, USA), and calculated as a ratio against untreated cells. To investigate the mechanism of PRM on cell proliferation, fibroblast growth factor (FGF) 2 and platelet-derived growth factor (PDGF) expression in TGF-β1-stimulated HLF-1 cells was analyzed in Western blots.
Determination of apoptosis in human A549 and AT I cells
A549 cells were cultured to approximately 70% confluency, starved in serum-free DMEM overnight, and then treated with TGF-β1 5 ng/mL for 48 h. The culture supernatant was collected. AT I cells were maintained in DMEM/F12 containing 10% (v/v) fetal bovine serum, 100 U/L penicillin and 100 mg/L streptomycin at 37°C in a humidified 5% CO2 atmosphere. When the cells were cultured to approximately 80% confluency, DMEM/F12 was replaced with the collected supernatant containing PRM (0, 0.1, 0.3, or 1 μg/mL), and the cells were cultured for 4 h. Apoptotic cells were evaluated with an annexin-V fluorescein isothiocyanate (FITC) apoptosis detection kit. The cells were harvested, washed, and incubated at 4°C for 30 min in the dark with annexin-V FITC and propidium iodide and assayed by flow cytometry (FACSVantage SE; Beckman Coulter, USA).
Western blot analysis in vitro
A549 cells were cultured for 24 h, and then washed twice with ice cold PBS and lysed in NP40 lysis buffer (Biosource, USA; 50 mM Tris, pH 7.4, 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 1% NP-40 and 0.02% NaN3) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1× protease inhibitor cocktail (Sigma). Aliquots of 50 μg total cell protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed in Western blots using antibodies specific to HMGB1, RAGE, vimentin, E-cadherin, and proliferating cell nuclear antigen (PCNA) as a loading control.
HLF-1 cells were cultured for 48 h, washed twice with ice-cold PBS, and lysed in NP40 lysis buffer supplemented with 1 mM PMSF and 1× protease inhibitor cocktail. Aliquots of 50 μg total cell protein were separated by SDS-PAGE and analyzed in Western blots using antibodies specific to PDGF, FGF2, and PCNA as a loading control. Optical densities of the bands were scanned and quantified with a Gel Doc 2000 (Bio-Rad Laboratories Ltd., USA). Data were normalized against that of the corresponding PCNA bands. Results are reported as percentage increase over the normal.
BLM-induced pulmonary fibrosis model
Thirty Sprague-Dawley rats with an average weight of 180 g were acclimatized for 7 continuous days. The pulmonary fibrosis model was established, with a minor modification, as previously described (8). Briefly, the rats were anesthetized by intraperitoneal (ip) injection of 0.5 mg/kg ketamine and 1 mg/kg xylazine, and lung injury was induced by a single intratracheal instillation of 6 mg/kg bleomycin (BLM) hydrochloride (Nippon Kayaku, Japan) in 0.3 mL saline. The control rats received an equal volume of saline only. Beginning on day 21, the pulmonary fibrosis rats were randomly divided into two groups by body weight: a BLM group (vehicle-treated model) and a PRM 3.0 g/kg (as total crude drug) group. PRM was administered orally once daily. The lungs of all rats were removed on day 35, and divided into two parts for further analysis. One part was frozen in liquid nitrogen, and the other was fixed in 10% formalin.
Histopathological examination
Lung tissue fixed in 10% formalin was embedded in paraffin, sectioned and stained with hematoxylin and eosin (HE). Each examined field was individually assessed for degree of interstitial fibrosis and graded as previously described (9) by an investigator who was blinded to the experimental groups.
Collagen deposition was evaluated in 4-µm paraffin sections that were stained with Masson's trichrome. Collagen deposition in the lung was determined by counting the number of pixels in collagen stained areas in digital images using Adobe Photoshop CS4 as previously described (10).
Immunohistochemistry
The tissue sections (4 µm) were deparaffinized, rehydrated, and treated in 0.01 M citric acid at 400 W in a microwave for 10 min. The endogenous peroxidase was inactivated with 5% H2O2 in methanol for 30 min at room temperature in the dark. The sections were then sealed with a serum cap for 30 min and incubated with anti-α-SMA or anti-HMGB1 for 16 h at 4°C. They were then washed and incubated with anti-rabbit horseradish peroxidase-conjugated antibody for 60 min at 37°C. The samples were observed by light microscopy.
Measurement of hydroxyproline (Hyp)
Lung specimens were washed with normal saline, hydrolyzed in 6 mL/L hydrochloric acid at 100°C for 5 h, and then neutralized with sodium hydroxide. The Hyp level in the hydrolysates was assessed at 560 nm by a colorimetric assay using p-dimethylaminobenzaldehyde and was reported as mg/g wet tissue.
Western blot analysis of lung tissue
Lung samples were suspended in a buffer containing 10 mM Tris, pH 7.5, 1.5 mM MgCl2, 10 mM KCl, and 0.1% Triton X-100 and lysed by homogenization. The supernatant was collected and stored at -80°C for Western blot analysis. Protein concentration of the extracts was measured by a bicinchoninic acid (BCA) assay. Aliquots of 50 µg protein were separated by SDS-PAGE and analyzed in Western blot using antibodies specific to α-SMA, PDGF, FGF2, HMGB1, RAGE and PCNA. Optical densities of the bands were scanned and quantified with a Gel Doc 2000. Densities were normalized against those of the corresponding PCNA bands, and the results are reported as percentage increase over the control group.
Statistical analysis
The grades of fibrosis between the groups were compared using the rank-sum test. Quantitative data are reported as means±SD and the significance of differences was determined by one-way analysis of variance (ANOVA) followed by the Dunnett's test. P<0.05 was considered to be statistically significant.
Results
Effects of PRM on EMT in A549 cells
As shown in Figure 1A, EMT was significantly enhanced in A549 cells treated with 5 ng/mL TGF-β1 for 48 h. When the TGF-β1-stimulated cells were exposed to 0.3 μg/mL PRM for 48 h, EMT was attenuated. To investigate how PRM might reduce EMT, its effects on the expression of the epithelial cell marker E-cadherin and the mesenchymal cell marker vimentin were analyzed in Western blots. Figure 1B and C shows that PRM treatment of TGF-β1-stimulated A549 cells attenuated both a decrease of E-cadherin and an increase of vimentin.
Figure 1. Effects of pulmonary rehabilitation mixture (PRM) on epithelial-mesenchymal transition (EMT) in A549 cells. A, Representative light microscopic appearance of A549 cells for the normal (A1), transforming growth factor (TGF)-β1-treated (A2), and TGF-β1+0.3 µg/mL PRM (A3). B-D, A549 cells were incubated with TGF-β1 (5 ng/mL) for 24 h. E-cadherin, vimentin, high mobility group protein B1 (HMGB1), and receptor for advanced glycation end-product (RAGE) expression were analyzed by Western blot. Results are reported as percent increase over the normal. Data are reported as means±SD. PCNA: proliferating cell nuclear antigen. #P<0.01 vs the normal group; *P<0.05 vs the TGF-β1 group (one-way ANOVA followed by Dunnett's test).

Effects of PRM on HMGB1 and RAGE expression in TGF-β1-stimulated A549 cells
TGF-β1 significantly promoted HMGB1 expression, and reduced RAGE expression in the TGF-β1-stimulated A549 cells. However, PRM attenuated the increase of HMGB1, whereas it increased its receptor RAGE expression in the TGF-β1-stimulated A549 cells, as shown in Figure 1B and D.
To investigate the mechanism of PRM on EMT, HMGB1 and RAGE expression were both analyzed. In TGF-β1-stimulated A549 cells plus HMGB1 siRNA, HMGB1 expression was decreased significantly. In addition, the EMT was attenuated accompanied with a higher E-cadherin expression and a lower vimentin expression, as shown in Figure 2A. Compared to the TGF-β1-stimulated A549 cells plus HMGB1 siRNA, PRM treatment with the TGF-β1-stimulated A549 cells plus HMGB1 siRNA had no additional influence on E-cadherin and vimentin expression, as shown in Figure 2B and C. It suggested that PRM might be due to blockage of EMT through a HMGB1 signaling pathway.
Figure 2. Effects of pulmonary rehabilitation mixture (PRM) on epithelial-mesenchymal transition (EMT) in high mobility group protein B1 (HMGB1) small interfering RNA (siRNA) A549 cells. Representative light microscopic appearance of HMGB1 siRNA A549 cells for HMGB1 siRNA A549 (A1), transforming growth factor (TGF)-β1-treated HMGB1 siRNA A549 (A2), and TGF-β1+PRM 0.3 µg/mL HMGB1 siRNA A549 (A3). B-D, HMGB1 siRNA A549 cells were incubated with TGF-β1 (5 ng/mL) for 24 h. E-cadherin, vimentin and HMGB1 expression were analyzed by Western blotting. Results are reported as means±SD percent increase over the normal. PCNA: proliferating cell nuclear antigen. #P<0.01 vs the HMGB1 siRNA group (one-way ANOVA followed by Dunnett's test).

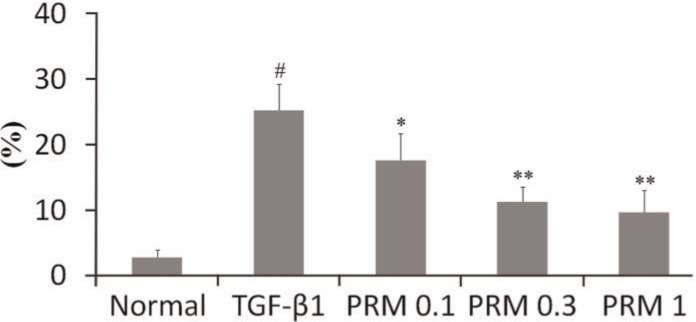
Effects of PRM on apoptosis in AT1 cells
The apoptosis of AT1 was detected by flow cytometry after AT1 was treated with the supernatant (5 ng/mL) of TGF-β1-stimulated A549 cells. The apoptotic rate of AT1 cells, which were treated with the supernatant of TGF-β1-stimulated A549 cells for 4 h, reached 25.2%, as shown in Figure 3. However, the rates decreased to 17.5, 11.2 and 9.6% after PRM (0.1-1 μg/mL) treatment, respectively.
Figure 3. Effects of pulmonary rehabilitation mixture (PRM) on cell apoptosis in type I alveolar epithelial cells (AT1). The apoptosis of AT1 was detected by flow cytometry when they were treated with the supernatant (5 ng/mL) of transforming growth factor (TGF)-β1-stimulated A549 cells for 4 h. Data are reported as means±SD. #P<0.01 vs the normal group; *P<0.05, **P<0.01 vs TGF-β1-stimulated supernatant group (one-way ANOVA followed by Dunnett's test).
Effects of PRM on HLF-1 proliferation, FGF2 and PDGF expression
HLF-1 cells were tested in the presence of TGF-β1 to mimic a pro-fibrotic environment. Under these conditions, TGF-β1 induced HLF-1 proliferation, as shown in Figure 4A. PRM inhibited proliferation of HLF-1 with an IC50 of 0.51 µg/mL.
Figure 4. Effects of pulmonary rehabilitation mixture (PRM) on human lung fibroblasts (HLF)-1 proliferation, fibroblast growth factor (FGF)-2 and platelet-derived growth factor (PDGF) expression. A, Effects of PRM on HLF-1 proliferation. Cell proliferation was tested by MTT when they were treated with 5 ng/mL transforming growth factor (TGF)-β1-stimulated HLF-1 for 48 h. B,C, HLF-1 was incubated with TGF-β1 (5 ng/mL) for 48 h and FGF2 and PDGF expression were analyzed by Western blotting. Data are reported as means±SD. PCNA: proliferating cell nuclear antigen. #P<0.01 vs the normal group; *P<0.05, **P<0.01 vs the TGF-β1 group (one-way ANOVA followed by Dunnett's test).

To demonstrate the mechanism of PRM on HLF-1 proliferation, FGF2 and PDGF expression were investigated. In TGF-β1-stimulated HLF-1 cells plus PRM 0.3 μg/mL, FGF2 and PDGF expression were decreased significantly and HLF-1 proliferation was attenuated, as shown in Figure 4C. This suggested that PRM might be due to block of HLF-1 proliferation through reduction of FGF2 and PDGF expression.
Effects of PRM on histopathological change and collagen content in vivo
The collagen content and collagen fibers in the lung tissues were observed by Masson's trichrome staining, as shown in Figure 5. The interstitial lung of the BLM-treated rat exhibited a large number of collagen fibers. The collagen and fiber contents were significantly reduced in the PRM-treated rats. In addition, another index of collagen accumulation, Hyp content, was assessed and the results showed that less Hyp was found in the PRM-treated lung (Table 1).
Figure 5. Effects of pulmonary rehabilitation mixture (PRM) on histopathological change and collagen content in bleomycin (BLM)-treated animals. Representative light microscopic appearance of HE staining and Masson staining in lung tissue for the normal (A1, B1), model (A2, B2), and 3 g/kg PRM (A3, B3). C, Effects of PRM on collagen content in BLM-treated animals. Data are reported as means±SD. #P<0.01 vs the normal group; *P<0.01 vs the model group (one-way ANOVA followed by Dunnett's test).

Table 1. Grade of lung fibrosis and lung hydroxyproline (Hyp) content.

In the semi-quantitative assessment of the lung sections, no inflammatory or fibrotic changes were observed in the control rats. BLM produced a significant increase in the pathology score compared to the control rats. PRM treatment attenuated the BLM-induced pathology score by 44.9%, as shown in Figure 5 and Table 1.
Effects of PRM on α-SMA, FGF2, PDGF, HMGB1 and RAGE expression in vivo
α-SMA was stained in the cytoplasm, while HMGB1 was stained in the nucleus in the lung tissue, which stained brown for immunohistochemistry, as shown in Figure 6. A smaller amount of α-SMA and HMGB1 was found in PRM-treated rats compared to the BLM-treated rats. FGF2, α-SMA, PDGF, HMGB1 and RAGE in lung tissue were analyzed by Western blot, as shown in Figure 6. The results showed that FGF2, PDGF, α-SMA, HMGB1 and RAGE were all decreased in the PRM rats. It showed that PRM inhibited the EMT and fibroblast proliferation in vitro and in vivo.
Figure 6. Effects of pulmonary rehabilitation mixture (PRM) on α-smooth muscle actin (α-SMA), fibroblast growth factor (FGF)-2, platelet-derived growth factor (PDGF), high mobility group protein B1 (HMGB1), and receptor for advanced glycation end-product (RAGE) expression in bleomycin (BLM)-treated animals. Representative light microscopic appearance of α-SMA and HMGB1 in lung tissue (immunostaining) for the normal (A1, B1), the model (A2, B2), and 3 g/kg PRM (A3, B3). C, Representative Western blots of α-SMA, FGF-2, PDGF, HMGB1 and RAGE. D, E, Effects of PRM on α-SMA, FGF-2, PDGF, HMGB1 and RAGE expression in BLM-treated lung by Western analysis. Data are reported as means±SD. PCNA: proliferating cell nuclear antigen. #P<0.01 vs the normal group; *P<0.01 vs the model group (one-way ANOVA followed by Dunnett's test).

Discussion
IPF is a chronic and ultimately fatal disease characterized by a progressive decline in lung function. IPF usually occurs in adult individuals between 50 and 70 years of age, particularly those with a history of cigarette smoking, and affects more men than women (1). The goals of treatment in IPF are essentially to reduce the symptoms, stop disease progression, prevent acute exacerbations, and prolong survival. Preventive care (e.g., vaccinations) and symptom-based treatment should be started early in every patient (11). However, the treatment options for IPF are very limited. The main aim of treatment is to relieve symptoms as much as possible and to slow down the progression of the disease; thus the identification of novel therapeutic targets is greatly needed.
PRM, which is extracted from eight kinds of traditional Chinese medicines, has very good lung protection in clinical use. In our previous toxicological study, Sprague-Dawley rats were administered with 10 and 30 g/kg PRM (calculated according to the total crude drug) for 4 consecutive weeks followed by a 4-week recovery phase. Transient slight body weight reduction was observed in male rats that received 30 g/kg. Except this, no apparent abnormalities were noted in food consumption, ocular exam, hematology, clinical chemistry, hormone, urine, absolute and relative organ weights, gross necropsy, and histopathology in the study. The no-observed-adverse-effect level (NOAEL) was established to be 30 g/kg for rats.
The fibrotic pathogenesis of the lung is driven by abnormal activated AECs. The secretion of type II pneumocytes and the activation of TGF-β1 promoted EMT and the differentiation of fibroblasts into myofibroblasts contributed to intra-alveolar fibrosis. The epithelial-dependent fibroblast activated process was confirmed in experimental and clinical pulmonary fibrosis (12-14). During the EMT, E-cadherin levels are reduced as well as vimentin levels are increased. As expected, PRM treatment resulted in EMT phenotypic reversion and normalization of vimentin and E-cadherin expression.
AT I covers more than 90% of the alveolar surface area of the peripheral lung, and interfaces with pulmonary capillaries, providing a surface readily permeable to gases. Increased AT I apoptosis leads to ineffective re-epithelialization and damaged alveolar walls (15), and there is higher loss of AT I in patients with IPF. Inhibition of the cell apoptosis of AT1 exerts anti-fibrotic effects in the experimental pulmonary fibrosis (16). Our results showed that the apoptotic rate of AT1 was reduced by the supernatant of TGF-β1-stimulated A549 cells plus PRM. This finding suggests that inhibition of AT1 apoptosis by PRM exerted anti-fibrotic effects and protected lung function.
Pulmonary fibrosis-derived fibroblasts or myofibroblasts synthesized more extracellular matrix components like collagen (17) than the normal lung tissue-derived fibroblasts. The Hyp content reflects the collagen deposition in fibrotic tissues (18). Therefore, we investigated the effect of PRM on Hyp content and the collagen deposition in the fibrotic lung tissue. Our results demonstrated that enhanced collagen deposition and higher Hyp content in the BLM-treated rats were obviously reduced by PRM treatment. This suggested that PRM exerted anti-fibrotic effects by reducing collagen deposition.
PDGF and FGF play important roles in IPF. Inhibition of PDGF and FGF expression attenuates fibrosis (19). The gradual increase of PDGF and FGF were both found in the TGF-β1-stimulated HLF-1 cells and the BLM-treated animals. Our study showed that PRM reduced PDGF and FGF in the TGF-β1-stimulated HLF-1 cells and the BLM-treated animals. These results supported that PRM reduced the proliferation and eased pulmonary fibrosis by suppressing PDGF and FGF expression.
HMGB1, a chromatin-associated nuclear protein and extracellular damage associated molecular pattern molecule (DAMP), is a critical regulator of cell death and survival. Overexpression of HMGB1 is associated with unlimited replicative potential, evasion of programmed cell death (apoptosis), self-sufficiency in growth signals and inflammation (20). HMGB1 also plays a key role in IPF, especially in the late phase of acute exacerbation (6). Reduced serum HMGB-1 levels in patients with IPF with acute exacerbation improved oxygenation (21). In BLM-induced pulmonary fibrosis in mice, HMGB1 protein was predominantly up-regulated in bronchiolar epithelial cells at early phase and in alveolar epithelial and inflammatory cells in fibrotic lesions at later phase. HMGB1 significantly induced proliferation on cultured fibroblasts (7). RAGE is a receptor, which demonstrated to bind HMGB1 (22). Significant down-regulation of RAGE is observed in lung homogenate and alveolar epithelial type II cells from IPF patients, as well as in the BLM-treated animals. RAGE is the signal transduction receptor, which senses a variety of signaling molecules including HMGB1 (23). RAGE plays a protective role in the lung, and loss of RAGE is related to functional changes of pulmonary cell types in pulmonary fibrosis (24). Studies on mouse models of pulmonary fibrosis and human IPF tissues indicate that loss of RAGE contributes to IPF pathogenesis (25). The gradual increase of HMGB1 and decrease of RAGE were both found in the TGF-β1-stimulated A549 cells and the BLM-treated animals. Our study showed that PRM treatment reduced HMGB1 as well as increased RAGE in the TGF-β1-stimulated A549 cells and the BLM-treated animals. These results supported that PRM ameliorates EMT and pulmonary fibrosis through HMGB1/RAGE signaling.
In summary, our results demonstrated that PRM prevented EMT and proliferation, reduced PDGF and FGF expression and ameliorated pulmonary fibrosis through modulating HMGB1/RAGE signaling, which highlights PRM as a new multi-target traditional Chinese medicine with low side effects for pulmonary fibrosis therapy.
Acknowledgments
Research supported by National Natural Science Foundation of China (#31270391 and #81273957) and Taishan Scholar Project to Fang Han.
Footnotes
First published online
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–1315. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 3.Datta A, Scotton CJ, Chambers RC. Novel therapeutic approaches for pulmonary fibrosis. Br J Pharmacol. 2011;163:141–172. doi: 10.1111/j.1476-5381.2011.01247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 5.Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–1316. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- 6.Ebina M, Taniguchi H, Miyasho T, Yamada S, Shibata N, Ohta H, et al. Gradual increase of high mobility group protein b1 in the lungs after the onset of acute exacerbation of idiopathic pulmonary fibrosis. Pulm Med. 2011;2011:916486. doi: 10.1155/2011/916486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamada N, Maeyama T, Kawaguchi T, Yoshimi M, Fukumoto J, Yamada M, et al. The role of high mobility group box1 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:440–447. doi: 10.1165/rcmb.2007-0330OC. [DOI] [PubMed] [Google Scholar]
- 8.Wang HD, Yamaya M, Okinaga S, Jia YX, Kamanaka M, Takahashi H, et al. Bilirubin ameliorates bleomycin-induced pulmonary fibrosis in rats. Am J Respir Crit Care Med. 2002;165:406–411. doi: 10.1164/ajrccm.165.3.2003149. [DOI] [PubMed] [Google Scholar]
- 9.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loughlin PM, Cooke TG, George WD, Gray AJ, Stott DI, Going JJ. Quantifying tumour-infiltrating lymphocyte subsets: a practical immuno-histochemical method. J Immunol Methods. 2007;321:32–40. doi: 10.1016/j.jim.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Lee JS, McLaughlin S, Collard HR. Comprehensive care of the patient with idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2011;17:348–354. doi: 10.1097/MCP.0b013e328349721b. [DOI] [PubMed] [Google Scholar]
- 12.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 13.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 14.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006;3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 15.Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y, Gochuico BR, Yu G, Tang X, Osorio JC, Fernandez IE, et al. Syndecan-2 exerts antifibrotic effects by promoting caveolin-1-mediated transforming growth factor-beta receptor I internalization and inhibiting transforming growth factor-beta1 signaling. Am J Respir Crit Care Med. 2013;188:831–841. doi: 10.1164/rccm.201303-0434OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bocchino M, Agnese S, Fagone E, Svegliati S, Grieco D, Vancheri C, et al. Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS One. 2010;5:e14003. doi: 10.1371/journal.pone.0014003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka K, Ishihara T, Azuma A, Kudoh S, Ebina M, Nukiwa T, et al. Therapeutic effect of lecithinized superoxide dismutase on bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;298:L348–L360. doi: 10.1152/ajplung.00289.2009. [DOI] [PubMed] [Google Scholar]
- 19.Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, Prasse A, Zissel G, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–985. doi: 10.1183/09031936.00152106. [DOI] [PubMed] [Google Scholar]
- 20.Tang D, Kang R, Zeh HJ, III, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abe S, Hayashi H, Seo Y, Matsuda K, Kamio K, Saito Y, et al. Reduction in serum high mobility group box-1 level by polymyxin B-immobilized fiber column in patients with idiopathic pulmonary fibrosis with acute exacerbation. Blood Purif. 2011;32:310–316. doi: 10.1159/000330325. [DOI] [PubMed] [Google Scholar]
- 22.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 23.Xie J, Mendez JD, Mendez-Valenzuela V, Aguilar-Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE) Cell Signal. 2013;25:2185–2197. doi: 10.1016/j.cellsig.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 24.Queisser MA, Kouri FM, Konigshoff M, Wygrecka M, Schubert U, Eickelberg O, et al. Loss of RAGE in pulmonary fibrosis: molecular relations to functional changes in pulmonary cell types. Am J Respir Cell Mol Biol. 2008;39:337–345. doi: 10.1165/rcmb.2007-0244OC. [DOI] [PubMed] [Google Scholar]
- 25.Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172:583–591. doi: 10.2353/ajpath.2008.070569. [DOI] [PMC free article] [PubMed] [Google Scholar]