

Abstract
Background
The World Health Organization (WHO) recommends Artemisinin‐based Combination Therapy (ACT) for treating uncomplicated Plasmodium falciparum malaria. This review aims to assist the decision‐making of malaria control programmes by providing an overview of the relative effects of dihydroartemisinin‐piperaquine (DHA‐P) versus other recommended ACTs.
Objectives
To evaluate the effectiveness and safety of DHA‐P compared to other ACTs for treating uncomplicated P. falciparum malaria in adults and children.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register; the Cochrane Central Register of Controlled Trials (CENTRAL) published in The Cochrane Library; MEDLINE; EMBASE; LILACS, and the metaRegister of Controlled Trials (mRCT) up to July 2013.
Selection criteria
Randomized controlled trials comparing a three‐day course of DHA‐P to a three‐day course of an alternative WHO recommended ACT in uncomplicated P. falciparum malaria.
Data collection and analysis
Two authors independently assessed trials for eligibility and risk of bias, and extracted data. We analysed primary outcomes in line with the WHO 'Protocol for assessing and monitoring antimalarial drug efficacy’ and compared drugs using risk ratios (RR) and 95% confidence intervals (CI). Secondary outcomes were effects on gametocytes, haemoglobin, and adverse events. We assessed the quality of evidence using the GRADE approach.
Main results
We included 27 trials, enrolling 16,382 adults and children, and conducted between 2002 and 2010. Most trials excluded infants aged less than six months and pregnant women.
DHA‐P versus artemether‐lumefantrine
In Africa, over 28 days follow‐up, DHA‐P is superior to artemether‐lumefantrine at preventing further parasitaemia (PCR‐unadjusted treatment failure: RR 0.34, 95% CI 0.30 to 0.39, nine trials, 6200 participants, high quality evidence), and although PCR‐adjusted treatment failure was below 5% for both ACTs, it was consistently lower with DHA‐P (PCR‐adjusted treatment failure: RR 0.42, 95% CI 0.29 to 0.62, nine trials, 5417 participants, high quality evidence). DHA‐P has a longer prophylactic effect on new infections which may last for up to 63 days (PCR‐unadjusted treatment failure: RR 0.71, 95% CI 0.65 to 0.78, two trials, 3200 participants, high quality evidence).
In Asia and Oceania, no differences have been shown at day 28 (four trials, 1143 participants, moderate quality evidence), or day 63 (one trial, 323 participants, low quality evidence).
Compared to artemether‐lumefantrine, no difference was seen in prolonged QTc (low quality evidence), and no cardiac arrhythmias were reported. The frequency of other adverse events is probably similar with both combinations (moderate quality evidence).
DHA‐P versus artesunate plus mefloquine
In Asia, over 28 days follow‐up, DHA‐P is as effective as artesunate plus mefloquine at preventing further parasitaemia (PCR‐unadjusted treatment failure: eight trials, 3487 participants, high quality evidence). Once adjusted by PCR to exclude new infections, treatment failure at day 28 was below 5% for both ACTs in all eight trials, but lower with DHA‐P in two trials (PCR‐adjusted treatment failure: RR 0.41 95% CI 0.21 to 0.80, eight trials, 3482 participants, high quality evidence). Both combinations contain partner drugs with very long half‐lives and no consistent benefit in preventing new infections has been seen over 63 days follow‐up (PCR‐unadjusted treatment failure: five trials, 2715 participants, moderate quality evidence).
In the only trial from South America, there were fewer recurrent parastaemias over 63 days with artesunate plus mefloquine (PCR‐unadjusted treatment failure: RR 6.19, 95% CI 1.40 to 27.35, one trial, 445 participants, low quality evidence), but no differences were seen once adjusted for new infections (PCR‐adjusted treatment failure: one trial, 435 participants, low quality evidence).
DHA‐P is associated with less nausea, vomiting, dizziness, sleeplessness, and palpitations compared to artesunate plus mefloquine (moderate quality evidence). DHA‐P was associated with more frequent prolongation of the QTc interval (low quality evidence), but no cardiac arrhythmias were reported.
Authors' conclusions
In Africa, dihydroartemisinin‐piperaquine reduces overall treatment failure compared to artemether‐lumefantrine, although both drugs have PCR‐adjusted failure rates of less than 5%. In Asia, dihydroartemisinin‐piperaquine is as effective as artesunate plus mefloquine, and is better tolerated.
11 April 2019
No update planned
Other
There is high‐certainty evidence that dihydroartemisinin‐piperaquine is effective, meaning further research is unlikely to change our confidence in the estimate of effect. All eligible published studies found in the last search (29 Jul, 2013) were included and one ongoing study was identified (see 'Characteristics of ongoing studies' section).
Plain language summary
Dihydroartemisinin‐piperaquine for treating uncomplicated malaria
This review summarises trials evaluating the effects of dihydroartemisinin‐piperaquine (DHA‐P) compared to other artemisinin‐based combination therapies recommended by the World Health Organization. After searching for relevant trials up to July 2013, we included 27 randomized controlled trials, enrolling 16,382 adults and children and conducted between 2002 and 2010.
What is uncomplicated malaria and how might dihydroartemisinin‐piperaquine work
Uncomplicated malaria is the mild form of malaria which usually causes a fever, with or without headache, tiredness, muscle pains, abdominal pains, nausea, and vomiting. If left untreated, uncomplicated malaria can develop into severe malaria with kidney failure, breathing difficulties, fitting, unconsciousness, and eventually death.
DHA‐P is one of five artemisinin‐based combination therapies the World Health Organization currently recommends to treat malaria. These combinations contain an artemisinin component (such as dihydroartemisinin) which works very quickly to clear the malaria parasite from the person's blood, and a longer acting drug (such as piperaquine) which clears the remaining parasites from the blood and may prevent new infections with malaria for several weeks.
What the research says
DHA‐P versus artemether lumefantrine
In studies of people living in Africa, both DHA‐P and artemether‐lumefantrine are very effective at treating malaria (high quality evidence). However, DHA‐P cures slightly more patients than artemether‐lumefantrine, and it also prevents further malaria infections for longer after treatment (high quality evidence). DHA‐P and artemether‐lumefantrine probably have similar side effects (moderate quality evidence).
DHA‐P versus artesunate plus mefloquine
In studies of people living in Asia, DHA‐P is as effective as artesunate plus mefloquine at treating malaria (moderate quality evidence). Artesunate plus mefloquine probably causes more nausea, vomiting, dizziness, sleeplessness, and palpitations than DHA‐P (moderate quality evidence).
Overall, in some people, DHA‐P has been seen to cause short term changes in electrocardiographs tracing the conduction of the heart rhythm (low quality evidence), but these small changes on the electrocardiograph resolved within one week without serious consequences.
Summary of findings
Summary of findings for the main comparison. Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine for uncomplicated P. falciparum malaria in Africa.
| Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine for uncomplicated P. falciparum malaria in Africa | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Malaria endemic settings in Africa Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artemether‐lumefantrine (AL6) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| AL6 | DHA‐P | ||||
|
Treatment failure Day 28 |
PCR‐unadjusted | RR 0.34 (0.30 to 0.39) | 6200 (9 trials) | ⊕⊕⊕⊕ high1,2,3,4 | |
| 23 per 100 | 8 per 100 (7 to 9) | ||||
| PCR‐adjusted | RR 0.42 (0.29 to 0.62) | 5417 (9 trials) | ⊕⊕⊕⊕ high1,2,3,5 | ||
| 3 per 100 | 1 per 100 (1 to 2) | ||||
|
Treatment failure Day 63 |
PCR‐unadjusted | RR 0.71 (0.65 to 0.78) | 3200 (2 trials) | ⊕⊕⊕⊕ high1,3,4,6,7 | |
| 45 per 100 | 32 per 100 (29 to 35) | ||||
| PCR‐adjusted | RR 0.72 (0.50 to 1.04) | 2097 (2 trials) | ⊕⊕⊕⊕ high1,3,7,8,9 | ||
| 6 per 100 | 4 per 100 (3 to 6) | ||||
| *The basis for the assumed risk (for example, the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate., Very low quality: We are very uncertain about the estimate. | |||||
1 No serious risk of bias: Trials are generally at low risk of bias. Exclusion of studies at high or unclear risk of selection bias or detection bias did not change the result. 2 No serious inconsistency: The trials all had similar results and statistical heterogeneity was low. 3 No serious indirectness: The trials were conducted in different transmission settings in East, West and Southern Africa. Most studies were limited to children. 4 No serious imprecision: Both limits of the 95% CI imply appreciable benefit, and the overall meta‐analysis is adequately powered to detect this result. 5 No serious imprecision: Although there is a benefit in favour of DHA‐P, PCR‐adjusted treatment failure was below 5% with both drugs. 6 No serious inconsistency: At this timepoint there is more inconsistency between trials. Both show a benefit with DHA‐P but there is variation in the size of this benefit. 7 Seven studies from East, West and Southern Africa reported outcomes at day 42. At this timepoint DHA‐P still had an advantage over AL6 on PCR‐unadjusted treatment failure (RR 0.60, 95% CI 0.53 to 0.67, seven studies, 3301, high quality evidence), and PCR‐adjusted treatment failure (RR 0.58, 95% CI 0.41 to 0.81, seven studies, 2559 participants, moderate quality evidence). 8 No serious inconsistency: Statistical heterogeneity was low. 9 No serious imprecision: Both ACTs performed well in these two trials with low levels of treatment failure.
Background
Description of the condition
Malaria is a febrile illness caused by infection with the protozoan parasite Plasmodium, which is transmitted from person to person by the bite of infected female Anopheles mosquitoes. Five Plasmodium species are capable of causing malaria in humans, of which P. falciparum is responsible for over 90% of malaria cases and almost all of the malaria deaths worldwide (WHO 2012).
Uncomplicated malaria is the mild form of the disease which typically presents as a fever, with or without associated headache, tiredness, muscle pains, abdominal pains, rigors (severe shivering), nausea, and vomiting. If left untreated P. falciparum malaria can rapidly develop into severe malaria with consequent renal failure (kidney failure), pulmonary oedema (fluid in the lungs), convulsions (fitting), coma, and eventually death (WHO 2010; Sinclair 2012). A clinical diagnosis of malaria can be confirmed by detection of the malaria parasite in the patient's blood. This has traditionally been done by light microscopy but increasingly rapid diagnostic tests are being used (Abba 2011).
Resistance of P. falciparum to the traditional antimalarial drugs (such as chloroquine, sulfadoxine‐pyrimethamine, amodiaquine, and mefloquine) is a growing problem and is thought to have contributed to increased malaria mortality in recent years (WHO 2010). Chloroquine resistance has now been documented in all regions except Central America and the Caribbean. There is high‐level resistance to sulfadoxine‐pyrimethamine throughout South East Asia and increasingly in Africa, and mefloquine resistance is common in the border areas of Cambodia, Myanmar, and Thailand (WHO 2010; WWARN 2013).
To combat the spread of resistance, the World Health Organization (WHO) now recommends that P. falciparum malaria is always treated using a combination of two drugs that act at different biochemical sites within the parasite (WHO 2010). If a parasite mutation producing drug resistance arises spontaneously during treatment, the parasite should then be killed by the partner drug, thus reducing or delaying the development of resistance and increasing the useful lifetime of the individual drugs (White 1996; White 1999). The current drug combinations all include a short‐acting artemisinin derivative (such as artesunate, artemether, or dihydroartemisinin), partnered with a longer‐acting drug in combinations known as 'Artemisinin‐based Combination Therapies' (ACTs).
Description of the intervention
The WHO recommends five ACTs for treating uncomplicated P. falciparum malaria: dihydroartemisinin‐piperaquine (DHA‐P); artesunate plus mefloquine (AS+MQ); artemether‐lumefantrine ‐ six doses regimen (AL6); artesunate plus amodiaquine (AS+AQ); and artesunate plus sulfadoxine‐pyrimethamine (AS+SP) (WHO 2010).
Dihydroartemisinin is the active metabolite of the artemisinin derivatives, and produces faster relief of clinical symptoms and faster clearance of parasites from the blood than other antimalarial drugs (McIntosh 2000; Adjuik 2004; WHO 2010). When used as a monotherapy, the short half‐life of the artemisinin derivatives (and rapid elimination from the blood) means that patients must take the drug for at least seven days (Meshnick 1996; Adjuik 2004). Failure to complete the course, due to the rapid improvement in clinical symptoms, can lead to high levels of treatment failure even in the absence of drug resistance. The long‐acting partner drug in ACTs therefore allows the artemisinin component to be taken for a shorter duration (White 1999), and the current recommendation is for three days of the artemisinin‐derivative to cover two asexual parasite life‐cycles (Adjuik 2004; WHO 2010).
The artemisinin derivatives also reduce the development of gametocytes (the sexual form of the P. falciparum parasite that is capable of infecting mosquitoes) and consequently the carriage of gametocytes in the peripheral blood (Price 1996; Targett 2001). This reduction in infectivity has the potential to reduce the post‐treatment transmission of malaria (particularly in areas of low or seasonal transmission), which may have important public health benefits (WHO 2010).
Artemisinin and its derivatives are generally reported as being safe and well‐tolerated, and the safety profile of ACTs may be largely determined by the partner drug (Nosten 2007; WHO 2010). Animal studies of artemisinin derivatives have reported neurotoxicity (brain damage), but this has not been seen in human studies (Price 1999). Animal studies have also shown adverse effects on the early development of the fetus, and consequently the use of artemisinin derivatives in pregnant women has so far been restricted to the second and third trimesters and continues to be evaluated (Nosten 2007). Other reported adverse events include gastrointestinal (GI) disturbance (stomach upset), dizziness, tinnitus (ringing in the ears), neutropenia (low levels of white blood cells), elevated liver enzymes (a marker for liver damage), and electrocardiographic (ECG) abnormalities (changes in cardiac conduction) (Nosten 2007). The incidence of type 1 hypersensitivity (allergic) reactions is reported to be approximately 1 in 3000 patients (Nosten 2007).
Piperaquine is a bisquinoline antimalarial whose mode of action is thought to be similar to that of chloroquine (a 4‐aminoquinolone) (Keating 2012). In vitro studies have shown it is effective against chloroquine‐resistant P. falciparum, although there are reports of some cross‐resistance (Keating 2012). Piperaquine has a very long elimination half‐life of between two to three weeks, similar to mefloquine but longer than lumefantrine or amodiaquine, and consequently could be expected to provide a long period of post‐treatment prophylaxis (Davis 2005; Keating 2012).
In a previous review of DHA‐P, Myint 2007 noted an association between DHA‐P and prolongation of the QT interval in two small observational trials (Karunajeewa 2004: N = 62, and Ashley 2004a THA; N = 32). Prolonged QT interval is a cardiac conduction defect which can sometimes lead to fatal arrhythmias.
Assessment of antimalarial drug efficacy
The WHO recommends that first‐line antimalarials should have a treatment failure rate of less than 10%, and that failure rates higher than 10% should trigger a change in treatment policy (WHO 2010). Treatment failure can be classified as:
Early treatment failure:
the development of danger signs or severe malaria on days 1, 2, or 3 in the presence of parasitaemia;
parasitaemia on day 2 higher than on day 0;
parasitaemia and axillary temperature > 37.5 °C on day three;
parasitaemia on day 3 > 20% of count on day 0.
or late treatment failure:
development of danger signs, or severe malaria, after day three with parasitaemia;
presence of P. falciparum parasitaemia and axillary temperature > 37.5 °C on or after day 4;
presence of P. falciparum parasitaemia after day 7.
The late reappearance of P. falciparum parasites in the blood can be due to failure of the drug to completely clear the original parasite (a recrudescence) or due to a new infection, which is especially common in areas of high transmission. A molecular genotyping technique called polymerase chain reaction (PCR) can be used in clinical trials to distinguish between recrudescence and new infection, giving a clearer picture of the efficacy of the drug and its post‐treatment prophylactic effect (White 2002; Cattamanchi 2003).
The WHO recommends a minimum follow‐up period of 28 days for antimalarial efficacy trials, but longer follow‐up may be required for antimalarials with long elimination half‐lives (White 2002; Bloland 2003). This is because treatment failure due to true recrudescence of malaria parasites may be delayed until the drug concentration falls below the minimum concentration required to inhibit parasite multiplication, which may be beyond 28 days. The WHO recommends 42 days follow‐up for trials involving lumefantrine and piperaquine and 63 days for mefloquine trials (WHO 2010).
Why it is important to do this review
This review aims to assist national decision‐making by providing a concise summary of the benefits and harms of DHA‐P in comparison to the other recommended ACTs. Other information that is also important when selecting national first or second‐line ACTs includes:
the appropriateness of the partner drug within a locality, based on regional and national overviews of drug resistance and the intensity of malaria transmission;
the simplicity of the treatment regimen (co‐formulated products are generally preferred as they reduce the availability and use of monotherapy, which may in turn reduce the development of resistance); and
the cost (since the ACT is likely to represent a large percentage of the annual health expenditure in highly endemic countries).
Objectives
To evaluate the effectiveness and safety of DHA‐P compared to other ACTs for the treatment of uncomplicated P. falciparum malaria in adults and children.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs). We excluded quasi‐RCTs.
Types of participants
Adults and children (including pregnant women and infants) with symptomatic, microscopically confirmed, uncomplicated P. falciparum malaria.
We also included trials that recruited participants with P. vivax co‐infection.
Types of interventions
Intervention
A three‐day course of DHA‐P.
Control
A three‐day course of an alternative WHO recommended ACT.
Types of outcome measures
Primary outcomes
Total failure at days 28, 42, and 63; PCR‐adjusted and PCR‐unadjusted.
Secondary outcomes
Gametocyte carriage at day 7 or 14 (preference for day 14 in data analyses);
Gametocyte development (negative at baseline and positive at follow‐up);
Change in haemoglobin from baseline (minimum 28 day follow‐up).
Adverse events
Deaths occurring during follow‐up;
Serious adverse events (life threatening, causing admission to hospital, or discontinuation of treatment);
Haematological and biochemical adverse effects (for example, neutropenia, liver toxicity);
Early vomiting;
Other adverse events.
Search methods for identification of studies
Electronic searches
We searched the following databases up to 29 July 2013 using the search terms detailed in Table 2: Cochrane Infectious Diseases Group Specialized Register; Cochrane Central Register of Controlled Trials (CENTRAL) published in The Cochrane Library; MEDLINE; EMBASE; LILACS. We also examined the metaRegister of Controlled Trials (mRCT) using the search terms 'malaria' and 'arte* OR dihydroarte*'.
1. Detailed search strategy.
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb |
| 1 | malaria | malaria | malaria | malaria | malaria |
| 2 | arte* | arte* | arte* | arte* | arte* |
| 3 | dihydroarte* | dihydroarte* | dihydroarte* | dihydroarte* | dihydroarte* |
| 4 | amodiaq* | amodiaq* | amodiaq* | amodiaq$ | amodiaq$ |
| 5 | lumefantrine | lumefantrine | lumefantrine | lumefantrine | lumefantrine |
| 6 | Coartem* | Coartem* | Coartem* | Coartem$ | Coartem$ |
| 7 | mefloquine | mefloquine | mefloquine | mefloquine | mefloquine |
| 8 | 2 or 3 | 2 or 3 | 2 or 3 | 2 or 3 | 2 or 3 |
| 9 | 4 or 5 or 6 or 7 | 4 or 5 or 6 or 7 | 4 or 5 or 6 or 7 | 4 or 5 or 6 or 7 | 4 or 5 or 6 or 7 |
| 10 | 1 and 8 and 9 | 1 and 8 and 9 | 1 and 8 and 9 | 1 and 8 and 9 | 1 and 8 and 9 |
| 11 | — | — | Limit 10 to humans | Limit 10 to human | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Lefebvre 2008); upper case: MeSH or EMTREE heading; lower case: free text term.
Searching other resources
We contacted individual researchers working in the field, organizations including the WHO, and pharmaceutical companies involved in the manufacture of DHA‐P (Atlantic, Guilin, Holleykin, HolleyPharm) for information on unpublished trials. In addition, reference lists of all trials identified by the methods described above were checked.
Data collection and analysis
Selection of studies
Babalwa Zani (BZ) and Michael Gathu (MG) independently reviewed the results of the literature search, obtained full‐text copies of all potentially relevant trials and checked each trial report for evidence of multiple publications from the same data set.
BZ and MG also independently assessed each trial for inclusion using an eligibility form based on the inclusion criteria and resolved any disagreements through discussion or, where necessary, by consultation with David Sinclair (DS). We contacted trial authors when further information was necessary. We listed the ineligible trials and the reasons for their exclusion in the 'Characteristics of excluded studies' table.
Data extraction and management
BZ and MG independently extracted data on trial characteristics including methods, participants, interventions, outcomes, dose, and drug ratios of the combinations using a pre‐tested data extraction form. We also recorded the number of participants randomized and analysed in each treatment group for each outcome and reported the loss to follow‐up in each group.
For dichotomous outcomes, we recorded the number of participants experiencing the event and the total number of participants in each treatment group. For continuous outcomes, the arithmetic means and standard deviations for each treatment group together with the numbers of participants in each group were extracted. Where trials reported the data using geometric means, we recorded this information and extracted standard deviations on the log scale. Where trials gave median values, we extracted medians and ranges.
Primary outcome
Our primary analyses drew on the WHO's protocol for assessing and monitoring antimalarial drug efficacy (Bloland 2003). This protocol has been used to guide most efficacy trials since its publication in 2003, even though it was designed to assess the level of antimalarial resistance in the study area rather than for comparative trials. As a consequence, a high number of randomized participants are excluded from the final efficacy outcome as losses to follow‐up or voluntary or involuntary withdrawals. For this reason we conducted a series of sensitivity analyses to restore the integrity of the randomization process and test the robustness of the results to this methodology (For a summary of the methodology and sensitivity analysis see Table 3).
2. Primary outcome measure (Total failure).
| Analysis | Participants | PCRb‐unadjusted | PCR‐adjusted | ||
| Numerator | Denominator | Numerator | Denominator | ||
| Primary analysis | Exclusions after enrolmenta | Excludedc | Excluded | Excluded | Excluded |
| Missing or indeterminate PCR | Included as failures | Included | Excluded | Excluded | |
| New infections | Included as failures | Included | Excluded | Excluded | |
| Sensitivity analysis 1d | As 'Primary analysis' except: missing or indeterminate PCR | — | — | Included as failures | Included |
| Sensitivity analysis 2e | As 'Sensitivity analysis 1' except: new infections | — | — | Included as successes | Included |
| Sensitivity analysis 3f | As 'Sensitivity analysis 2' except: exclusions after enrolment | Included as failures | Included | Included as failures | Included |
| Sensitivity analysis 4g | As 'Sensitivity analysis 2' except: exclusions after enrolment | Included as successes | Included | Included as successes | Included |
aNote: participants who were found to not satisfy the inclusion criteria after randomization are removed from all calculations. bPCR: polymerase chain reaction. c'Excluded' means removed from the calculation. dTo re‐classify all indeterminate or missing PCR results as treatment failures in the PCR‐adjusted analysis. eTo re‐classify all PCR‐confirmed new infections as treatment successes in the PCR‐adjusted analysis. (This analysis may overestimate efficacy as PCR is not wholly reliable and some recrudescences may be falsely classified as new infections. Also some participants may have proceeded to develop a recrudescence after the new infection.) fTo re‐classify all exclusions after enrolment (losses to follow‐up, withdrawn consent, other antimalarial use, or failure to complete treatment) as treatment failures. For PCR‐unadjusted total failure, this represents a true worse‐case scenario. gTo re‐classify all exclusions after enrolment (losses to follow‐up, withdrawn consent, other antimalarial use, or failure to complete treatment) as treatment successes.
PCR‐unadjusted total failure
We calculated PCR‐unadjusted total failure (P. falciparum) as the sum of early treatment failures and late treatment failures (without PCR adjustment). The denominator excluded participants for whom an outcome was not available (for example, those who were lost to follow‐up, withdrew consent, took other antimalarials, or failed to complete treatment) and those participants who were found not to fulfil the inclusion criteria after randomization.
PCR‐adjusted total failure
We defined PCR‐adjusted total failure (P. falciparum) as the sum of early treatment failures, and late treatment failures due to PCR‐confirmed recrudescence. Participants with indeterminate PCR results, missing PCR results, or PCR‐confirmed new infections were treated as involuntary withdrawals and excluded from the calculation. The denominator excluded participants for whom an outcome was not available (for example, those who were lost to follow‐up, withdrew consent, took other antimalarials, or failed to complete treatment) and those participants who did not fulfil the inclusion criteria after randomization.
These primary outcomes relate solely to failure due to P. falciparum. For both PCR‐unadjusted and PCR‐adjusted total failure, we retained participants who developed confirmed P. vivax infection during follow‐up in the calculation if they were treated with chloroquine and continued in follow‐up. They were classified as treatment successes if they did not subsequently develop P. falciparum parasitaemia. We excluded from the calculation those participants who developed P. vivax parasitaemia and were removed from the trial's follow‐up.
Assessment of risk of bias in included studies
BZ and MG independently assessed the risk of bias for each trial using 'The Cochrane Collaboration's tool for assessing the risk of bias' (Higgins 2008) and resolved differences of opinion through discussion with DS. We followed the guidance to assess whether adequate steps were taken to reduce the risk of bias across six domains: sequence generation; allocation concealment; blinding (of participants, personnel, and outcome assessors); incomplete outcome data; selective outcome reporting; and other sources of bias.
For sequence generation and allocation concealment, we reported the methods used. For blinding, we described who was blinded and the blinding method. For incomplete outcome data, we reported the percentage and proportion of participants lost to follow‐up. For selective outcome reporting, any discrepancies between the methods used and the results were stated in terms of the outcomes measured or the outcomes reported. For other biases, we described any other trial features that could have affected the trial result (for example, if the trial was stopped early).
We categorized our risk of bias judgements as 'low', 'high', or 'unclear'. Where risk of bias was unclear, we attempted to contact the trial authors for clarification and resolved any differences of opinion through discussion.
Measures of treatment effect
We analysed the data using Review Manager (RevMan) and presented and combined dichotomous data using risk ratios (RR). For continuous data summarized by arithmetic means and standard deviations, we combined data using mean differences. RRs and mean differences were accompanied by 95% confidence intervals (CI). We reported medians and ranges in tables.
Unit of analysis issues
We split trials including more than two comparison groups and analysed as individual pair‐wise comparisons. When conducting meta‐analysis, we ensured that participants and cases in the placebo group were counted only once, by dividing the placebo cases and participants evenly between the intervention groups.
Dealing with missing data
If data from the trial reports were insufficient, unclear, or missing, we attempted to contact the trial authors for additional information. If the missing data rendered the result uninterpretable, we excluded the data from the meta‐analysis and clearly stated the reason for exclusion. We explored the potential effects of missing data through a series of sensitivity analyses (Table 3).
Assessment of heterogeneity
We assessed heterogeneity between trials by inspecting the forest plots, applying the Chi² test with a 10% level of statistical significance, and using the I² statistic with a value of 50% to denote moderate levels of heterogeneity.
Assessment of reporting biases
The possibility of publication bias was assessed by examining funnel plots for asymmetry. We noted that funnel plot asymmetry could also be caused by differences in methodological quality or heterogeneity.
Data synthesis
To aid interpretation, we gave the included trials identity codes including the first author, the year of publication, and the three‐letter international country code or two‐letter continent code (for trials conducted in more than one country). We listed trials in forest plots in chronological order of the year the trial was completed.
Using pair‐wise comparisons we directly compared treatments. For outcomes that were measured at different time points, we stratified the analysis by the time point. The primary outcome analysis was also stratified by geographical region as a crude marker for differences in transmission and resistance patterns.
We performed meta‐analysis within geographic regions where appropriate after assessment and investigation of heterogeneity. In the first instance, we used a fixed‐effects model and applied a random‐effects model when the Chi² test P value was less than 0.1 or the I² statistic greater than 50%.
Quality of evidence
We assessed the quality of evidence across each outcome measure using the GRADE approach. The quality rating across studies has four levels: high, moderate, low, or very low. RCTs are initially categorized as high quality but can be downgraded after assessment of five criteria: risk of bias, consistency, directness, imprecision, and publication bias. Similarly, observational studies are initially categorized as low quality and can be downgraded by the same criteria, but in exceptional circumstances may be upgraded by three further criteria; large effect size, all plausible confounders would act to reduce the effect size, and evidence of a dose‐response effect (Guyatt 2008).
Subgroup analysis and investigation of heterogeneity
We investigated potential sources of heterogeneity through a series of analyses, sub‐grouping the trials by: geographical region, intensity of malaria transmission (low to moderate versus high malaria transmission), known parasite resistance, allocation concealment, participant age, and drug dose (comparing regimens where there are significant variations in drug dose).
Sensitivity analysis
We conducted a series of sensitivity analyses to investigate the robustness of the methodology used in the primary analysis. The aim was to restore the integrity of the randomization process by adding excluded groups back into the analysis in a stepwise fashion (see Table 3 for details).
Results
Description of studies
See the Characteristics of included studies and Characteristics of excluded studies section.
Results of the search
We conducted the searches up to 29 July 2013 and identified 90 trials in total. After screening titles and abstracts, we obtained full text copies of 49 trials. Of these, 34 trials met the inclusion criteria and we excluded 15 trials (Figure 1). We included 27 trials as primary references and retained seven trials as secondary references for additional data on secondary outcomes and adverse events. One of the 26 trials had two different recruitment settings which we split and considered as two separate trials (Ashley 2004a THA; Ashley 2004b THA). One trial (Borrmann 2011 KEN (a)) is pending as we await data for a separate recruitment period from the trial authors.
1.

Study flow diagram.
Included studies
We included 27 trials, enrolling 16,382 participants and conducted between 2002 and 2010.
Twelve trials were conducted in Africa; Uganda (three trials), Kenya (three trials), Sudan (one trial), Rwanda (one trial), Burkina Faso (one trial), and three multi‐centre trials with sites in Kenya, Uganda, Rwanda, Mozambique, Zambia, Gabon, Burkina Faso, Nigeria, Senegal, Côte d’Ivoire, and Cameroon (Bassat 2009 AF; The 4ABC Study 2011 AF; Yavo 2011 AF). Fourteen trials were conducted in Asia and Oceania; Thailand (five trials), Myanmar (two trials), Laos (one trial), Vietnam (one trial), Cambodia (one trial), Indonesia (two trials), Papua New Guinea (one trial); and one multi‐centre trial had sites in Thailand, Laos, and India (Valecha 2010 AS). Only one trial was from South America (Peru).
The African trials focused on children, while Asian trials included older populations and excluded children below one year of age. All trials excluded pregnant and lactating women.
Eleven trials compared DHA‐P with AS+MQ, 16 trials compared DHA‐P with AL, four trials compared DHA‐P with ASAQ, and one trial compared DHA‐P with AS+SP. Some trials had more than two arms and compared multiple ACTs.
Three trials (Hasugian 2007 IDN; Ratcliff 2007 IDN; Karunajeewa 2008 PNG) conducted in Asia and Oceania included participants with P. vivax mono‐infection at baseline. For our primary analysis we obtained data from the trial authors for only those participants who had P. falciparum or mixed infection (P. falciparum and P. vivax) at baseline. Arinaitwe 2009 UGA had an unusual trial design where participants were followed up for more than one episode of malaria. We used data from all malaria episodes in our primary analysis.
We listed the trial details of the included studies in the 'Characteristics of included studies' table.
Excluded studies
The reasons for exclusion are in the 'Characteristics of excluded studies' table.
Risk of bias in included studies
For a summary of the 'Risk of bias' assessments, see Figure 2.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included trial.
Allocation
Nine trials were at low risk of selection bias, with adequate methods for both generation of the randomization sequence and allocation concealment. A further 18 studies were at unclear risk of selection bias due to inadequate descriptions of their methods. For primary outcomes we conducted a sensitivity analysis including only the trials with adequate allocation concealment.
Blinding
Eighteen trials blinded the microscopists to treatment allocation and so were at low risk of performance and detection bias for the primary outcomes. Only four of the included trials blinded the outcome assessors for adverse events.
Incomplete outcome data
We reported the proportion of participants in each treatment arm for whom an outcome was not available and conducted sensitivity analyses to test the possible effect of these losses. Four trials were at high risk of bias due to high dropout rates (> 15%).
Selective reporting
Due to the varying half‐lives of drugs, the choice of which day to measure outcomes can influence the comparative effects of the ACTs. If an ACT with a long half‐life (DHA‐P or AS+MQ) is compared to a drug with a short half‐life (AS+AQ or AS+SP), day 28 outcomes may underestimate PCR‐adjusted failure with the long half‐life drug. At later time points (day 42 and 63), drugs with long half‐lives are likely to appear superior in preventing new infections (PCR‐unadjusted failure) which represents a prophylactic effect. We noted this while interpreting the data but did not consider this a source of trial bias.
Other potential sources of bias
Pharmaceutical companies provided financial support or study drugs in 13 trials. In the two large trials of the new Eurartesim® formulation (Bassat 2009 AF & Valecha 2010 AS), the pharmaceutical company was fully involved in the design, conduct and analysis of the trials, In one of these (Bassat 2009 AF), it is stated that an independent author had access to the primary dataset and took responsibility for the analyses. We judged this trial to be at unclear risk of bias. In the second trial, this additional safety measure was not described and we judged the trial to be at high risk of bias.
Effects of interventions
See: Table 1
Comparison 1. DHA‐P versus artesunate plus mefloquine
We found 11 trials, 10 in Asia and one in South America, that assessed this comparison; conducted between 2002 and 2009.
Allocation concealment was at 'low risk of bias' in only two trials (Mayxay 2006 LAO; Grande 2007 PER). Five trials blinded laboratory staff (outcome assessors) to treatment allocation (Ashley 2004a THA; Ashley 2004b THA; Ashley 2005 THA; Smithuis 2010 MMR; Valecha 2010 AS). Patients were unblinded in all trials, and only one trial blinded outcome assessors for adverse effects (The 4ABC Study 2011 AF).
Total failure
In Asia over 63 days follow‐up, recurrent parasitaemias (including both recrudescences and new infections) occurred in less than 15% of all participants, with no differences in PCR‐unadjusted treatment failure between groups (day 28: eight trials, 3487 participants, Analysis 1.1; day 42: seven trials, 3421 participants, Analysis 1.3; Day 63: five trials, 2715 participants, Analysis 1.5).
1.1. Analysis.
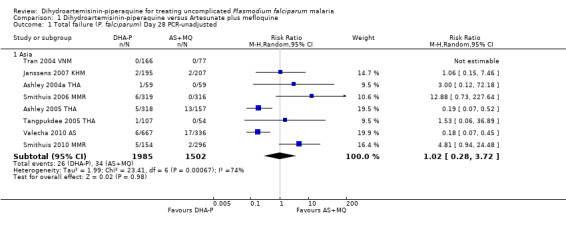
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted.
1.3. Analysis.
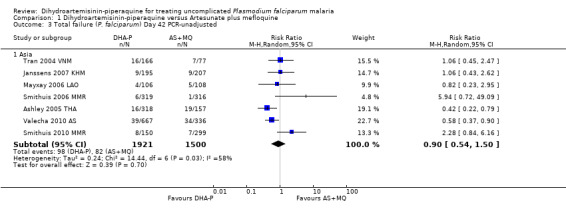
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted.
1.5. Analysis.

Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 5 Total failure (P. falciparum) Day 63 PCR‐unadjusted.
Once adjusted by PCR to exclude new infections, treatment failure at day 28 was below 5% for both ACTs in all eight trials for which data was available (eight trials, 3482 participants, Analysis 1.2). Two of the eight trials, conducted in Thailand at trial sites with multi‐drug resistant P. falciparum, found slightly higher levels of recrudescence following AS+MQ and statistically significant benefits with DHA‐P (Ashley 2005 THA; Valecha 2010 AS). Recrudescences remained low in both groups over 63 days of follow‐up (day 42: six trials, 2901 participants, Analysis 1.4; day 63: five trials, 2500 participants, Analysis 1.6).
1.2. Analysis.
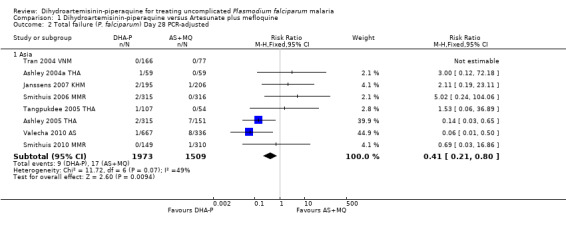
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 2 Total failure (P. falciparum) Day 28 PCR‐adjusted.
1.4. Analysis.
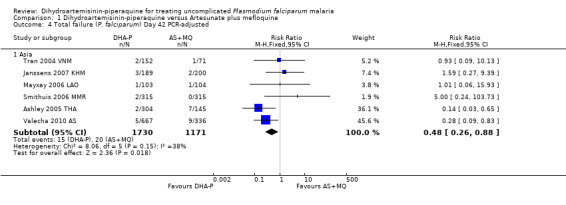
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 4 Total failure (P. falciparum) Day 42 PCR‐adjusted.
1.6. Analysis.
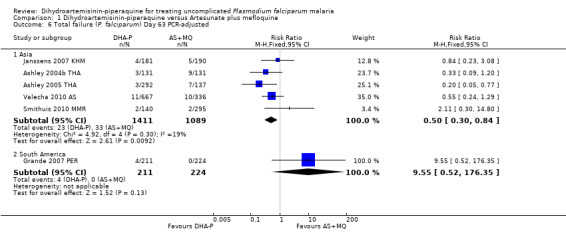
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 6 Total failure (P. falciparum) Day 63 PCR‐adjusted.
In the one trial from South America, only day 63 data was available (Analysis 1.5; Analysis 1.6). Recrudescences and new infections were very rare with both treatments, but new infections were lower with AS+MQ (RR 6.19, 95% CI 1.40 to 27.35, one trial, 445 participants, Analysis 1.5).
Gametocytes
AS+MQ appears to clear gametocytes from the peripheral blood quicker than DHA‐P (Gametocyte carriage on Day 7: RR 1.99, 95% CI 1.57 to 2.51, three trials, 2270 participants, Analysis 1.7; Day 14: RR 5.11, 95% CI 3.26 to 7.99, three trials, 2249 participants, Analysis 1.7). In addition, the number of participants who developed detectable gametocytes (after being negative at baseline) was low in both groups, but lowest with AS+MQ (RR 3.06, 95% CI 1.13 to 8.33, three trials, 1234 participants, Analysis 1.8). Five trials reported additional data on gametocyte carriage which could not be pooled and are presented in Table 4.
1.7. Analysis.
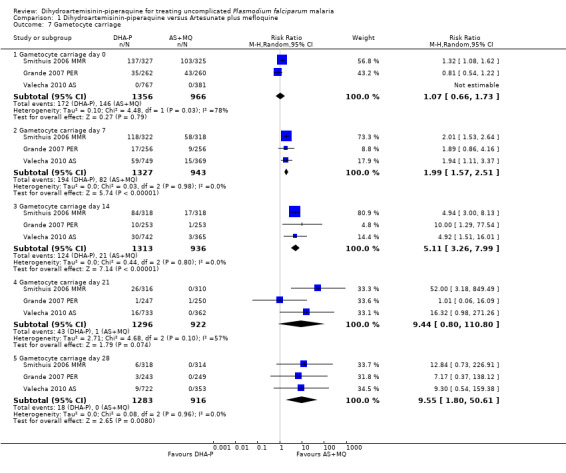
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 7 Gametocyte carriage.
1.8. Analysis.
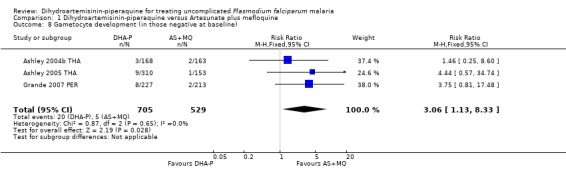
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 8 Gametocyte development (in those negative at baseline).
3. DHA‐P versus AS+MQ: Additional secondary outcomes data.
| Outcome | Study ID | Measure | DHA‐P | AS+MQ | P value | Comment |
| Gametocyte carriage | Grande 2007 PER | Person gametocytemia weeks per 1000 person weeks | 32.5 | 24.9 | 0.31 | |
| Mayxay 2006 LAO | Proportion with gametocytes at any timepoint after treatment (on or after day 7) | 9/110 | 3/110 | 0.07 | ||
| Person gametocytemia weeks per 1000 person weeks | ‐ | ‐ | > 0.05 | Mean across all groups was 0.10 (95% CI 0.03 to 0.20). No difference between groups (published data). | ||
| Smithuis 2006 MMR | Gametocyte incidence at day 7 | 18/188 | 5/218 | 0.01 | ||
| Gametocyte incidence at day 14 | 3/168 | 1/212 | 0.12 | |||
| Person gametocytemia weeks per 1000 person weeks | ‐ | ‐ | 0.03 | Gametocyte carriage in DHA‐P group was higher than in AS+MQ group (published data). Figures not given. | ||
| Smithuis 2010 MMR | Person gametocytemia weeks per 1000 person weeks | 112.8 | 29.5 | Not stated | Data presented are for fixed‐dose AS+MQ combination. | |
| Valecha 2010 AS | Person gametocytemia weeks per 1000 person weeks | 20.2 (130/6420) |
7.4 (23/3108) |
0.01 | Published data in paper presented as "person gametocytemia weeks per 100 person weeks". | |
| Anaemia | Ashley 2004b THA | Median decrease in HCT by day 7 | 6.3% | 9.4% | 0.21 | "Mean decrease in HCT up to day 7 then recovery in all groups". |
| Ashley 2005 THA | Absolute changes in HCT | ‐ | ‐ | ‐ | "Mean decrease in HCT up to day 7 then recovery in all groups". | |
| Janssens 2007 KHM | Mean HCT at day 63 | 40.0 % (3.7) | 40.2% (3.8) | Not stated | "Patients in both treatment groups showed similar haematological recovery during the 63‐day follow‐up period". | |
| Mayxay 2006 LAO | Mean HCT days 7 to 42 | Not stated | Not stated | > 0.05 | "the mean hematocrit after treatment did not significantly differ between groups". | |
| Smithuis 2006 MMR | Mean haemoglobin at day 28 | 10.4 g/dL | 10.5 g/dL | 0.65 | Data presented are for supervised treatment groups. | |
| Proportion anaemic (Hb < 10 g/dL) on day 28 | 56/152 | 59/156 | 0.85 | Data presented are for supervised treatment groups. | ||
| Smithuis 2010 MMR | Mean increase in haemoglobin | Not stated | Not stated | > 0.05 | "The mean increase of haemoglobin was similar among the treatment groups". | |
| Valecha 2010 AS | Mean increase in Hb from day 0 to day 63 in g/dL | 1.28 ± 2.22 | 1.42 ± 2.12 | 0.30 |
Hb ‐ Haemoglobin HCT ‐ Haematocrit
Anaemia
Seven trials reported a variety of measures of haematological changes between baseline and the last day of follow‐up which we could not pool. None of the individual trials reported differences between groups (see Table 4).
Adverse events
There was no difference in the frequency of serious adverse events (eight trials, 3522 participants, Analysis 1.9; see Appendix 2 for details of serious adverse events).
1.9. Analysis.
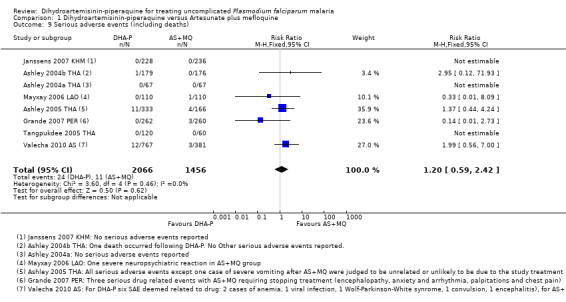
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 9 Serious adverse events (including deaths).
Nine trials reported some measure of early vomiting (vomiting related to drug administration) and there was no difference shown in any trial (nine trials, 4114 participants, Analysis 1.10). However, subsequent nausea and vomiting were consistently more common with AS+MQ (Nausea: RR 0.68, 95% CI 0.60 to 0.78, nine trials, 4531 participants; vomiting: RR 0.59, 95% CI 0.47 to 0.75, five trials, 2744 participants, Analysis 1.10). Diarrhoea was more common with DHA‐P (RR 1.46, 95% CI 1.05 to 2.04, five trials, 2217 participants, Analysis 1.10).
1.10. Analysis.
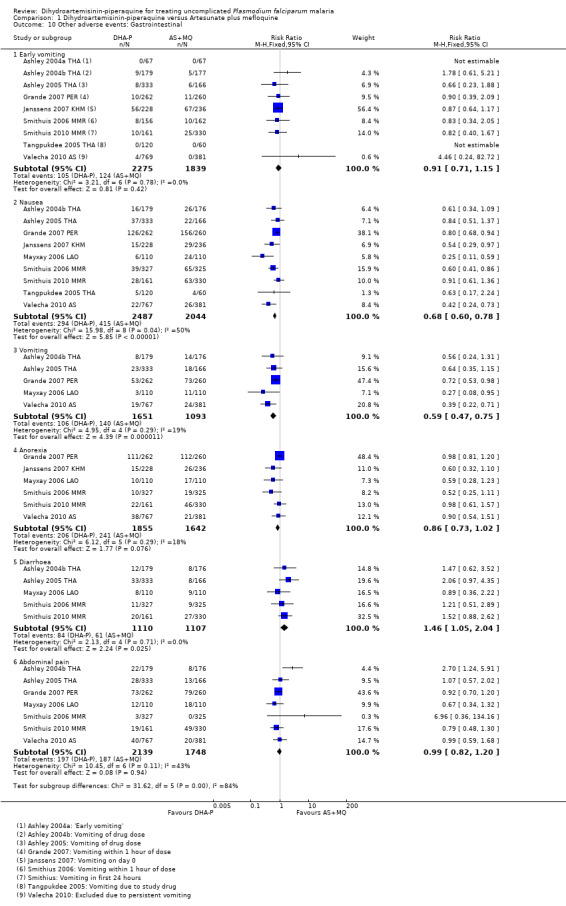
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 10 Other adverse events: Gastrointestinal.
AS+MQ was consistently associated with increased dizziness (RR 0.72, 95% CI 0.66 to 0.78, nine trials, 4531 participants), and sleeplessness (RR 0.49, 95% CI 0.40 to 0.60, six trials, 2551 participants, Analysis 1.11), and increases in headache (four trials), fatigue (two trials), nightmares (one trial), and anxiety (one trial) are reported in the few trials that recorded them (Analysis 1.11).
1.11. Analysis.
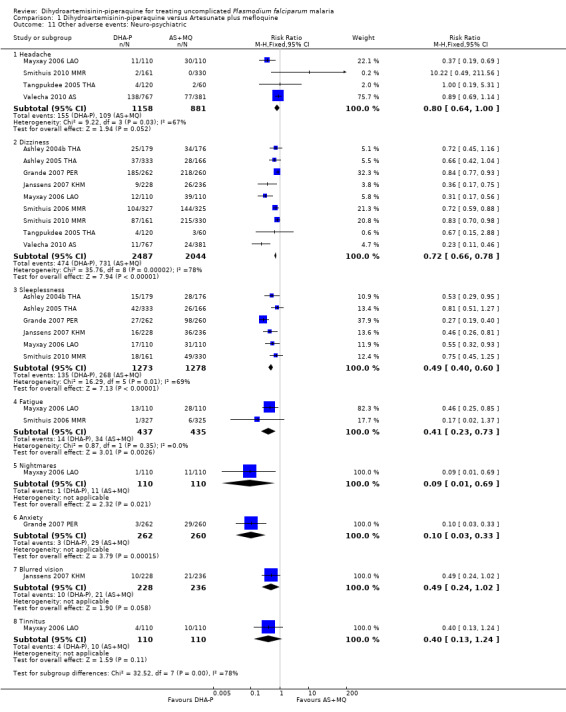
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 11 Other adverse events: Neuro‐psychiatric.
Palpitations were also more common with AS+MQ (RR 0.61, 95% CI 0.45 to 0.82, three trials, 1175 participants, Analysis 1.12), but only one trial performed routine ECGs in both treatment groups (Valecha 2010 AS). In this trial there was a baseline imbalance in the prevalence of borderline prolonged QTc (431 to 450 ms in children and adult men/451 to 470 ms in adult women), using Bazett's correction method (16.6% DHA‐P versus 12.2% AS+MQ, P = 0.066; authors' own figures), but not Fridericia's method (2.9% DHA‐P versus 1.6% AS+MQ, P > 0.05; authors' own figures).
1.12. Analysis.
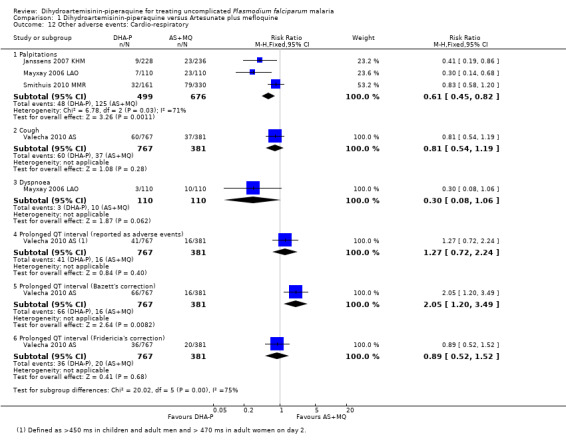
Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 12 Other adverse events: Cardio‐respiratory.
On day 2, a higher proportion of participants treated with DHA‐P had borderline prolonged QTc by both correction methods (Bazett's: 21.4% DHA‐P versus 16.3% AS+MQ, P = 0.043; Fridericia's: 13.0% DHA‐P versus 5.3% AS+MQ, P < 0.001; authors' own figures). There was also a statistically significant increase in the prevalence of prolonged QTc with DHA‐P (> 450 ms in children and adult men, and > 470 ms in adult women), using Bazett's method but not Fridericia's method (one trial, 1148 participants, Analysis 1.12). No consequent arrhythmias were noted, and these differences were no longer present at day seven (for additional data see Table 5).
4. DHA‐P versus AS+MQ/AL6: QTc measurements.
| Study ID | No. of participants | Day | Outcome | Correction method | DHA‐P (%) | AS+MQ (%) | P value |
| Valecha 2010 AS | 1150 | At baseline | Borderline QTc (431 to 450ms) | QTcB | 16.6 | 12.1 | 0.066 |
| QTcF | 2.9 | 1.6 | >0.05 | ||||
| Day 2 | Borderline QTc (431 to 450ms) | QTcB | 21.4 | 16.3 | 0.043 | ||
| QTcF | 13.0 | 5.3 | <0.001 | ||||
| Prolonged QTc (> 450ms) | QTcB | 8.6 | 4.2 | 0.007 | |||
| QTcF | 4.7 | 5.3 | < 0.0011 | ||||
| QTc increase > 60 ms | QTcB | 0.9 | 0.8 | > 0.05 | |||
| QTcF | 4.6 | 2.9 | < 0.001 | ||||
| Day 7 | No differences between groups | N/R | N/R | > 0.05 | |||
| DHA‐P (%) | AL6 (%) | P value | |||||
| Bassat 2009 AF | 1553 | At baseline | Borderline QTc (431‐450ms) | N/R | N/R | ||
| Day 2 | Borderline QTc (431‐450ms) | QTcB | 29.1 | 19.8 | < 0.001 | ||
| QTcF | 1.0 | 1.2 | 0.76 | ||||
| Prolonged QTc (> 450ms) | QTcB | 9.1 | 6.9 | 0.152 | |||
| QTcF | 0.2 | 0.2 | 0.992 | ||||
| QTc > 500 ms | QTcB | 0.19 | 0.39 | > 0.05 | |||
| QTcF | N/R | N/R | |||||
| QTc increase > 60 ms | QTcB | 2.7 | 2.0 | > 0.05 | |||
| QTcF | N/R | N/R | |||||
| Day 7 | No differences between groups | N/R | N/R | > 0.05 | |||
1 In this analysis the direction of effect is reversed. These figures have been confirmed as correct by the study authors. 2 Figures not presented in paper: Taken from Analysis 4.13. N/R ‐ Not reported QTcB ‐ QT interval corrected for rate and gender using Bazett's method. QTcF ‐ Qt interval corrected for rate and gender using Fridericia's method.
Four trials conducted biochemical monitoring for either renal or hepatic adverse events. Monitoring was adequate in three trials (Ashley 2004a THA; Tran 2004 VNM; Grande 2007 PER), and inadequate in one (Valecha 2010 AS), but incompletely reported in all four trials. No clinically important toxicities were reported (see Table 6).
5. DHA‐P versus AS+MQ: Biochemical monitoring and adverse events.
| Study ID | No. of participants | Tests | Days tested | Days reported | Days tested adequate to detect adverse events? | For adequate testing, was reporting complete? | Results as presented in the paper |
| Ashley 2004a THA | 134 | U&E, LFTs | Days 0 and 7 | None | Adequate1 | Incomplete 2 | "No biochemical evidence of toxicity was observed". |
| Grande 2007 PER | 522 | U&E, LFTs | Days 0 and 7 | None | Adequate1 | Incomplete2 | "No patient had abnormal liver and renal function test results of clinical significance, both at entry and at day 7". |
| Tran 2004 VNM | 243 | LFTs | Days 3,7,28 | None | Adequate1 | Incomplete2 | "There were no significant differences between the three groups in the results of liver function tests done on all patients on days 3, 7, and 28". |
| Valecha 2010 AS | 1150 | Not clearly stated | Days 0, 28, 63 | None | Inadequate3 | Incomplete2 | "Other than elevated liver parameters, as might be expected in this population, there were no relevant changes in biochemistry parameters".' |
1 Judged as adequate given that no clinically important abnormalities were seen at day 7. Longer follow‐up is therefore probably unnecessary. 2 Judged as incomplete as data were not presented. Only a text summary was given. 3 Judged as inadequate as biochemical abnormalities are likely to occur earlier than day 28.
LFT = Liver Function Tests
U&E = Urea and electrolytes
Sensitivity analysis
As described in the methods section, we undertook a series of sensitivity analyses to test the robustness of our results to different analysis plans. An example of these is given in Analysis 1.14 & Analysis 1.15. In general, the method of analysis did not change the significance of results and so the remaining sensitivity analyses were deleted.
1.14. Analysis.

Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 14 Sensitivity analysis: Total failure Day 63 PCR‐unadjusted.
1.15. Analysis.

Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 15 Sensitivity analysis: Total failure Day 63 PCR‐adjusted.
Comparisons 2 and 3: DHA‐P dosing concerns
Two dosing regimens were commonly used in clinical trials of DHA‐P versus AS+MQ, which give the same total dose, but divided into three or four doses, given over three days (see Table 7). One trial (Ashley 2005 THA) directly compared the three‐dose regimen with the four‐dose regimen and found no difference at any time point (one trial, 318 participants, Analysis 2.1; Analysis 2.2).
6. DHA‐P versus AS+MQ dosing regimens.
| Study ID | Year of study | Age limits | DHA‐P | AS+MQ |
| Ashley 2004a THA | 2003 | > 14 yrs | 6 mg/kg DHA + 48 mg/kg P in 4 divided doses at 0, 8, 24, and 48 hrs | 4 mg/kg AS once daily for 3 days + 8 mg/kg MQ once daily for 3 days. |
| Ashley 2004b THA | 2003 | 1 to 65 yrs | 6 mg/kg DHA + 48 mg/kg P in 4 divided doses at 0, 8, 24, and 48 hrs | 4 mg/kg AS once daily for 3 days + 8 mg/kg MQ once daily for 3 days. |
| Ashley 2005 THA | 2005 | 1 to 65 yrs | 6.4 mg/kg DHA and 51.2 mg/kg P in 4 divided doses at 0, 8, 24, and 48 hrs or 3 divided doses at 0, 24, and 48 hrs | 4 mg/kg AS once daily for 3 days + 8 mg/kg MQ once daily for 3 days. |
| Grande 2007 PER | 2005 | 5 to 60 yrs | Total dose: 6.3 mg/kg DHA and 50.4 mg/kg PQP in 3 divided doses, given once daily for 3 days | 4 mg/kg AS once daily for 3 days + 8 mg/kg MQ once daily for 3 days. |
| Janssens 2007 KHM | 2003 | > 1 yr |
Adult total dose: 6 mg/kg DHA and + 48 mg/kg P in 4 divided doses at 0, 8, 24, and 48 hrs Children total dose: 6.4 mg/kg DHA + 51.2 mg/kg P in 4 divided doses at 0, 8, 24, and 48 hrs |
Adults: 100 mg AS plus 500 mg MQ twice daily on day 0, 200 mg AS once daily on days 1 and 2. Children: 4 mg/kg AS once daily for 3 days + 25 mg/kg MQ in 2 equal doses on day 0. |
| Mayxay 2006 LAO | 2004 | > 1 yr | 6.3 mg/kg DHA + 50.4 mg/kg P in 3 divided doses, once daily for 3 days | 4 mg/kg AS once daily for 3 days + 15 mg MQ base/kg on day 1 and 10 mg base/kg on day 2. |
| Smithuis 2006 MMR | 2004 | > 1 yr | 6.3 mg/kg DHA + 50.4 mg/kg P in 3 divided doses, once daily for 3 days | 4 mg/kg AS once daily for 3 days. 25 mg MQ base/kg as a single dose on day 0. |
| Smithuis 2010 MMR | 2009 | > 1 yr | 2.5 mg/kg DHA + 20 mg/kg P daily, given once daily for 3 days |
Fixed combination: 4 mg/kg AS + 8.8 mg/kg MQ daily, once daily for 3 days. Loose combination: 4 mg/kg AS once daily for 3 days + 25 mg base/kg MQ as a single dose on day 0. |
| Tangpukdee 2005 THA | Not stated | > 14 yrs | 6 mg/kg DHA + 45 mg/kg P in 3 divided doses, given once daily for 3 days | 4 mg/kg AS once daily for 3 days + 8 mg/kg MQ once daily for 3 days. |
| Tran 2004 VNM | 2002 | > 2 yrs | 40 mg/320 mg tablets
Adults: 2 tablets at 0, 6, 24, and 48 hrs Children < 15 yrs: 1 tablet at 0, 6, 24, and 48 hrs |
4 mg/kg AS once daily for 3 days + 25 mg base/kg MQ as 2 divided doses 6 hrs apart on day 3. |
| Valecha 2010 AS | 2007 | 3m to 65 yrs (≥18 yrs in India) | DHA: 2.25 mg/kg DHA + 18 mg/kg P daily dose for 3 days | 4mg/kg AS once daily for 3 days + MQ none on day 0, then 15 mg/kg once on day 1 and 10 mg/kg once on day 2 |
2.1. Analysis.

Comparison 2 Dihydroartemisinin‐piperaquine dose analysis: 3 dose versus 4 dose regimen, Outcome 1 Total failure PCR‐unadjusted.
2.2. Analysis.
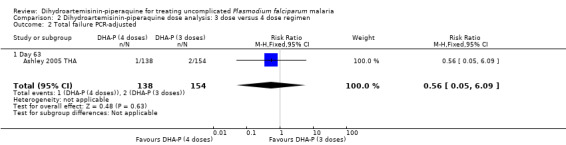
Comparison 2 Dihydroartemisinin‐piperaquine dose analysis: 3 dose versus 4 dose regimen, Outcome 2 Total failure PCR‐adjusted.
In comparisons comparing DHA‐P to AS+MQ, six trials used the three‐dose regimen, four trials used the four‐dose regimen, and one trial used both. Stratifying the analysis by dosing regimen did not reveal any important differences in efficacy between the two regimens (Analysis 3.1 to Analysis 3.6).
3.1. Analysis.
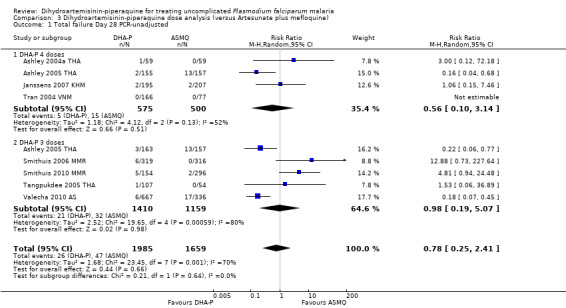
Comparison 3 Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine), Outcome 1 Total failure Day 28 PCR‐unadjusted.
3.6. Analysis.
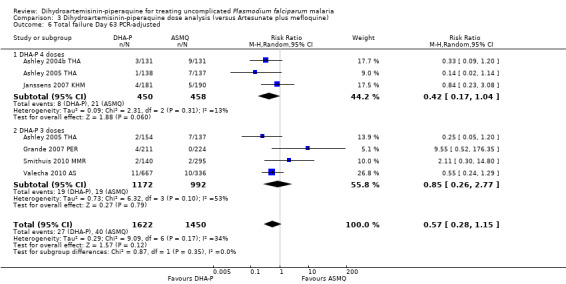
Comparison 3 Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine), Outcome 6 Total failure Day 63 PCR‐adjusted.
Comparison 4. DHA‐P versus artemether‐lumefantrine (six doses)
We found fifteen trials which assessed this comparison; eleven in Africa, three in Asia and one in Oceania; conducted between 2005 and 2011. Eleven of the fifteen trials included children only.
Allocation concealment was at low risk of bias in eight trials (Kamya 2007 UGA; Ratcliff 2007 IDN; Zongo 2007 BFA; Yeka 2008 UGA; Arinaitwe 2009 UGA; Bassat 2009 AF; The 4ABC Study 2011 AF; Yavo 2011 AF). Ten out of 14 trials blinded laboratory staff to treatment allocation.
Total failure
In Africa, PCR‐unadjusted treatment failure at day 28 was consistently lower with DHA‐P (RR 0.34, 95% CI 0.30 to 0.39, nine trials, 6200 participants, Analysis 4.1). After PCR adjustment to exclude new infections, treatment failure at Day 28 was below 5% with both ACTs in all nine trials, but was consistently lowest with DHA‐P (RR 0.42, 95% CI 0.29 to 0.62, nine trials, 5417 participants, Analysis 4.2). Six trials continued follow‐up until day 42, and two until day 63. DHA‐P appears to have a longer post‐treatment prophylactic effect than AL6 in keeping with its longer elimination half‐life (Day 42 PCR‐unadjusted treatment failure: RR 0.60, 95% CI 0.53 to 0.67, seven trials, 3301 participants, Analysis 4.3; Day 63 PCR‐unadjusted treatment failure: RR 0.71, 95% CI 0.65 to 0.78, two trials, 3200 participants, Analysis 4.5).
4.1. Analysis.
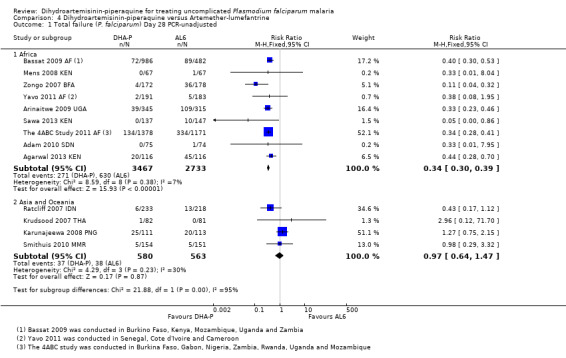
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted.
4.2. Analysis.
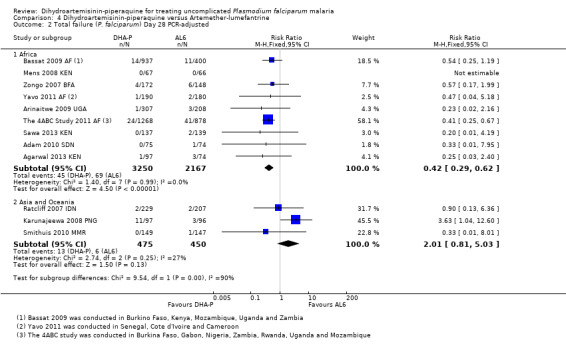
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 2 Total failure (P. falciparum) Day 28 PCR‐adjusted.
4.3. Analysis.

Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted.
4.5. Analysis.
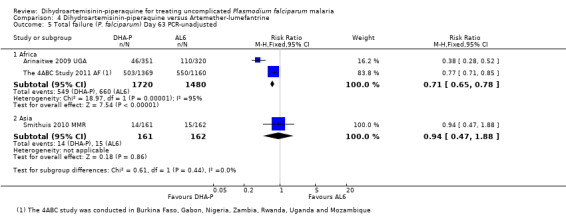
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 5 Total failure (P. falciparum) Day 63 PCR‐unadjusted.
In Asia and Oceania, PCR‐unadjusted treatment failure at day 28 was similar between treatments (four trials, 1143 participants, Analysis 4.1), and with no statistically significant differences after PCR adjustment (three trials, 925 participants, Analysis 4.2). Of note, PCR‐adjusted treatment failure at day 28 was above 10% in those treated with DHA‐P in the one trial from Papua New Guinea (Karunajeewa 2008 PNG), but this has not been seen elsewhere. No differences were seen in PCR‐unadjusted or PCR‐adjusted treatment failure at day 42 (two trials, 572 participants, Analysis 4.3; Analysis 4.4), or day 63 (one trial, 323 participants, Analysis 4.5; Analysis 4.6).
4.4. Analysis.
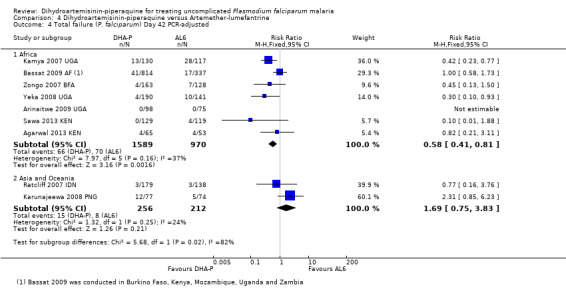
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 4 Total failure (P. falciparum) Day 42 PCR‐adjusted.
4.6. Analysis.
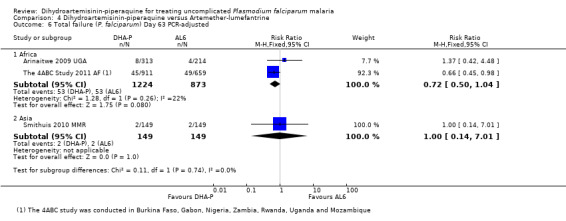
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 6 Total failure (P. falciparum) Day 63 PCR‐adjusted.
Gametocytes
SIx trials, all from Africa, reported the development of gametocytes in those negative at baseline. The results were highly heterogenous and we could not pool them (six trials, 1968 participants) heterogeneity: Chi² test, P = 0.001, I² = 78%, Analysis 4.7). Carriage of gametocytes during the first two weeks was higher with DHA‐P (RR 4.32, 95% 1.48 to 12.63, four trials, 1537 participants, Analysis 4.8), but lower with DHA‐P during weeks three to six; a finding which may reflect the lower treatment failure rates with DHA‐P at later time points (Analysis 4.8). Bassat 2009 AF reports that person‐gametocyte weeks was higher in those treated with DHA‐P (see Table 8).
4.7. Analysis.

Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 7 Gametocyte development (in those negative at baseline).
4.8. Analysis.
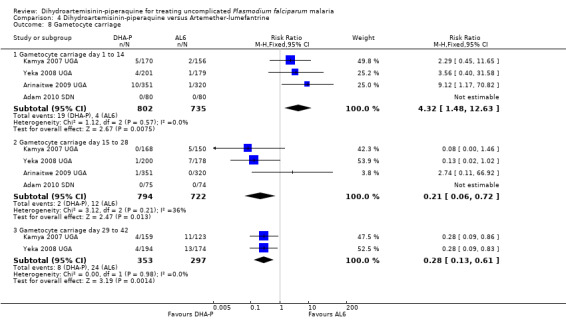
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 8 Gametocyte carriage.
7. DHA‐P versus AL6: Additional secondary outcomes data.
| Outcome | Trial ID | Measure | DHA‐P | AL6 | P value | Comment |
| Gametocyte carriage | Bassat 2009 AF | Person gametocytemia weeks per 1000 person weeks | 43.97 | 21.43 | 0.005 | |
| Smithuis 2010 MMR | Person gametocytemia weeks per 1000 person weeks | 112.8 | 58.2 | Not stated | ||
| Ratcliff 2007 IDN | Person gametocytemia weeks per 1000 person weeks | ‐ | ‐ | Not significant | Figures not given. | |
| Karunajeewa 2008 PNG | Post‐treatment gametocytemia | ‐ | ‐ | No difference | Figures not given. | |
| Anaemia | Yeka 2008 UGA | Mean increase in Hb (g/dL) | 1.75 ± 1.8 | 1.66 ± 2.0 | 0.63 | |
| Mens 2008 KEN | Final mean Hb level (mmol/L) | 7.15 ± 1.07 | 6.79 ± 1.24 | Not significant | ||
| Arinaitwe 2009 UGA | Mean Hb recovery (g/dL) | 0.62 ± 1.68 | 0.56 ± 1.58 | 0.41 | ||
| Agarwal 2013 KEN | Mean Hb increase from baseline in patients not re‐infected | 11.6 g/dL | 9.8 g/dL | Not stated | P value for difference in mean Hb increase in re‐infected patients and those not re‐infected is given as 0.9. | |
| Mean Hb increase from baseline in re‐infected patients | 11.1 g/dL | 9.9 g/dL | Not stated |
Hb ‐ Haemoglobin
In Asia, Karunajeewa 2008 PNG and Ratcliff 2007 IDN report no differences in gametocyte carriage between groups but did not give figures, while Smithuis 2010 MMR reports higher gametocyte carriage with DHA‐P (see Table 8).
Anaemia
Six trials reported changes in haemoglobin from baseline to the last day of follow‐up (day 28 or 42). There is a trend towards a small benefit with DHA‐P which may not be of clinical significance (six trials, 3529 participants, Analysis 4.9).
4.9. Analysis.
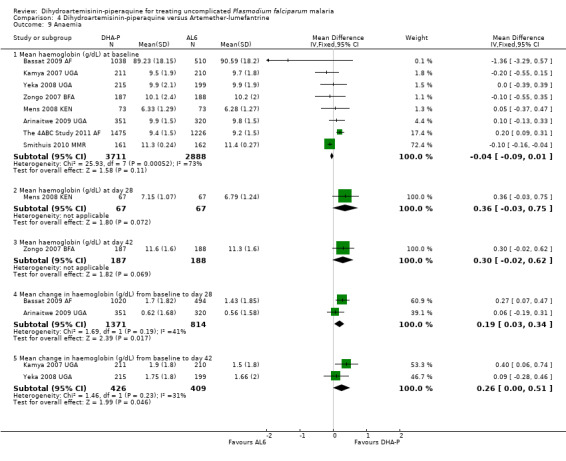
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 9 Anaemia.
Adverse events
No difference has been shown in the frequency of serious adverse events, although the trend is towards a small increase in serious adverse events with DHA‐P (nine trials, 7246 participants, Analysis 4.10, see Appendix 2 for details of serious adverse events).
4.10. Analysis.

Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 10 Serious adverse events (including deaths).
DHA‐P is associated with a higher frequency of early vomiting (drug‐related vomiting), which just reached statistical significance (RR 1.69, 95% CI 1.00 to 2.83, four trials, 2969 participants, Analysis 4.11), but there was no difference in vomiting overall (nine trials, 6761 participants, Analysis 4.11).
4.11. Analysis.
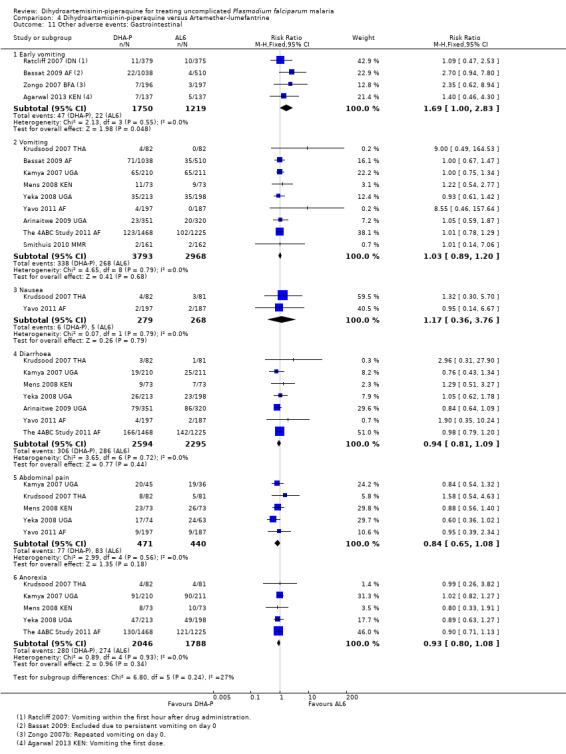
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 11 Other adverse events: Gastrointestinal.
Compared to AL6, DHA‐P was also associated with a slightly higher frequency of pruritis (RR 1.74, 95% CI 1.03 to 2.92, five trials, 2033 participants, Analysis 4.14), but there were no differences in any other clinical side effects (Analysis 4.11 to Analysis 4.14).
4.14. Analysis.
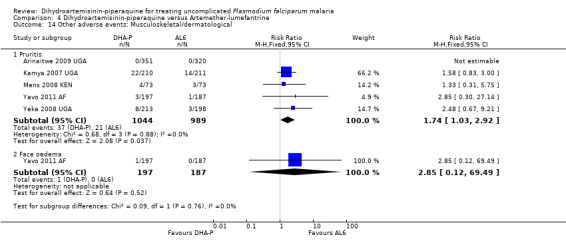
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 14 Other adverse events: Musculoskeletal/dermatological.
Only one trial conducted ECG monitoring on participants in both treatment groups (Bassat 2009 AF). On day 2, a higher proportion of participants treated with DHA‐P had borderline raised QTc intervals (431 to 450 ms) when corrected by Bazett's method (29.1% DHA‐P versus 19.8% AL6, P < 0.001; authors' own figures), but not Fridericia's method (1.0% DHA‐P versus 1.2% AL6, P = 0.76; authors' own figures). There were no differences in the proportion of patients with prolonged QTc interval (> 450 ms), using either Bazett's or Fridericia's method (one trial, 1548 participants, Analysis 4.13) or reported at day 7 (see Table 5 for additional data).
4.13. Analysis.
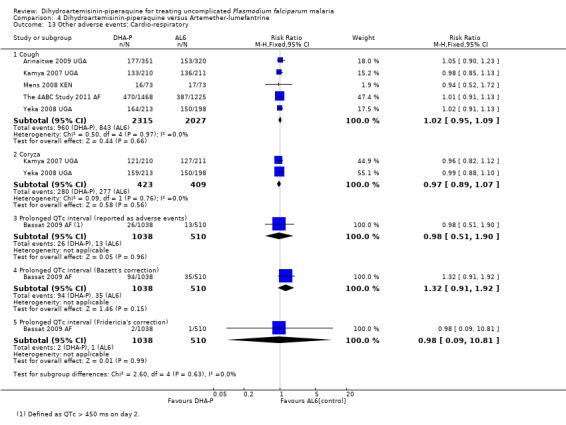
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 13 Other adverse events: Cardio‐respiratory.
Three trials conducted biochemical monitoring for either renal or hepatic adverse events (Bassat 2009 AF; The 4ABC Study 2011 AF; Yavo 2011 AF). Monitoring was adequate in all three trials but incompletely reported in one trial. No clinically important toxicities were reported (see Table 9).
8. DHA‐P versus AL6: Biochemical monitoring and adverse events.
| Trial ID | No. of participants | Tests | Days tested | Days reported | Days tested adequate to detect adverse events? | For adequate testing, was reporting complete? | Results as presented in the paper |
| Bassat 2009 AF | 1553 | LFTs and renal function | Days 3, 28, and 42 | None | Adequate1 | Incomplete 2 | "...altered liver enzymes (ALT)... was similar between the two treatment groups". |
| The 4ABC Study 2011 AF | 2701 | LFT and renal function | Days 7 and 28 | Days 7 and 28 | Adequate1 | Complete3 | "The median levels of alanine aminotransferase and creatinine before treatment, as well as the proportion of patients with values above the normal range (both clinically and non‐clinically significant, the latter not shown), were similar between the four study arms, and this did not change during the follow‐ups at day 7 and 28". |
| Yavo 2011 AF | 384 | LFTs | Days 0 and 4 | Baseline and day 4 | Adequate1 | Complete3 | "In DP group from the beginning of the treatment to day 4, there was a decrease of the mean of AST and a small increase of ALT mean, while in the AL group, AST and ALT means increased. However, these variations were not significantly different. The decrease of the mean of creatinin from the beginning of the treatment to day 4 was not significant in the DP group but was significant in the AL group. In the two groups, the bilirubin decrease was significant". |
1 Adequate given that no clinically important abnormalities were seen. 2 Incomplete as trial authors did not present data and only gave a text summary. 3 Complete as trial authors presented data for the two days tested.
Comparison 5. DHA‐P versus artesunate plus amodiaquine
We found four trials which assessed this comparison; two in Africa and two in Asia; conducted between 2004 and 2009.
Allocation concealment was assessed as low risk of bias in two trials (Hasugian 2007 IDN; The 4ABC Study 2011 AF) and unclear in the other two. In all four trials laboratory staff were blinded to treatment allocation.
Total failure
In Africa, PCR‐unadjusted treatment failure at day 28 was lower following treatment with DHA‐P in both trials (RR 0.49, 95% CI 0.41 to 0.59, two trials, 2800 participants, Analysis 5.1). After PCR‐adjustment to exclude new infections, the difference between treatments was no longer statistically significant, but treatment failure was below 5% following treatment with DHA‐P in both trials, and above 5% following AS+AQ in Rwanda (two trials, 2486 participants, Analysis 5.2). One trial followed participants up to day 63 (The 4ABC Study 2011 AF), and found no differences in PCR‐unadjusted or PCR‐adjusted treatment failure at this time point (one trial, 2292 participants, Analysis 5.5; Analysis 5.6).
5.1. Analysis.
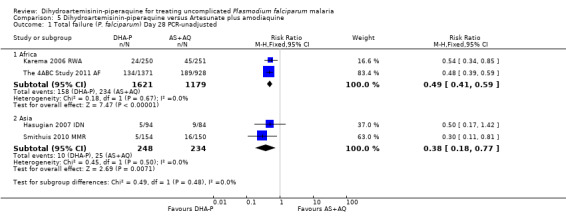
Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted.
5.2. Analysis.

Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 2 Total failure (P. falciparum) Day 28 PCR‐adjusted.
5.5. Analysis.
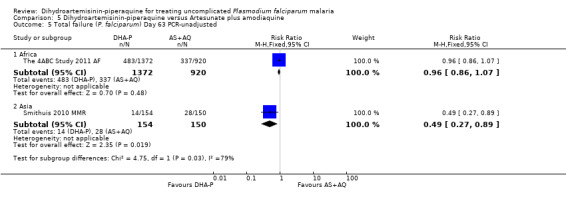
Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 5 Total failure (P. falciparum) Day 63 PCR‐unadjusted.
5.6. Analysis.

Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 6 Total failure (P. falciparum) Day 63 PCR‐adjusted.
In Asia, PCR‐unadjusted treatment failure at day 28 was lower following treatment with DHA‐P (RR 0.38 95% CI 0.18 to 0.77, two trials, 482 participants, Analysis 5.1), and remained lower after PCR‐adjustment although the number of events was very low (RR 0.08, 95% CI 0.01 to 0.40, two trials, 466 participants, Analysis 5.2). One trial followed participants up to day 42 (Hasugian 2007 IDN), and one to day 63 (Smithuis 2010 MMR), when the differences remained statistically significant in favour of DHA‐P (Analysis 5.3 to Analysis 5.6).
5.3. Analysis.
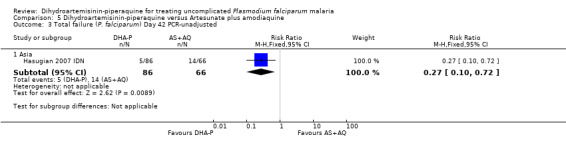
Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted.
Gametocytes
Two trials reported no statistically significant differences in gametocyte carriage during follow‐up but did not report the data (see Table 10).
9. DHA‐P versus AS+AQ: Additional secondary outcomes data.
| Outcome | Trial ID | Measure | DHA‐P | AS+AQ | P value | Comment |
| Gametocyte carriage | Karema 2006 RWA | Gametocyte prevalence | ‐ | ‐ | Not significant | Figures not given. |
| Hasugian 2007 IDN | Post‐treatment gametocyte carriage | ‐ | ‐ | No difference | Figures not given. | |
| Anaemia | Karema 2006 RWA | Mean PCV at baseline | 31.5 ± 4.9 | 31.0 ± 5.5 | No difference | |
| Mean PCV at day 14 | 33.4 ± 3.6 | 34.0 ± 3.7 | 0.08 | |||
| Hasugian 2007 IDN | Proportion of patients with anaemia at day 7 | ‐ | ‐ | 0.02 | "Although there was no significant difference in haemoglobin levels between treatment groups at the time of admission, the rates of anemia at days 7 and 28 were significantly higher in AS+AQ recipients". | |
| Proportion of patients with anaemia at day 28 | ‐ | ‐ | 0.006 | |||
| Smithuis 2010 MMR | Mean increase in haemoglobin | Not stated | Not stated | > 0.05 | "The mean increase of haemoglobin was similar among the treatment groups". |
Hb ‐ Haemoglobin PCV ‐ Packed cell volume Neutropenia ‐ neutrophil count < 1000/µL
Anaemia
Two trials reported no difference between PCV and haemoglobin levels respectively between the treatment groups (Karema 2006 RWA; Smithuis 2010 MMR; see Table 10). Hasugian 2007 IDN found that the prevalence of anaemia at day 7 (P = 0.02) and 28 (P = 0.006) was higher with AS+AQ (authors' own figures); this may be attributed to the recurrence of parasitaemia with both P. falciparum and P. vivax being higher in the AS+AQ group.
Adverse events
The frequency of serious adverse events was lower with DHA‐P, and despite few events, this reached statistical significance (RR 0.40 95% CI 0.19 to 0.87, two trials, 2805 participants, Analysis 5.7, see Appendix 2 for details of serious adverse events). The 4ABC Study 2011 AF reported 15 serious adverse events in 1003 participants treated with AS+AQ versus 10/1468 with DHA‐P. The exact nature of these serious adverse events was unclear, but the authors reported no differences in serious adverse events classified as possibly, probably, or definitely related to the trial drug (4/1003 versus 4/1468).
5.7. Analysis.
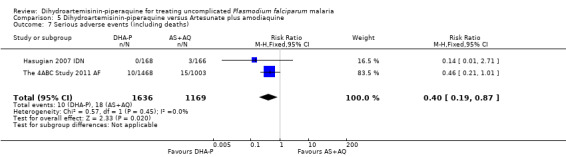
Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 7 Serious adverse events (including deaths).
Hasugian 2007 IDN and Smithuis 2010 MMR found no difference in the number of participants with early vomiting (two trials, 650 participants, Analysis 5.8).
5.8. Analysis.
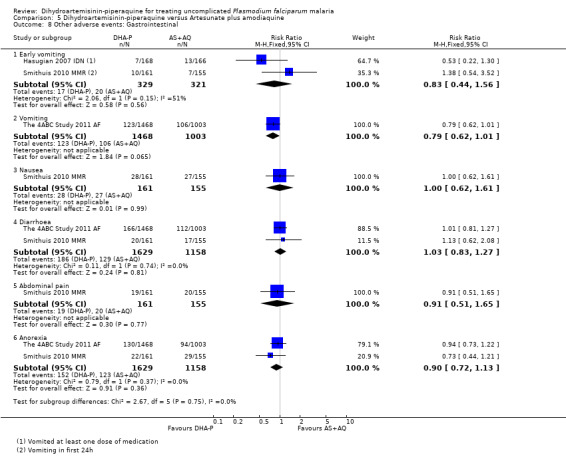
Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 8 Other adverse events: Gastrointestinal.
Pyrexia was the only adverse event that was statistically more common with DHA‐P (RR 1.18 95% CI 1.02 to 1.37, one trial, 2471 participants, Analysis 5.9).
5.9. Analysis.

Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 9 Other adverse events: Neuro‐psychiatric.
Two trials conducted biochemical monitoring for renal or hepatic adverse events (Karema 2006 RWA; Smithuis 2010 MMR). Monitoring was adequate in both trials but incompletely reported in one trial. No clinically important toxicities were reported (see Table 11).
10. DHA‐P versus AS+AQ: Biochemical monitoring and adverse events.
| Study ID | No. of participnts | Tests | Days tested | Days reported | Days tested adequate? | For adequate testing, was reporting complete? | Results as presented in the paper |
| Karema 2006 RWA | 762 | LFTs at one site only | Days 0 and 14 | None | Adequate1 | Incomplete 2 | "No hepatotoxicity was observed, although analyses were performed at one site only (data not shown)". |
| The 4ABC Study 2011 AF | 2701 | LFT and renal function | Days 7 and 28 | Days 7 and 28 | Adequate1 | Complete3 | "The median levels of alanine aminotransferase and creatinine before treatment, as well as the proportion of patients with values above the normal range (both clinically and non‐clinically significant, the latter not shown), were similar between the four study arms, and this did not change during the follow‐ups at day 7 and 28". |
1 Adequate given that no clinically important abnormalities were seen. 2 Incomplete as trial authors did not present data and only gave a text summary. 3 Complete as trial authors presented data for the two days tested.
Comparison 6. DHA‐P versus artesunate plus sulfadoxine‐pyrimethamine
One trial conducted in Oceania in 2007 assessed this comparison (Karunajeewa 2008 PNG). The trial authors did not describe any attempt to conceal allocation. Laboratory staff were blinded to treatment allocation.
Total failure
At day 28 PCR‐adjusted treatment failure was > 10% in both groups (Analysis 6.2).
6.2. Analysis.
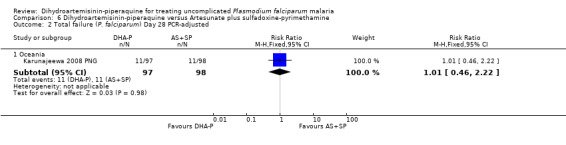
Comparison 6 Dihydroartemisinin‐piperaquine versus Artesunate plus sulfadoxine‐pyrimethamine, Outcome 2 Total failure (P. falciparum) Day 28 PCR‐adjusted.
There were no statistically significant differences in treatment failure between the two arms (one trial, 223 participants, Analysis 6.1 to Analysis 6.4)
6.1. Analysis.
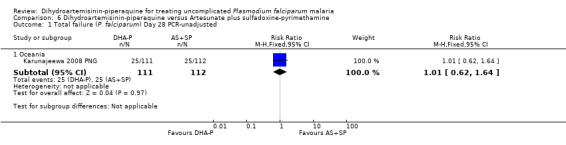
Comparison 6 Dihydroartemisinin‐piperaquine versus Artesunate plus sulfadoxine‐pyrimethamine, Outcome 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted.
6.4. Analysis.

Comparison 6 Dihydroartemisinin‐piperaquine versus Artesunate plus sulfadoxine‐pyrimethamine, Outcome 4 Total failure (P. falciparum) Day 42 PCR‐adjusted.
Gametocytes
No significant differences in gametocyte carriage during follow‐up were reported (figures not reported).
Anaemia
Trial authors reported haemoglobin levels remained similar in both groups throughout follow‐up (figures not reported).
Adverse events
Monitoring for adverse events was undertaken but no differences between the groups were reported.
Discussion
For summaries of the main results for efficacy see; Table 1; Table 12; Table 13; Table 14; Table 15; Table 16), and for adverse effects see Appendix 3.
11. Dihydroartemisinin‐piperaquine compared to Artemether‐lumefantrine for uncomplicated P. falciparum malaria in Asia and Oceania.
| Dihydroartemisinin‐piperaquine compared to Artemether‐lumefantrine for uncomplicated P. falciparum malaria in Asia | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Asia and Oceania Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artemether‐lumefantrine (AL6) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| AL6 | DHA‐P | ||||
|
Treatment failure Day 28 |
PCR‐unadjusted | RR 0.97 (0.64 to 1.47) | 1143 (4 trials) | ⊕⊕⊕⊝ moderate1,2,3,4 | |
| 7 per 100 | 7 per 100 (4 to 10) | ||||
| PCR‐adjusted | RR 2.01 (0.81 to 5.03) | 925 (3 trials) | ⊕⊕⊕⊝ moderate1,2,3,4 | ||
| 1 per 100 | 3 per 100 (1 to 7) | ||||
|
Treatment failure Day 63 |
PCR‐unadjusted | RR 0.94 (0.47 to 1.88) | 323 (1 trial) | ⊕⊕⊝⊝ low4,5,6,7 | |
| 9 per 100 | 8 per 100 (4 to 17) | ||||
| PCR‐adjusted | RR 1.00 (0.14 to 7.01) | 298 (1 trial) | ⊕⊕⊝⊝ low4,5,6,7 | ||
| 1 per 100 | 1 per 100 (0 to 7) | ||||
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 No serious risk of bias: Trials are generally at low or unclear risk of bias. Exclusion of trials as high risk of selection bias or detection bias did not change the result. 2 No serious inconsistency: Statistical heterogeneity was low. 3 No serious indirectness: The trials were conducted in adults and children in Indonesia, Thailand, Papua New Guinea, and Myanmar. 4 Downgraded by 1 for serious imprecision: The 95% CI is wide and includes appreciable differences between drugs. 5 No serious risk of bias: This single trial is generally at low risk of bias. 6 Downgraded by 1 for serious indirectness: This single trial is from Myanmar. The results may not be easily generalized to elsewhere. 7 Two trials from Indonesia and Papua New Guinea reported outcomes at Day 42. At this timepoint there was no difference in PCR unadjusted or PCR adjusted treatment failure (two trials, 572 participants, low quality evidence).
12. Dihydroartemisinin‐piperaquine compared to Artesunate plus mefloquine for treating uncomplicated P. falciparum malaria in Asia.
| Dihydroartemisinin‐piperaquine compared to Artesunate plus mefloquine for treating uncomplicated P. falciparum malaria in Asia | |||||
| Patient or population: Patients with treating uncomplicated P. falciparum malaria Settings: Endemic settings in Asia Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artesunate plus mefloquine (AS+MQ) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| AS+MQ | DHA‐P | ||||
|
Treatment failure Day 28 |
PCR‐unadjusted | RR 1.02 (0.28 to 3.72) | 3487 (8 trials) | ⊕⊕⊕⊕ high1,2,3,4 | |
| 2 per 100 | 2 per 100 (1 to 8) | ||||
| PCR‐adjusted | RR 0.41 (0.21 to 0.8) | 3467 (8 trials) | ⊕⊕⊕⊕ high1,2,3,5 | ||
| 1 per 100 | 0 per 100 (0 to 1) | ||||
|
Treatment failure Day 63 |
PCR‐unadjusted | RR 0.84 (0.69 to 1.03) | 2715 (5 trials) | ⊕⊕⊕⊝ moderate1,6,7,8 | |
| 12 per 100 | 10 per 100 (8 to 13) | ||||
| PCR‐adjusted | RR 0.5 (0.3 to 0.84) | 2500 (5 trials) | ⊕⊕⊕⊕ high1,7,8,9 | ||
| 3 per 100 | 2 per 100 (1 to 3) | ||||
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 No serious risk of bias: Trials are generally at low risk of selection bias and detection bias. Exclusion of trials as high or unclear risk of bias did not change the result. 2 No serious inconsistency: Six trials found very few recurrent parasitaemia in both groups. Two trials primarily conducted in Thailand in areas with multi‐drug resistance found an increased risk of recurrent parasitaemia with AS+MQ. 3 No serious indirectness: The trials were conducted in adults and children in Vietnam, Thailand, Cambodia, Myanmar, India, and Laos. 4 No serious imprecision: The overall result is of no significant difference between treatments. However, where there is P. falciparum resistance to mefloquine, DHA‐P may be superior. 5 No serious imprecision: The overall result is of a statistically significant benefit with DHA‐P although this benefit may only be present where there is resistance to mefloquine. 6 Downgraded by one for serious inconsistency: Of the five trials, one from Thailand in 2005 found a statistically significant benefit with DHA‐P, one from Myanmar in 2009 found a benefit with DHA‐P, and three found no difference. 7 No serious indirectness: The trials were conducted in adults and children in Thailand, Cambodia, Myanmar, India, and Laos. 8 No serious imprecision: The overall result is of no significant difference between treatments. Although some trials found statistically significant differences, these may not be clinically important. 9 No serious inconsistency: There is a small amount of variability between trials, with only one trial showing a statistically significant benefit with DHA‐P.
13. Dihydroartemisinin‐piperaquine compared to Artesunate plus mefloquine for uncomplicated P. falciparum malaria in South America.
| Dihydroartemisinin‐piperaquine compared to Artesunate plus mefloquine for uncomplicated P. falciparum malaria in South America | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Endemic settings in South America Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artesunate plus mefloquine (AS+MQ) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| AS+MQ | DHA‐P | ||||
|
Treatment failure Day 28 |
PCR‐unadjusted | ‐ | Data unavailable | ‐ | |
| ‐ | ‐ | ||||
| PCR‐adjusted | ‐ | Data unavailable | ‐ | ||
| ‐ | ‐ | ||||
|
Treatment failure Day 63 |
PCR‐unadjusted | RR 6.19 (1.4 to 27.35) | 445 (1 trial) | ⊕⊕⊝⊝ low1,2,3 | |
| 1 per 100 | 6 per 100 (1 to 24) | ||||
| PCR‐adjusted | RR 9.55 (0.52 to 176.35) | 435 (1 trial) | ⊕⊕⊝⊝ low1,2,4 | ||
| 0 per 100 | 0 per 100 (0 to 0) | ||||
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 No serious risk of bias: This trial is at low risk of selection bias and unclear risk of detection bias. 2 Downgraded by 1 for serious indirectness: This findings of this single trial are not easily generalized to other South American countries. 3 Downgraded by 1 for serious imprecision: Although this result reached statistical significance the number of events is very low, and there is a high possibility that this is a chance finding. 4 Downgraded by 1 for serious imprecision: There were too few events in this single trial to confidently exclude important effects.
14. Dihydroartemisinin‐piperaquine compared to Artesunate plus amodiaquine for uncomplicated P. falciparum malaria in Africa.
| Dihydroartemisinin‐piperaquine compared to Artesunate plus amodiaquine for uncomplicated P. falciparum malaria in Africa | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Africa Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artesunate plus amodiaquine (AS+AQ) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| AS+AQ | DHA‐P | ||||
|
Treatment failure Day 28 |
PCR‐unadjusted | RR 0.49 (0.41 to 0.59) | 2800 (2 trials) | ⊕⊕⊕⊕ high1,2,3,4 | |
| 20 per 100 | 10 per 100 (8 to 12) | ||||
| PCR‐adjusted | RR 0.67 (0.42 to 1.06) | 2486 (2 trials) | ⊕⊕⊕⊝ moderate1,2,3,5 | ||
| 4 per 100 | 2 per 100 (2 to 4) | ||||
|
Treatment failure Day 63 |
PCR‐unadjusted | RR 0.96 (0.86 to 1.07) | 2292 (1 trial) | ⊕⊕⊕⊝ moderate3,6,7,8 | |
| 37 per 100 | 35 per 100 (32 to 39) | ||||
| PCR‐adjusted | RR 1.8 (0.85 to 3.83) | 1506 (1 trial) | ⊕⊕⊕⊝ moderate3,6,7,8 | ||
| 2 per 100 | 3 per 100 (1 to 6) | ||||
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 No serious risk of bias: Trials are generally at low risk of bias. Exclusion of trials as high or unclear risk of selection bias or detection bias did not change the result. 2 No serious inconsistency: The trials had similar results and statistical heterogeneity was low. 3 No serious indirectness: The trials were conducted in children in different transmission settings in Burkina Faso, Gabon, Nigeria, Rwanda, Uganda, Zambia, and Mozambique. 4 No serious imprecision: Both limits of the 95% CI imply appreciable benefit. 5 Downgraded by 1 for serious imprecision: The findings did not reach statistical significance. 6 No serious risk of bias: This finding is only reported in one trial which was at low risk of bias. 7 Downgraded by 1 for serious imprecision: There 95% CI are wide and include what might be important differences. 8 No data were presented for day 42.
15. Dihydroartemisinin‐piperaquine compared to Artesunate plus amodiaquine for treating uncomplicated P. falciparum malaria in Asia.
| Dihydroartemisinin‐piperaquine compared to Artesunate plus amodiaquine for treating uncomplicated P. falciparum malaria in Asia | |||||
| Patient or population: Patients with treating uncomplicated P. falciparum malaria Settings: Asia Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artesunate plus amodiaquine (AS+AQ) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| AS+AQ | DHA‐P | ||||
|
Treatment failure Day 28 |
PCR‐unadjusted | RR 0.38 (0.18 to 0.77) | 482 (2 trials) | ⊕⊕⊕⊝ moderate1,2,3,4 | |
| 11 per 100 | 4 per 100 (2 to 8) | ||||
| PCR‐adjusted | RR 0.08 (0.01 to 0.4) | 466 (2 trials) | ⊕⊕⊕⊝ moderate1,2,3,4 | ||
| 8 per 100 | 1 per 100 (0 to 3) | ||||
|
Treatment failure Day 63 |
PCR‐unadjusted | RR 0.49 (0.27 to 0.89) | 304 (1 trial) | ⊕⊕⊝⊝ low4,5,6,7 | |
| 19 per 100 | 9 per 100 (5 to 17) | ||||
| PCR‐adjusted | RR 0.14 (0.03 to 0.59) | 278 (1 trial) | ⊕⊕⊝⊝ low4,5,6,7 | ||
| 10 per 100 | 1 per 100 (0 to 6) | ||||
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 No serious risk of bias: Trials are generally at low risk of bias. Exclusion of trials as high or unclear risk of selection bias or detection bias did not change the result. 2 No serious inconsistency: The trials all had similar results and statistical heterogeneity was low. 3 No serious indirectness: The trials were conducted in adults and children in Indonesia and Myanmar. 4 Downgraded by 1 for serious imprecision: Although this result reached statistical significance there are limited data, with few events. Larger trials are needed to have full confidence in this result. 5 No serious risk of bias: This finding is only reported in one trial which was generally at low risk of bias. 6 Downgraded by 1 for serious indirectness: This trial was from a single setting in Myanmar, and may not be easily generalized to elsewhere. 7 One trial from Indonesia conducted in 2007 presented day 42 outcomes and at this timepoint there was still an advantage on PCR‐unadjusted treatment failure with DHA‐P (RR 0.27, 95% CI 0.10 to 0.72, one trial, 152 participants, moderate quality evidence), and PCR‐adjusted treatment failure (RR 0.10, 95% CI 0.01 to 0.81, one trial, 141 participants, moderate quality evidence).
Summary of main results
DHA‐P versus artemether lumefantrine
In Africa, during 28 days follow‐up, DHA‐P is superior to AL6 at preventing further parasitaemia (high quality evidence), and although PCR‐adjusted treatment failure was below 5% for both ACTs it was consistently lower with DHA‐P (high quality evidence). DHA‐P has a longer prophylactic effect on new infections which may last for up to 63 days (high quality evidence).
In Asia and Oceania, no differences in treatment failure have been shown at day 28 (moderate quality evidence), or day 63 (low quality evidence).
DHA‐P and AL6 appear to have similar adverse effect profiles (moderate quality evidence). DHA‐P was associated with borderline prolongation of QTc interval but no difference was seen in prolonged QTc (low quality evidence) and no cardiac arrhythmias were reported.
DHA‐P versus artesunate plus mefloquine
In Asia, during 28 days follow‐up, DHA‐P is as effective as AS+MQ at preventing further parasitaemia (high quality evidence). Once adjusted by PCR to exclude new infections, treatment failure at day 28 was below 5% for both ACTs in all eight trials, but lower with DHA‐P in two trials from sites with multi‐drug resistant P. falciparum (high quality evidence). Both combinations contain partner drugs with very long half‐lives and no consistent difference in preventing new infections has been seen between drugs over 63 days follow‐up (moderate quality evidence).
In the only trial from South America, there were fewer recurrent parasitaemias over 63 days with AS+MQ (low quality evidence), but there was no difference between treatments once adjusted by PCR for new infections (low quality evidence).
Compared to AS+MQ, DHA‐P is associated with reduced nausea, vomiting, dizziness, sleeplessness, and palpitations (moderate quality evidence). The only notable adverse event associated with DHA‐P was an increased frequency of prolongation of the QTc interval (low quality evidence), however no cardiac arrhythmias were reported in these trials.
Overall completeness and applicability of evidence
DHA‐P is one of the most studied ACTs, and we included 27 trials in this review, which enrolled 16,382 adults and children with uncomplicated malaria. Notably, these trials excluded infants aged less than six months and pregnant women, and further safety data is required for these groups.
The efficacy of DHA‐P against uncomplicated P. falciparum malaria in adults and children however is now well established, and although there is only limited data from South America, it is likely that the findings of this review can be applied worldwide.
Despite the high efficacy against asexual parasitaemia, DHA‐P appears to have a reduced efficacy against gametocytes. Compared to DHA‐P, both AS+MQ and AL6 reduce the carriage of gametocytes during the first 14 days post‐treatment. This deficiency has been discussed in the literature, and it is likely due to a relative underdosing of the artemisinin derivative in the combination. The clinical significance of this effect remains unclear as gametocyte carriage is only an indirect measure of transmission potential. Furthermore, any increased risk of transmission in the early period after treatment may be offset by the later improved prophylactic effect of DHA‐P.
DHA‐P has been available and in use for several years despite the lack of a WHO prequalified formulation manufactured according to Good Manufacturing Practices Standards (GMP), and concerns about the stability and shelf‐life of the dihydroartemisinin combination (Jansen 2010; Schmatz 2010). However, the Eurartesim® formulation evaluated by Bassat 2009 AF and Valecha 2010 AS has now been registered and approved for use by the European Medicines Agency (EMA) (European Medicines Agency 2011).
The potential for prolongation of the QTc interval is the most notable adverse effect. This was noted in the EMA's report where they advised that DHA‐P should not be used in people who have, or are at risk of, QTc interval prolongation or cardiac arrhythmias, and should not be taken with other drugs which prolong the QTc interval (European Medicines Agency 2011). No participants were reported to have experienced confirmed cardiac arrhythmias in these studies.
Systematic reviews in infectious diseases also need to consider the possibility of changing drug effects over time as drug resistance patterns change and develop. In this review, we partially explored this possibility by presenting all forest plots with trials arranged in chronological order, but we found no evidence of a decline in efficacy over time. However, a systematic review may not be the most appropriate way to examine these effects, as RCTs tend to be conducted for new drugs with little research interest once they are well established. Selection of first and second line antimalarials should therefore take into account other knowledge on antimalarial resistance, such as that produced by the WorldWide Antimalaria Resistance Network (WWARN 2013).
Quality of the evidence
We assessed the quality of the evidence in this review using the GRADE approach and presented the evidence in six summary of findings tables for efficacy (Table 1; Table 13; Table 14; Table 12; Table 15; Table 16), and in three summary of findings tables for adverse events (in Appendix 3).
The evidence that DHA‐P is at least as effective as AS+MQ in Asia was of high quality. There was some statistical heterogeneity, with two trials (predominantly from trial sites in Thailand) finding slightly higher levels of treatment failure with AS+MQ. This may be a consequence of resistance to mefloquine in the area and was not considered sufficient to downgrade the evidence.
The evidence for superiority of DHA‐P over AL6 in Africa was of high quality, with no reason to downgrade for risk of bias, inconsistency, indirectness, or imprecision. It should be noted that both DHA‐P and AL6 performed better than the WHO standard of 5% PCR‐adjusted treatment failure at day 28 in all trials. The choice between DHA‐P and AL6 may therefore be based more on considerations of adherence and cost, rather than efficacy.
We also assessed the quality of evidence on comparative adverse effects and presented these in Appendix 3. In general, the evidence was of moderate to low quality, meaning we can have reasonable confidence in some of these effects.
Agreements and disagreements with other studies or reviews
We found three recent systematic reviews of DHA‐P (Keating 2012; Naing 2013 & WWARN 2013b), and reviewed the public assessment report of Eurartesim® by the EMA (European Medicines Agency 2011).
The most recent systematic review (Naing 2013) includes 26 of the 27 trials we included in this review and reaches very similar conclusions: "DHA‐P is non‐inferior to other currently used ACTs such as AS+MQ and AL6" and "the better safety profile of DHA‐P and once‐daily dosage improves adherence. For these reasons, DHA‐P has the potential to become a first‐line antimalarial drug".
The second review (Keating 2012) is more narrative and focuses on the Eurartesim® formulation registered with the EMA. The review contains an extensive discussion of the effects of the formulation on the QTc interval, and the author concludes that "there are currently no data signalling that DHA‐P is associated with clinically significant arrhythmias". Similarly, the EMA public report concludes that "Treatment emergent QTc prolongation was asymptomatic in all cases", and "The magnitude of QTc prolongation is reduced if dosing occurs between meals". Conditional for registration, the pharmaceutical company agreed to undertake further post‐marketing evaluation of the effects of DHA‐P on the QTc interval and the potential for arrhythmias (European Medicines Agency 2011).
The third review (WWARN 2013b) was based on individual patient data from 24 published and two unpublished studies. The analysis paid particular attention to the relationship between age, the drug dose administered, and treatment efficacy. The authors report that treatment failure following DHA‐P was highest in young children (aged between one and five years), and conclude that this is related to significant underdosing of both dihydroartemisinin and piperaquine in this age group, and to different pharmacokinetics in young children. On this basis, some further dose optimization of this combination is underway.
Authors' conclusions
Implications for practice.
In Africa, DHA‐P seems to reduce treatment failure compared to AL6, although it should be noted that AL6 also performed above the WHO standard of 95% cure rate in all these trials. DHA‐P therefore represents an effective alternative with a simplified dosing regimen, and a longer post‐treatment prophylactic effect.
In Asia, DHA‐P appears to be as effective as the widely used AS+MQ, and is better tolerated. This may promote DHA‐P to become the first line treatment option.
Implications for research.
The efficacy of DHA‐P is now well established. Future research should concentrate on safety surveillance, particularly in infants and pregnant women, and further appraisal of the potential effects on cardiac conduction.
Acknowledgements
We acknowledge the contributions of Paul Garner and Hasifa Bukirwa to the development of the original protocol and thank Vittoria Lutje for conducting the searches.
The authors are grateful to the Cochrane Editorial Unit, who helped with data extraction during a review updating pilot programme in 2010. This document is an output from a project funded by UKaid from the UK Government for the benefit of developing countries. The views expressed are not necessarily those of the Department for International Development (DFID).
Appendices
Appendix 1. Adverse event monitoring
| DHA‐P versus Artesunate plus mefloquine | ||||||
| Trial ID | Sample Size | Blinding | Clinical symptoms monitoring | Biochemistry | Haematological | ECG |
| Ashley 2004a THA | 134 | Open label | Inpatient monitoring until day 28 | U&E, LFT on days 0 and 7 | FBC on days 0 and 7 | None |
| Ashley 2004b THA | 356 | Open label | Daily review until parasites cleared then weekly until day 63 | U&E, LFT on days 0 and 7 (DHA‐P group only) | FBC on days 0 and 7 (DHA‐P group only) |
Days 0 and 7 (DHA‐P group only) |
| Ashley 2005 THA | 499 | Open label | Clinical examination and symptom enquiry daily until parasites cleared then weekly until day 63 | None | Haematocrit daily until parasites cleared then weekly until day 63 | None |
| Grande 2007 PER | 522 | Open label | Clinical assessment daily until day 3 then weekly until day 63 | U&E, LFT on days 0 and 7, | FBC, PCV days 0 and 7, PCV days 14 and 63 | None |
| Janssens 2007 KHM | 464 | Open label | Clinical examination and symptom questionnaire days 0, 1, 2, and 3 | None | None | None |
| Mayxay 2006 LAO | 220 | Open label | Daily review until parasites cleared then weekly until day 42 | None | None | None |
| Smithuis 2006 MMR | 652 | Open label | Symptom questionnaire at days 0, 1, 2, 3, and 7 | None | None | None |
| Smithuis 2010 MMR | 491 | Open label | Review weekly for 9 weeks | None | None | None |
| Tangpukdee 2005 THA | 180 | Open label | Inpatient monitoring until day 28. Assessed using non‐suggestive questioning. | None | None | None |
| Tran 2004 VNM | 243 | Open label | Review at days 0, 2, and 7 | LFTs on days 3, 7 and 28 | None | None |
| Valecha 2010 AS | 1150 | Open label | Clinical review until parasites cleared then weekly until day 63. | Blood and urine samples 0, 28, 63 and on the day of any recurrent parasitaemia. | None | Days 0, 2, 7, 28, 63 and on the day of any recurrent parasitaemia. |
| DHA‐P versus Artemether‐lumefantrine | ||||||
| Trial ID | Sample Size | Blinding | Clinical symptoms monitoring | Biochemistry | Haematological | ECG |
| Adam 2010 SDN | 160 | Open label |
Review at days 1, 2, 3, 7, 14, 21, and 28. | None | None | None |
| Agarwal 2013 KEN | 274 | Open label | Clinical assessment on days 1, 2, 3, 7, 14, 21, 28, 35, and 42 after enrolment or at any day if ill. | None | None | None |
| Arinaitwe 2009 UGA | 671 treated episodes |
Open label |
Review at days 0, 2, 3, 7, 14, 21, 28 after each episode for 1 year. | None | None | None |
| Bassat 2009 AF | 1548 | Open label |
In‐patient review throughout the dosing period then weekly till day 42. | LFT and renal function at days 3, 28, and 42 and at clinician's request | FBC at days 3, 28 and 42 | 12‐lead ECG at days 0, 2, and 7 |
| Kamya 2007 UGA | 421 | Double blind |
Clinical assessment daily till day 3 then weekly until day 42. | None | None | None |
| Karunajeewa 2008 PNG | 250 | Open label | Clinical assessment on days 0, 1, 2, 3, 7, 14, 28, and 42. | None | None | None |
| Krudsood 2007 THA | 191 | Open label | In‐patient review daily until day 28. | None | None | None |
| Mens 2008 KEN | 146 | Open label | Review on days 0, 1, 2, 3, 7, 14, and 28. | None | None | None |
| Ratcliff 2007 IDN | 774 | Open label | Review and symptom questionnaire daily until fever and parasites cleared then weekly until day 42. | None | None | None |
| Sawa 2013 KEN | 298 | Single blind | Clinical examination on days 1, 2, 3, 7, 14, 28, and 42. | None | None | None |
| The 4ABC Study 2011 AF | 2,701 | Single blind | Clinical assessment on days 1, 2, 3, 7, 14, 21, and 28. | LFT and renal function at days 7 and 28 | FBC at days 3, 7, 14, and 28 | None |
| Yavo 2011 AF | 384 | Open label | Clinical assessment on days 1, 2, 3, 4, 7, 14, 21, and 28. | LFT at baseline and day 4 | None | None |
| Yeka 2008 UGA | 414 | Single blind | Review daily till day 3 then weekly until day 42. | None | None | None |
| Zongo 2007 BFA | 375 | Open label | Assessment daily until day 3 then weekly until day 42. | None | None | None |
| DHA‐P versus Artemether plus amodiaquine | ||||||
| Trial ID | Sample Size | Blinding | Clinical symptoms monitoring | Biochemistry | Haematological | ECG |
| Hasugian 2007 IDN | 334 | Open label | Clinical assessment daily until afebrile then weekly until day 42. | None | None | None |
| Karema 2006 RWA | 504 | Open label | Clinical assessment on days 0, 1, 2, 3, 7, 14, 21, and 28. | Differential WBC count (and LFT at one site only) at days 0 and 14 | None | None |
| Smithuis 2010 MMR | 316 | Open label | Review weekly for 9 weeks. | None | None | None |
| The 4ABC Study 2011 AF | 2477 | Single blind | Clinical assessment on days 1, 2, 3, 7, 14, 21, and 28. | Liver and renal function tests at days 7 and 28 | FBC at days 3, 7, 14, and 28 | None |
| DHA‐P versus artesunate plus sulfadoxine‐pyrimethamine | ||||||
| Trial ID | Sample Size | Blinding | Clinical symptoms monitoring | Biochemistry | Haematological | ECG |
| Karunajeewa 2008 PNG | 245 | Open label | Clinical assessment on days 0, 1, 2, 3, 7, 14, 28, and 42 | None | None | None |
Appendix 2. Serious adverse event descriptions
| DHA‐P versus Artesunate plus mefloquine | |||
| Trial ID | Number of participants | Blinding | Comment |
| Ashley 2004a THA | 134 | Open label | No serious adverse events observed. |
| Ashley 2004b THA | 356 | Open label | No serious adverse events observed. |
| Ashley 2005 THA | 499 | Open label | 4/166 serious events with AS+MQ (death, severe anaemia, febrile convulsion, coagulopathy) and 11/333 with DHA‐P (2 deaths, bacterial sepsis, febrile convulsion, leptospirosis, haematemesis, nephritic syndrome, severe anaemia, respiratory infection, epigastric pain, and vomiting). |
| Grande 2007 PER | 522 | Open label | 3 serious drug related events with AS+MQ requiring stopping treatment (encephalopathy, anxiety and arrhythmia, palpitations, and chest pain). |
| Janssens 2007 KHM | 464 | Open label | No serious adverse events observed. |
| Mayxay 2006 LAO | 220 | Open label | One neuropsychiatric reaction in AS+MQ group. |
| Smithuis 2006 MMR | 652 | Open label | No serious adverse events reported in the first 7 days. |
| Smithuis 2010 MMR | 491 | Open label | Not reported. |
| Tangpukdee 2005 THA | 180 | Open label | No serious adverse events observed. |
| Tran 2004 VNM | 243 | Open label | 12 events (10 vomiting, 2 dizziness) described as significant in AS+MQ group and none with DHA‐P (P = 0.002). |
| Valecha 2010 AS | 1150 | Open label | 12/767 events described as serious in DHA‐P group (6 deemed related to drug: 2 cases of anaemia, 1 viral infection, 1 Wolf‐Parkinson‐White syndrome, 1 convulsion, 1 encephalitis), 3/381 in AS+MQ group (all deemed related to drug: 1 anaemia, 1 convulsion, 1 encephalitis). |
| DHA‐P versus Artemether‐lumefantrine | |||
| Trial ID | Number of participants | Blinding | Comment |
| Adam 2010 SDN | 160 | Open label |
All AE described as mild. |
| Agarwal 2013 KEN | 274 | Open label | 1/137 with DHA‐P, 2/137 with AL (all severe malaria and attributed to new infections). |
| Arinaitwe 2009 UGA | 671 treated episodes | Open label |
3/320 with DHA‐P, 1/351 with AL6 (all due to development of severe anaemia). |
| Bassat 2009 AF | 1548 | Open label |
18/1038 with DHA‐P and 5/510 with AL6 (P = 0.2490. One death occurred in each group but the other SAEs are not described. |
| Kamya 2007 UGA | 421 | Double blind |
4/211 with DHA‐P, 2/210 with AL, all judged to be unrelated to study meds (3 febrile convulsions, otitis media, asthma attack, pyomyositis). |
| Karunajeewa 2008 PNG | 250 | Open label | Overall comment: No treatment withdrawals were attributable to adverse events related to a study drug. |
| Krudsood 2007 THA | 191 | Open label | Overall comment: No significant differences are noted between the treatments. |
| Mens 2008 KEN | 146 | Open label | One patient treated with DHA‐P died on day 14. Assessed as unrelated to treatment. |
| Ratcliff 2007 IDN | 774 | Open label | One death 60 days after treatment. Cause not known. |
| Sawa 2013 KEN | 298 | Single blind | Overall comment: No adverse events reported. |
| The 4ABC Study 2011 AF | 2701 | Single blind | 10/1468 with DHA‐P compared to 6/1225 with AL6. The only ones described are one death due to diarrhoeal disease in the DHA‐P group and three deaths in the AL group (two severe malaria and one unknown cause). None were related to treatment. |
| Yavo 2011 AF | 384 | Open label | None reported. |
| Yeka 2008 UGA | 414 | Single blind | 2/215 with AL, 5/199 with DHA‐P, all judged unrelated to study meds (2 convulsions, 2 pyomyositis, vomiting, severe anaemia, dehydration). |
| Zongo 2007 BFA | 375 | Open label | None observed. |
| Bassat 2009 AF | 1548 | Open label |
No difference in QT prolongation. |
| DHA‐P versus Artesunate plus amodiaquine | |||
| Trial ID | Number of participants | Blinding | Comment |
| Hasugian 2007 IDN | 334 | Open label | 3 with AS+AQ (2 vomiting, 1 ataxia), none with DHA‐P. |
| Karema 2006 RWA | 504 | Open label | Not reported (one seizure with AS+AQ). |
| Smithuis 2010 MMR | 316 | Open label | Not reported. |
| The 4ABC Study 2011 AF | 2477 | Single Blind | Occurence more frequent in AS+AQ group (15/1003 in the AS+AQ group versus 10/1468 in the DHA‐P group). The only ones described are one severe malaria case in AS+AQ group and one death due to diarrhoeal disease in DHA‐P group. |
| DHA‐P versus artesunate plus sulfadoxine‐pyrimethamine | |||
| Trial ID | Number of participants | Blinding | Comment |
| Karunajeewa 2008 PNG | 245 | Open label | Overall comment: No treatment withdrawals were attributable to adverse events related to a study drug. |
Footnotes
AE = adverse event; AL6 = artemether‐lumefantrine; AQ = amodiaquine; AS = artesunate; DHA‐P = dihydroartemisinin‐piperaquine; ECG: electrocardiogram; MQ = mefloquine; QT = interval between the Q and T waves of an ECG; SAE = serious adverse event; SP = sulfadoxine‐pyrimethamine.
Appendix 3. Adverse events GRADE tables
| Dihydroartemisinin‐piperaquine compared to Artesunate plus mefloquine for treating uncomplicated P. falciparum malaria | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Malaria endemic areas Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artesunate plus mefloquine (AS+MQ) | |||||
| Outcomes | Number of participants having adverse events (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | ||
| AS+MQ | DHA‐P | ||||
| Serious adverse events (including deaths) | 8 per 1000 | 9 per 1000 (4 to 18) | 3522 (8 trials) | moderate1,2,3,4 | |
| Gastroenterological | Early vomiting | 7 per 100 | 6 per 100 (5 to 8) | 4114 (9 trials) | moderate2,3,5,6 |
| Nausea | 20 per 100 | 14 per 100 (12 to 16) | 4531 (9 trials) | moderate3,5,7,8 | |
| Vomiting | 13 per 100 | 8 per 100 (6 to 10) | 2744 (5 trials) | moderate3,5,7,8 | |
| Anorexia | 15 per 100 | 13 per 100 (11 to 15) | 3497 (6 trials) | low3,5,7,9 | |
| Diarrhoea | 6 per 100 | 8 per 100 (6 to 11) | 2217 (5 trials) | moderate3,5,7,8 | |
| Abdominal pain | 11 per 100 | 11 per 100 (9 to 13) | 3887 (7 trials) | moderate3,5,7,10 | |
| Neuro‐psychiatric | Headache | 12 per 100 | 10 per 100 (8 to 12) | 2039 (4 trials) | low3,5,8,11 |
| Dizziness | 36 per 100 | 26 per 100 (24 to 28) | 4531 (9 trials) | moderate3,5,7,8 | |
| Sleeplessness | 21 per 100 | 10 per 100 (8 to 13) | 2551 (6 trials) | moderate3,5,7,8 | |
| Fatigue | 8 per 100 | 3 per 100 (2 to 6) | 872 (2 trials) | low5,7,12 | |
| Nightmares | 10 per 100 | 1 per 100 (0 to 7) | 220 (1 trial) | low5,12 | |
| Anxiety | 11 per 100 | 1 per 100 (0 to 4) | 522 (1 trial) | low5,12 | |
| Blurred vision | 9 per 100 | 4 per 100 (2 to 9) | 464 (1 trial) | low5,12 | |
| Tinnitus | 9 per 100 | 4 per 100 (1 to 11) | 220 (1 trial) | low5,12 | |
| Cardio‐respiratory | Palpitations | 18 per 100 | 11 per 100 (8 to 15) | 1175 (3 trials) | moderate3,5,7,8 |
| Cough | 10 per 100 | 8 per 100 (5 12) | 1148 (1 trial) | low5,9 | |
| Dyspnoea | 9 per 100 | 3 per 100 (1 to 10) | 220 (1 trial) | low5,13 | |
| Prolonged QT interval (adverse event) | 4 per 100 | 5 per 100 (3 to 9) | 1148 (1 trial) | low9,14,15 | |
| Prolonged QT interval (Bazett's correction) | 4 per 100 | 9 per 100 (5 to 15) | 1148 (1 trial) | low5,9,15 | |
| Prolonged QT interval (Fridericia's correction) | 5 per 100 | 4 per 100 (3 to 8) | 1148 (1 trial) | low5,9,15 | |
| Musculoskeletal/ dermatological | Arthralgia | 6 per 100 | 5 per 100 (3 to 9) | 1148 (1 trial) | moderate5,10,14 |
| Myalgia | 6 per 100 | 6 per 100 (4 to 10) | 1148 (1 trial) | moderate5,10,14 | |
| Urticaria | 2 per 100 | 1 per 100 (0 to 4) | 719 (2 trials) | low5,13 | |
| Pruritis | 3 per 100 | 2 per 100 (1 to 4) | 872 (2 trials) | low5,13 | |
| Rash | 1 per 100 | 0 per 100 (0 to 7) | 220 (1 trial) | low5,13 | |
| The assumed risk of adverse events in the AS+MQ group is an average risk across trials. The corresponding risk with DHA‐P (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
| 1 No serious risk of bias: Only eight of the 11 trials made any comment on serious adverse events. None of these eight trials were blinded. 2 No serious inconsistency: None of the eight trials found statistically significant differences. 3 No serious indirectness: These trials recruited both adults and children, and were conducted in Asia and South America. 4 Downgraded by 1 for imprecision: These trials do not exclude the possibility of rare but clinically important adverse effects. 5 Downgraded by 1 for serious risk of bias: All trials were open label. 6 No serious imprecision: The 95% CI around the absolute effect is narrow and excludes clinically important differences. 7 No serious inconsistency: This finding was consistent across trials with no significant statistical heterogeneity. 8 No serious imprecision: The result is statistically significant and the meta‐analysis is adequately powered to detect this effect. 9 Downgraded by 1 for serious imprecision: This result does not reach statistical significance. 10 No serious imprecision: The finding is of no difference between treatments and the sample size is adequately powered to detect differences if they existed. 11 Downgraded by 1 for serious inconsistency: There is moderate heterogeneity between trials. 12 Downgraded by 1 for serious indirectness: Only two trials have assessed this outcome. 13 Downgraded by 1 for imprecision: Limited data available and the result is not statistically significant. 14 Downgraded by 1 for serious risk of bias: This trial is unblinded. Only a few of the recorded prolonged QT intervals were registered as adverse events which removed the statistical significance. The reasons for this are unclear. 15 No serious indirectness: This single large trial was conducted in adults and children in Thailand, Laos, and India. | |||||
| Dihydroartemisinin‐piperaquine compared to Artemether‐lumefantrine for uncomplicated P. falciparum malaria | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Malaria endemic areas Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artemether‐lumefantrine (AL6) | |||||
| Outcomes | Number of participants having adverse events (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | ||
| AL6 | DHA‐P | ||||
| Serious adverse events (including deaths) | 6 per 1000 | 10 per 1000 (6 to 17) | 7022 (8 trials) | moderate1,2,3,4 | |
| Gastroenterological | Early vomiting | 2 per 100 | 3 per 100 (2 to 5) | 2695 (3 trials) | moderate2,3,5,6 |
| Vomiting | 9 per 100 | 9 per 100 (8 to 11) | 6761 (9 trials) | moderate2,3,5,6 | |
| Nausea | 2 per 100 | 2 per 100 (1 to 7) | 547 (2 trials) | low2,3,5,7 | |
| Diarrhoea | 12 per 100 | 12 per 100 (10 to 14) | 4889 (7 trials) | moderate2,3,5,6 | |
| Abdominal pain | 19 per 100 | 16 per 100 (12 to 20) | 911 (5 trials) | low2,3,5,8 | |
| Anorexia | 15 per 100 | 14 per 100 (12 to 17) | 3834 (5 trials) | moderate2,3,5,6 | |
| Neuro‐psychiatric | Headache | 27 per 100 | 33 per 100 (25 to 44) | 309 (2 trials) | low2,3,5,8 |
| Sleeplessness | 1 per 100 | 3 per 100 (1 to 9) | 547 (2 trials) | low2,3,5,7 | |
| Dizziness | 3 per 100 | 4 per 100 (2 to 11) | 547 (2 trials) | low3,5,7 | |
| Sleepiness | 0 per 100 | 0 per 100 (0 to 0) | 384 (1 trial) | low2,3,5,7 | |
| Weakness | 17 per 100 | 18 per 100 (15 to 21) | 1812 (5 trials) | moderate2,3,5,6 | |
| Cardio‐respiratory | Cough | 42 per 100 | 42 per 100 (40 to 45) | 4342 (5 trials) | moderate2,3,5,6 |
| Coryza | 68 per 100 | 66 per 100 (60 to 72) | 832 (2 trials) | low1,2,3,8 | |
| Prolonged QT interval (adverse event) | 3 per 100 | 2 per 100 (1 to 5) | 1548 (1 trial) | low8,10,11 | |
| Prolonged QT interval (Bazett's correction) | 7 per 100 | 9 per 100 (6 to 11) | 1548 (1 trial) | low5,8,11 | |
| Prolonged QT interval (Fridericia's correction) | 0 per 100 | 0 per 100 (0 to 2) | 1548 (1 trial) | low5,8,11 | |
| Musculoskeletal/ dermatological | Pruritis | 2 per 100 | 4 per 100 (2 to 6) | 2033 (5 trials) | moderate2,3,5,6 |
| Facial oedema | 0 per 100 | 0 per 100 (0 to 0) | 384 (1 trial) | low2,3,5,7 | |
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
|
1 No serious risk of bias: All but one of the trials are open label. However, we did not down grade for this outcome.
2 No serious inconsistency: The finding is consistent across all trials. Statistical heterogeneity is low.
3 No serious indirectness: Trials were mainly conducted in children in Africa, with few trials in Asia or in adults.
4 Downgraded by 1 for serious imprecision: No statistically significant difference was detected between treatments. However the current sample size does not exclude the possibility of rare but clinically important differences.
5 Downgraded by 1 for risk of bias: The majority of trials are open label.
6 No serious imprecision: The finding is of no effect and the CIs around the absolute effect excludes clinically important differences.
7 Downgraded by 1 for serious imprecision: There are limited data. 8 Downgraded by 1 for serious imprecision: The result does not reach statistical significance. 9 No serious imprecision: The total number of participants is high and findings are precise. 10 Downgraded by 1 for serious risk of bias: This trial is unblinded. Only a few of the recorded prolonged QT intervals were registered as adverse events which removed the statistical significance. The reasons for this are unclear. 11 No serious indirectness: This single trial was conducted in children in Uganda, Kenya, Mozambique, Zambia, and Burkina Faso. | |||||
| Dihydroartemisinin‐piperaquine compared to Artesunate plus amodiaquine for uncomplicated P. falciparum malaria | |||||
| Patient or population: Patients with uncomplicated P. falciparum malaria Settings: Malaria endemic areas Intervention: Dihydroartemisinin‐piperaquine (DHA‐P) Comparison: Artesunate plus amodiaquine (AS+AQ) | |||||
| Outcomes | Number of participants having adverse events (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | ||
| AS+AQ | DHA‐P | ||||
| Serious adverse events (including deaths) | 2 per 100 | 1 per 100 (0 to 1) | 2805 (2 trials) | moderate1,2,3,4 | |
| Gastrointestinal | Early vomiting | 6 per 100 | 5 per 100 (3 to 10) | 650 (2 trials) | low3,5,6,7 |
| Vomiting | 11 per 100 | 8 per 100 (7 to 11) | 2471 (1 trial) | moderate7,8,9,10 | |
| Nausea | 17 per 100 | 17 per 100 (11 to 28) | 316 (1 trial) | moderate5,9,11,12 | |
| Diarrhoea | 11 per 100 | 11 per 100 (9 to 14) | 2787 (2 trials) | moderate2,3,12,13 | |
| Abdominal pain | 13 per 100 | 12 per 100 (7 to 21) | 316 (1 trial) | low5,7,9,11 | |
| Anorexia | 11 per 100 | 10 per 100 (8 to 12) | 2787 (2 trials) | low2,3,7,13 | |
| Neuro‐psychiatric | Headache | 1 per 100 | 1 per 100 (0 to 9) | 316 (1 trial) | low5,9,11,14 |
| Sleeplessness | 14 per 100 | 11 per 100 (6 to 20) | 316 (1 trial) | low5,7,9,11 | |
| Cardio‐respiratory | Cough | 31 per 100 | 32 per 100 (28 to 36) | 2471 (1 trial) | moderate7,8,9,10 |
| Palpitations | 23 per 100 | 20 per 100 (13 to 30) | 316 (1 trial) | low5,7,9,11 | |
| *The basis for the assumed risk (for example, the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
| 1 No serious risk of bias: Only one of the two trials was blinded. However, we did not downgrade for this outcome. 2 No serious inconsistency: The finding is consistent across all trials. Statistical heterogeneity is low. 3 No serious indirectness: Trials were mainly conducted in children in Africa and Asia, with few Asian adults. 4 Downgraded by 1 for serious imprecision: The number of events is low despite the findings reaching statistical significance and the total number of participants being high. 5 Downgraded by 1 for risk of bias: The trial that reported this finding was open‐label 6 No serious inconsistency: The finding is consistent across all trials. Statistical heterogeneity can be explained by difference in definition of early vomiting. 7 Downgraded by 1 for serious imprecision: The result does not reach statistical significance. 8 No serious risk of bias: The trial that reported this outcome had low risk of bias for blinding of adverse events. 9 No serious inconsistency: This outcome was only reported in one trial. 10 No serious indirectness: This trial was mainly conducted in children in Africa. 11 No serious indirectness: The trial was mainly conducted in children and adults in Asia. 12 No serious imprecision: The finding is of no effect but the CIs around the absolute effect excludes clinically important differences. 13 Downgraded by 1 for risk of bias: Only one trial was blinded for adverse events. 14 Downgraded by 1 for serious imprecision: There are limited data and the 95% CI is wide. | |||||
Data and analyses
Comparison 1. Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Asia | 8 | 3487 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.28, 3.72] |
| 2 Total failure (P. falciparum) Day 28 PCR‐adjusted | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Asia | 8 | 3482 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.41 [0.21, 0.80] |
| 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Asia | 7 | 3421 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.54, 1.50] |
| 4 Total failure (P. falciparum) Day 42 PCR‐adjusted | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Asia | 6 | 2901 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.26, 0.88] |
| 5 Total failure (P. falciparum) Day 63 PCR‐unadjusted | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Asia | 5 | 2715 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.69, 1.03] |
| 5.2 South America | 1 | 445 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.19 [1.40, 27.35] |
| 6 Total failure (P. falciparum) Day 63 PCR‐adjusted | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Asia | 5 | 2500 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.30, 0.84] |
| 6.2 South America | 1 | 435 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.55 [0.52, 176.35] |
| 7 Gametocyte carriage | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Gametocyte carriage day 0 | 3 | 2322 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.66, 1.73] |
| 7.2 Gametocyte carriage day 7 | 3 | 2270 | Risk Ratio (M‐H, Random, 95% CI) | 1.99 [1.57, 2.51] |
| 7.3 Gametocyte carriage day 14 | 3 | 2249 | Risk Ratio (M‐H, Random, 95% CI) | 5.11 [3.26, 7.99] |
| 7.4 Gametocyte carriage day 21 | 3 | 2218 | Risk Ratio (M‐H, Random, 95% CI) | 9.44 [0.80, 110.80] |
| 7.5 Gametocyte carriage day 28 | 3 | 2199 | Risk Ratio (M‐H, Random, 95% CI) | 9.55 [1.80, 50.61] |
| 8 Gametocyte development (in those negative at baseline) | 3 | 1234 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.06 [1.13, 8.33] |
| 9 Serious adverse events (including deaths) | 8 | 3522 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.59, 2.42] |
| 10 Other adverse events: Gastrointestinal | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Early vomiting | 9 | 4114 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.71, 1.15] |
| 10.2 Nausea | 9 | 4531 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.60, 0.78] |
| 10.3 Vomiting | 5 | 2744 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.47, 0.75] |
| 10.4 Anorexia | 6 | 3497 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.73, 1.02] |
| 10.5 Diarrhoea | 5 | 2217 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [1.05, 2.04] |
| 10.6 Abdominal pain | 7 | 3887 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.82, 1.20] |
| 11 Other adverse events: Neuro‐psychiatric | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 Headache | 4 | 2039 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.64, 1.00] |
| 11.2 Dizziness | 9 | 4531 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.66, 0.78] |
| 11.3 Sleeplessness | 6 | 2551 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.40, 0.60] |
| 11.4 Fatigue | 2 | 872 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.41 [0.23, 0.73] |
| 11.5 Nightmares | 1 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.01, 0.69] |
| 11.6 Anxiety | 1 | 522 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.03, 0.33] |
| 11.7 Blurred vision | 1 | 464 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.24, 1.02] |
| 11.8 Tinnitus | 1 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.13, 1.24] |
| 12 Other adverse events: Cardio‐respiratory | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 Palpitations | 3 | 1175 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.45, 0.82] |
| 12.2 Cough | 1 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.54, 1.19] |
| 12.3 Dyspnoea | 1 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.3 [0.08, 1.06] |
| 12.4 Prolonged QT interval (reported as adverse events) | 1 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.72, 2.24] |
| 12.5 Prolonged QT interval (Bazett's correction) | 1 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [1.20, 3.49] |
| 12.6 Prolonged QT interval (Fridericia's correction) | 1 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.52, 1.52] |
| 13 Other adverse events: Musculoskeletal/dermatological | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 Arthralgia | 1 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.60, 1.65] |
| 13.2 Myalgia | 1 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.63, 1.70] |
| 13.3 Urticaria | 2 | 719 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.15, 2.35] |
| 13.4 Pruritis | 2 | 872 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.26, 1.60] |
| 13.5 Rash | 1 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 8.09] |
| 14 Sensitivity analysis: Total failure Day 63 PCR‐unadjusted | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 Total failure (P. falciparum) Day 63 PCR‐unadjusted | 4 | 1627 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.52, 1.70] |
| 14.2 Total failure Day 63 PCR‐unadjusted (losses to follow‐up included as failures) | 4 | 1801 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.65, 1.38] |
| 14.3 Total failure Day 63 PCR‐unadjusted (losses to follow‐up included as successes) | 4 | 1801 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.52, 1.68] |
| 15 Sensitivity analysis: Total failure Day 63 PCR‐adjusted | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Total failure (P. falciparum) Day 63 PCR‐adjusted | 4 | 1497 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.17, 1.83] |
| 15.2 Total failure Day 63 PCR‐adjusted (indeterminate PCR included as failures) | 4 | 1508 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.32, 1.39] |
| 15.3 Total failure Day 63 PCR‐adjusted (new infections included as successes) | 4 | 1627 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.34, 1.35] |
| 15.4 Total failure Day 63 PCR‐adjusted (losses to follow‐up included as failures) | 4 | 1801 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.67, 1.30] |
| 15.5 Total failure Day 63 PCR‐adjusted (losses to follow‐up included as successes) | 4 | 1801 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.34, 1.33] |
1.13. Analysis.

Comparison 1 Dihydroartemisinin‐piperaquine versus Artesunate plus mefloquine, Outcome 13 Other adverse events: Musculoskeletal/dermatological.
Comparison 2. Dihydroartemisinin‐piperaquine dose analysis: 3 dose versus 4 dose regimen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total failure PCR‐unadjusted | 1 | 318 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [0.84, 3.53] |
| 1.1 Day 63 | 1 | 318 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [0.84, 3.53] |
| 2 Total failure PCR‐adjusted | 1 | 292 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.05, 6.09] |
| 2.1 Day 63 | 1 | 292 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.05, 6.09] |
Comparison 3. Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total failure Day 28 PCR‐unadjusted | 8 | 3644 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.25, 2.41] |
| 1.1 DHA‐P 4 doses | 4 | 1075 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.10, 3.14] |
| 1.2 DHA‐P 3 doses | 5 | 2569 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.19, 5.07] |
| 2 Total failure Day 28 PCR‐adjusted | 8 | 3633 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.15, 1.65] |
| 2.1 DHA‐P 4 doses | 4 | 1067 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.10, 6.11] |
| 2.2 DHA‐P 3 doses | 5 | 2566 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.08, 1.94] |
| 3 Total failure Day 42 PCR‐unadjusted | 7 | 3578 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.51, 1.31] |
| 3.1 DHA‐P 4 doses | 3 | 957 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.50, 1.28] |
| 3.2 DHA‐P 3 doses | 5 | 2621 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.40, 1.99] |
| 4 Total failure Day 42 PCR‐adjusted | 6 | 3046 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.20, 1.18] |
| 4.1 DHA‐P 4 doses | 3 | 903 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.14, 2.82] |
| 4.2 DHA‐P 3 doses | 4 | 2143 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.12, 1.48] |
| 5 Total failure Day 63 PCR‐unadjusted | 6 | 3317 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.62, 1.40] |
| 5.1 DHA‐P 4 doses | 3 | 1019 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.59, 1.10] |
| 5.2 DHA‐P 3 doses | 4 | 2298 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.52, 2.90] |
| 6 Total failure Day 63 PCR‐adjusted | 6 | 3072 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.28, 1.15] |
| 6.1 DHA‐P 4 doses | 3 | 908 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.17, 1.04] |
| 6.2 DHA‐P 3 doses | 4 | 2164 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.26, 2.77] |
3.2. Analysis.
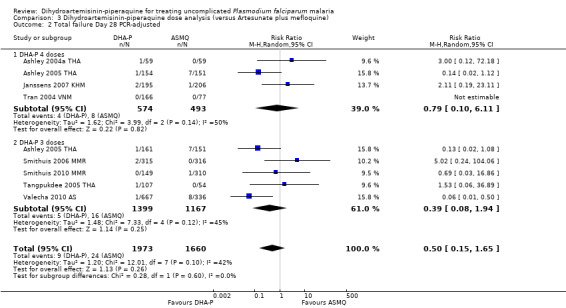
Comparison 3 Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine), Outcome 2 Total failure Day 28 PCR‐adjusted.
3.3. Analysis.
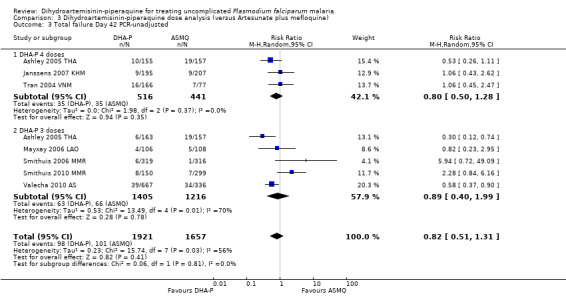
Comparison 3 Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine), Outcome 3 Total failure Day 42 PCR‐unadjusted.
3.4. Analysis.
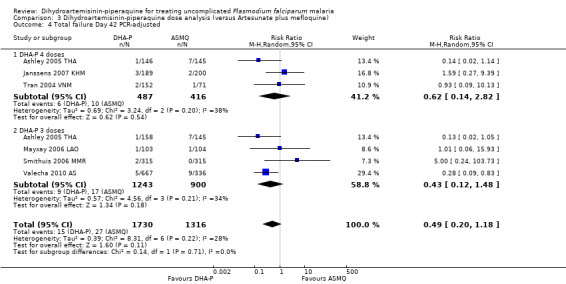
Comparison 3 Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine), Outcome 4 Total failure Day 42 PCR‐adjusted.
3.5. Analysis.

Comparison 3 Dihydroartemisinin‐piperaquine dose analysis (versus Artesunate plus mefloquine), Outcome 5 Total failure Day 63 PCR‐unadjusted.
Comparison 4. Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted | 13 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Africa | 9 | 6200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.30, 0.39] |
| 1.2 Asia and Oceania | 4 | 1143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.64, 1.47] |
| 2 Total failure (P. falciparum) Day 28 PCR‐adjusted | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Africa | 9 | 5417 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.29, 0.62] |
| 2.2 Asia and Oceania | 3 | 925 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [0.81, 5.03] |
| 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Africa | 7 | 3301 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.53, 0.67] |
| 3.2 Asia and Oceania | 2 | 572 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.65, 1.17] |
| 4 Total failure (P. falciparum) Day 42 PCR‐adjusted | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Africa | 7 | 2559 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.41, 0.81] |
| 4.2 Asia and Oceania | 2 | 468 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.75, 3.83] |
| 5 Total failure (P. falciparum) Day 63 PCR‐unadjusted | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Africa | 2 | 3200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.65, 0.78] |
| 5.2 Asia | 1 | 323 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.47, 1.88] |
| 6 Total failure (P. falciparum) Day 63 PCR‐adjusted | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Africa | 2 | 2097 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.50, 1.04] |
| 6.2 Asia | 1 | 298 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.14, 7.01] |
| 7 Gametocyte development (in those negative at baseline) | 6 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Gametocyte carriage | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Gametocyte carriage day 1 to 14 | 4 | 1537 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.32 [1.48, 12.63] |
| 8.2 Gametocyte carriage day 15 to 28 | 4 | 1516 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.06, 0.72] |
| 8.3 Gametocyte carriage day 29 to 42 | 2 | 650 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.13, 0.61] |
| 9 Anaemia | 8 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 Mean haemoglobin (g/dL) at baseline | 8 | 6599 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04 [‐0.09, 0.01] |
| 9.2 Mean haemoglobin (g/dL) at day 28 | 1 | 134 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐0.03, 0.75] |
| 9.3 Mean haemoglobin (g/dL) at day 42 | 1 | 375 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.02, 0.62] |
| 9.4 Mean change in haemoglobin (g/dL) from baseline to day 28 | 2 | 2185 | Mean Difference (IV, Fixed, 95% CI) | 0.19 [0.03, 0.34] |
| 9.5 Mean change in haemoglobin (g/dL) from baseline to day 42 | 2 | 835 | Mean Difference (IV, Fixed, 95% CI) | 0.26 [0.00, 0.51] |
| 10 Serious adverse events (including deaths) | 9 | 7246 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.93, 2.68] |
| 11 Other adverse events: Gastrointestinal | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 Early vomiting | 4 | 2969 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [1.00, 2.83] |
| 11.2 Vomiting | 9 | 6761 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.89, 1.20] |
| 11.3 Nausea | 2 | 547 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.36, 3.76] |
| 11.4 Diarrhoea | 7 | 4889 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.81, 1.09] |
| 11.5 Abdominal pain | 5 | 911 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.65, 1.08] |
| 11.6 Anorexia | 5 | 3834 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.80, 1.08] |
| 12 Other adverse events: Neuro‐psychiatric | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 Headache | 2 | 309 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.90, 1.63] |
| 12.2 Sleeplessness | 2 | 547 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.07 [0.69, 6.16] |
| 12.3 Dizziness | 2 | 547 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.67, 4.15] |
| 12.4 Sleepiness | 1 | 384 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.85 [0.12, 69.49] |
| 12.5 Weakness | 5 | 1812 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.89, 1.27] |
| 13 Other adverse events: Cardio‐respiratory | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 Cough | 5 | 4342 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.95, 1.09] |
| 13.2 Coryza | 2 | 832 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.89, 1.07] |
| 13.3 Prolonged QTc interval (reported as adverse events) | 1 | 1548 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.51, 1.90] |
| 13.4 Prolonged QTc interval (Bazett's correction) | 1 | 1548 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.91, 1.92] |
| 13.5 Prolonged QTc interval (Fridericia's correction) | 1 | 1548 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.09, 10.81] |
| 14 Other adverse events: Musculoskeletal/dermatological | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 Pruritis | 5 | 2033 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [1.03, 2.92] |
| 14.2 Face oedema | 1 | 384 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.85 [0.12, 69.49] |
4.12. Analysis.
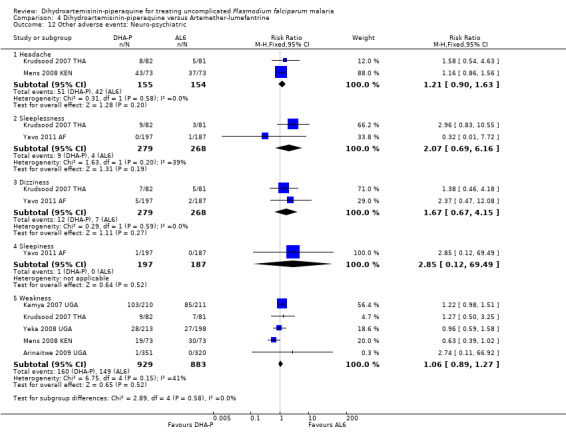
Comparison 4 Dihydroartemisinin‐piperaquine versus Artemether‐lumefantrine, Outcome 12 Other adverse events: Neuro‐psychiatric.
Comparison 5. Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Africa | 2 | 2800 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.41, 0.59] |
| 1.2 Asia | 2 | 482 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.18, 0.77] |
| 2 Total failure (P. falciparum) Day 28 PCR‐adjusted | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Africa | 2 | 2486 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.42, 1.06] |
| 2.2 Asia | 2 | 466 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.08 [0.01, 0.40] |
| 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Asia | 1 | 152 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.10, 0.72] |
| 4 Total failure (P. falciparum) Day 42 PCR‐adjusted | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Asia | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.01, 0.81] |
| 5 Total failure (P. falciparum) Day 63 PCR‐unadjusted | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Africa | 1 | 2292 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.86, 1.07] |
| 5.2 Asia | 1 | 304 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.27, 0.89] |
| 6 Total failure (P. falciparum) Day 63 PCR‐adjusted | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Africa | 1 | 1506 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.85, 3.83] |
| 6.2 Asia | 1 | 278 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.03, 0.59] |
| 7 Serious adverse events (including deaths) | 2 | 2805 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.19, 0.87] |
| 8 Other adverse events: Gastrointestinal | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Early vomiting | 2 | 650 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.44, 1.56] |
| 8.2 Vomiting | 1 | 2471 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.62, 1.01] |
| 8.3 Nausea | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.62, 1.61] |
| 8.4 Diarrhoea | 2 | 2787 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.83, 1.27] |
| 8.5 Abdominal pain | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.51, 1.65] |
| 8.6 Anorexia | 2 | 2787 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.72, 1.13] |
| 9 Other adverse events: Neuro‐psychiatric | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Headache | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.14, 6.75] |
| 9.2 Sleeplessness | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.44, 1.41] |
| 10 Other adverse events: Cardio‐respiratory | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Cough | 1 | 2471 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.91, 1.15] |
| 10.2 Palpitations | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.58, 1.35] |
5.4. Analysis.

Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 4 Total failure (P. falciparum) Day 42 PCR‐adjusted.
5.10. Analysis.
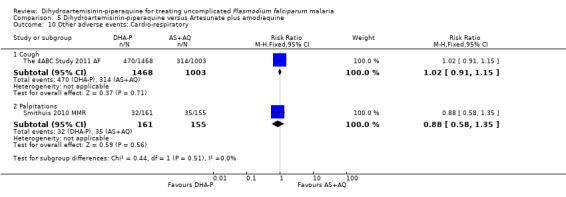
Comparison 5 Dihydroartemisinin‐piperaquine versus Artesunate plus amodiaquine, Outcome 10 Other adverse events: Cardio‐respiratory.
Comparison 6. Dihydroartemisinin‐piperaquine versus Artesunate plus sulfadoxine‐pyrimethamine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total failure (P. falciparum) Day 28 PCR‐unadjusted | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Oceania | 1 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.62, 1.64] |
| 2 Total failure (P. falciparum) Day 28 PCR‐adjusted | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Oceania | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.46, 2.22] |
| 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Oceania | 1 | 215 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.74, 1.45] |
| 4 Total failure (P. falciparum) Day 42 PCR‐adjusted | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Oceania | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.39, 1.51] |
6.3. Analysis.
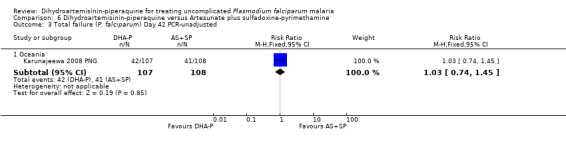
Comparison 6 Dihydroartemisinin‐piperaquine versus Artesunate plus sulfadoxine‐pyrimethamine, Outcome 3 Total failure (P. falciparum) Day 42 PCR‐unadjusted.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adam 2010 SDN.
| Methods | Trial design: An open label RCT Follow‐up: Temperature and blood smears on days 1, 2, 3, 7, 14, 21, and 28. Haemoglobin concentrations (Hb) measured on days 0 and 28. Adverse event monitoring: Participants requested to attend the health centre any time they felt unwell. During follow‐up participants were asked about the presence of adverse effects that might be expected from treatment (for example, nausea, vomiting). These were considered treatment‐related if they had not been reported at the participant's first presentation. |
|
| Participants | Number of participants: 160 Inclusion criteria: Age ≥ 6 months, uncomplicated P. falciparum mono‐infection, axillary temperature > 37.5°C or a history of fever within the preceding 24 hrs, able to take oral treatment, informed consent. Exclusion criteria: Severe or complicated malaria, severe concomitant pathology or other illness needing medical follow‐up incompatible with the trial, allergy to one of the trial drugs, use of one of the trial drugs in the preceding 28 days, pregnancy. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg adult tablets, 20 mg/160 mg children's tablets (Duo‐Cotecxin: Beijing)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg (Coartem: Novartis)
All doses were supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Sudan Setting: Elmouraf health centre, Sinnar Transmission: Unstable transmission Resistance: "Multiple drug resistance" Dates: Dec 2009 to Feb 2010 Funding: Beijing Holley‐Cotec Pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Block‐randomization using a concealed envelope system was used to allocate each patient to one of the two treatment arms". Block size was unclear. |
| Allocation concealment (selection bias) | Unclear risk | Used concealed envelopes, unclear if they were sequentially numbered or opaque. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "all the slides were double‐checked blindly". |
| Blinding for adverse events (performance and detection bias) | High risk | Trial described as "open". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in both groups (6.3% DHA‐P versus 7.5% AL6). |
| Selective reporting (reporting bias) | Low risk | The WHO recommends 42 day follow‐up in studies of AL6. Day 28 outcomes may underestimate treatment failure with AL6. |
| Other bias | Unclear risk | The role of the trial sponsor was not described. |
Agarwal 2013 KEN.
| Methods | Trial design: An open label RCT Follow‐up: Followed up for 42 days and asked to return on days 1, 2, 3, 7, 14, 21, 28, 35, and 42 after enrolment or at any day if ill. Clinical assessment and blood smear at each visit. Hb measured on days 0, 7, 14, 28, and 42. Adverse event monitoring: Not reported. "Adverse events investigated and addressed". |
|
| Participants | Number of participants: 274 Inclusion criteria: Children aged 6 to 59 months with axillary temperature ≥ 37.5 °C or history of fever in preceding 48 hrs, weight ≥ 5.0 kg, parasitaemia, residing within 10 km of Siaya District Hospital, written informed consent. Exclusion criteria: Lethargy, convulsions, persistent vomiting, inability to drink, signs of severe malaria, severe anaemia (Hb < 5 g/dL), known hypersensitivity to trial drugs, presence of chronic medical conditions, treatment with any anti‐malarial in preceding two weeks, previous enrolment in any malaria trial, severe malnutrition (weight‐for‐age ≤ 3 standard deviations below mean for gender according to WHO standards). |
|
| Interventions | 1. DHA‐P, fixed dose combination, 20 mg/160 mg tablets (DuoCotexin: Beijing Holley‐Cotec)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg (Coartem: Novartis)
All doses, except AL evening doses, administered under direct supervision. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Kenya Setting: district hospital in western Kenya Transmission: Holoendemic with high transmission and two seasonal peaks, April to July and November to December Resistance: Not reported Dates: Oct 2010 to Aug 2011 Funding: KEMRI/CDC Research and Public Health Collaboration, Beijing Holley‐Cotec provided DHA‐P free of charge. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "children were block randomized in fixed blocks of ten to treatment". |
| Allocation concealment (selection bias) | Unclear risk | None described. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "All microscopists were blinded to the treatment arm". |
| Blinding for adverse events (performance and detection bias) | Unclear risk | No other blinding reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Percentage withdrawn from analysis high in both treatment groups (17.5% DHA‐P versus 18.2% AL). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. |
| Other bias | Low risk | No other forms of bias identified. |
Arinaitwe 2009 UGA.
| Methods | Trial design: An open label RCT Follow‐up: Blood smears taken on days 0, 2, 3, 7, 14, 21, and 28 after each episode and any time they felt ill. Follow‐up continued for up to one year. Adverse event monitoring: Clinicians assessed participants for adverse events using standardized criteria at each follow‐up visit. Passive monitoring was carried out for up to 63 days after treatment. Adverse events were defined as any untoward medical occurrence, regardless of its suspected relationship to the trial drugs, as per International Conference of Harmonization guidelines. Adverse events were graded as mild, moderate, severe, or life threatening. |
|
| Participants | Number of participants: 232, with 671 episodes of uncomplicated falciparum malaria treated. All episodes treated were included in the analysis. Inclusion criteria: For enrolment in the trial cohort: age 6 weeks to 12 months, HIV status of mother and child documented, living within a 30 km radius of the trial clinic, currently breast‐feeding if HIV exposed, and informed consent including consent to come to the trial clinic for any illness and avoid medications given outside of the trial. For randomization: uncomplicated malaria diagnosed by positive blood smear after documented fever of ≥ 38.0 °C or history of fever in the past 24 hrs, age ≥ 4 months, weight ≥ 5 kg. Exclusion criteria: Active medical problems requiring inpatient evaluation. For withdrawal from trial cohort: movement outside trial area, inability to tolerate trial drugs, withdrawal of informed consent, inability to be located for > 60 days, or inability to adhere to trial procedures and schedule. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Duocotecxin: Holley Pharm)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg (Coartem: Novartis)
Only the first daily dose supervised. Subsequent episodes of malaria occurring > 14 days after a previous episode were treated with the assigned trial drug. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Uganda Setting: Enrolled from local antenatal clinics in Tororo Transmission: High transmission Resistance: Not reported Dates: Aug 2007 to Jul 2008 Funding: Doris Duke Charitable Foundation and Puget Sound Partners in Global Health. Holleypharm provided DHA‐P free of charge. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomization list was computer generated by an off‐site investigator". |
| Allocation concealment (selection bias) | Low risk | "Sequentially numbered, sealed envelopes containing the treatment group assignments were prepared from the randomization list. The study nurse assigned treatment numbers sequentially and allocated treatment by opening the envelope corresponding to the treatment number". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | No blinding of microscopists reported, but all slides were re‐read by a second microscopist, and a third microscopist resolved discrepancies. |
| Blinding for adverse events (performance and detection bias) | High risk | Described as "open‐label". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar low drop out in both groups (1.7% DHA‐P versus 1.6% AL6). |
| Selective reporting (reporting bias) | Low risk | All listed outcomes reported. |
| Other bias | Low risk | This trial randomized individuals to an ACT and then followed them up through multiple treatment episodes. The data presented is for the all malaria episodes reported during the trial period. |
Ashley 2004a THA.
| Methods | Trial design: A 3‐arm RCT Follow‐up: All patients admitted to hospital for 28 days, oral temperature taken every 6 hrs, parasite counts 12‐hourly until negative then daily for 28 days Adverse event monitoring: Adverse events defined as signs or symptoms that occurred or became more severe after treatment started. All patients had full blood counts, urea, electrolytes, creatinine, and liver function tests at days 0 and 7. |
|
| Participants | Number of participants: 134 Inclusion criteria: Age > 14 yrs, weight > 40 kg, symptoms of malaria, P. falciparum parasitaemia, informed consent. Exclusion criteria: Pregnancy or lactation, signs or symptoms of severe malaria, > 4% of red blood cells parasitized, contraindication to mefloquine, treatment with mefloquine in the previous 60 days, sulphonamides or 4‐aminoquinolones present in urine on admission. |
|
| Interventions | 1. DHA‐P, fixed dose combination (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination (Artesunate: Guilin, Mequin: Atlantic)
All doses were supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Thailand Setting: Bangkok Hospital for Tropical Diseases Transmission: Low transmission Resistance: Multiple‐drug resistance Dates: Jul 2002 to Apr 2003 Funding: Mahidol University, Tak Malaria Initiative Project, supported by Bill and Melinda Gates Foundation, Wellcome Trust of Great Britain. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation was computer generated (STATA; version 7; Statacorp)". Randomized in blocks of six. |
| Allocation concealment (selection bias) | Unclear risk | "The treatment allocation was concealed in sealed envelopes labelled with the study code", unclear if these were sequentially numbered or opaque. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Laboratory staff reading the blood smears had no knowledge of the treatment received". |
| Blinding for adverse events (performance and detection bias) | Unclear risk | No other blinding described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar loss to follow‐up in all groups (10.6% DHA‐P versus 11.9% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | The WHO recommends 63 days follow‐up in studies of AS+MQ. Day 28 outcomes are likely to underestimate treatment failure with AS+MQ and DHA‐P. |
| Other bias | Low risk | No other sources of bias identified. |
Ashley 2004b THA.
| Methods | Trial design: A RCT Follow‐up: Temperature and blood smears daily until clearance of fever and parasites, then weekly attendance until day 63 Adverse event monitoring: Adverse events defined as signs or symptoms that occurred or became more severe after treatment started. A subset of 55 patients in the DHA‐P group had full blood counts, urea, electrolyte, creatinine and liver function tests at days 0 and 7. Thirty‐two patients from the DHA‐P group also had ECG monitoring before and after treatment. |
|
| Participants | Number of participants: 355 Inclusion criteria: Age 1 to 65 yrs, symptomatic P. falciparum parasitaemia, informed consent Exclusion criteria: Pregnancy or lactation, signs or symptoms of severe malaria, > 4% of red blood cells parasitized, contraindication to mefloquine, treatment with mefloquine in the previous 60 days |
|
| Interventions | 1. DHA‐P, fixed dose combination (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination (Artesunate: Guilin, Mequin: Atlantic)
All doses were supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Thailand Setting: Four clinics on the Thai‐Myanmar border Transmission: Unstable low and seasonal transmission Resistance: Multiple‐drug resistance Dates: Jul 2002 to Apr 2003 Funding: Wellcome Trust of Great Britain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation was computer generated (STATA; version 7; Statacorp)". Randomized in blocks of 9. |
| Allocation concealment (selection bias) | Unclear risk | "The treatment allocation was concealed in sealed envelopes labelled with the study code", unclear if these were sequentially numbered or opaque. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Laboratory staff reading the blood smears had no knowledge of the treatment received". |
| Blinding for adverse events (performance and detection bias) | Unclear risk | No other blinding described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar losses to follow‐up in all groups (12.8% DHA‐P versus 13.6% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. |
| Other bias | Low risk | No other sources of bias identified. |
Ashley 2005 THA.
| Methods | Trial design: A 3‐arm RCT Follow‐up: Temperature and blood smears daily until clearance of fever and parasites, then weekly attendance for examination, symptom enquiry, malaria smear and haematocrit until day 63 Adverse event monitoring: Adverse events defined as signs or symptoms that occurred or became more severe after treatment started. Symptoms were screened at each visit |
|
| Participants | Number of participants: 499 Inclusion criteria: Age 1 to 65 yrs, symptomatic P. falciparum mono‐infection or mixed infections, informed consent Exclusion criteria: Pregnancy or lactation, signs or symptoms of severe malaria, > 4% of red blood cells parasitized, treatment with mefloquine in the previous 60 days |
|
| Interventions | 1. DHA‐P, fixed dose combination (Artekin: Holleykin)
2. DHA‐P, fixed dose combination (Artekin: Holleykin)
3. Artesunate plus mefloquine, loose combination (Artesunate: Guilin, Mequin: Atlantic)
All doses supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Thailand Setting: Six clinics on the Thai‐Myanmar border Transmission: Unstable low and seasonal transmission Resistance: Multiple‐drug resistance Dates: Nov 2004 to Jun 2005 Funding: DnDi, European Union International Co‐operation programme, Médecins sans Frontières, WHO/TDR, Wellcome Trust of Great Britain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation list was generated using STATA; version 7 (Stata)". Randomized in blocks of nine. |
| Allocation concealment (selection bias) | Unclear risk | "The treatment allocation was concealed in sealed envelopes labelled with the study code", unclear if these were sequentially numbered or opaque. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Laboratory staff reading the blood smears had no knowledge of the treatment received". |
| Blinding for adverse events (performance and detection bias) | Unclear risk | No other blinding described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Losses to follow‐up were low in all groups (4.2% DHA‐P versus 4.8% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Two patients were considered to be early treatment failures by the reviewers and reclassified as such. This was not clearly stated in the paper. |
| Other bias | Low risk | No other sources of bias identified. |
Bassat 2009 AF.
| Methods | Trial design: An open label RCT (non‐inferiority). Follow‐up: Children were kept at the health facility for the three day treatment period, and then returned on days 7, 14, 21, 28, 35, and 42, and any time symptoms occurred, for clinical assessment and blood smears. Haematological and biochemical assessments were carried out at enrolment, days 3, 28, and 42 and at clinician request. Adverse event monitoring: Monitoring and recording of adverse events was carried out throughout the trial. A 12‐lead ECG was performed at enrolment and on days 2 and 7 to assess any QT/QTc interval prolongation. |
|
| Participants | Number of participants: 1553 Inclusion criteria: Uncomplicated malaria, age 6 to 59 months, body weight > 5 kg, fever (axillary temperature ≥ 37.5 °C) or history of fever in the preceding 24 hrs, microscopically confirmed P. falciparum mono‐infection, asexual parasite densities between 2,000 and 200,000/μL, informed consent Exclusion criteria: severe malaria or other danger signs, acute malnutrition (weight for height < 70% of the median National Center for Health Statistics/WHO reference), any other concomitant illness or underlying disease, contra‐indication to trial drugs, ongoing antimalarial prophylaxis, ECG abnormality requiring urgent management |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets and 20 mg/160 mg tablets (Eurartesim®, Sigma‐Tau)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg (Coartem: Novartis)
All doses were supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Burkina Faso, Kenya, Mozambique, Uganda, and Zambia Setting: Four rural sites and one peri‐urban site. Transmission: Malaria mesoendemic at all sites. Two sites had high transmission in one period of the year (Jun to Dec or Nov to May), three others had perennial malaria with two sites having two peak seasons and one with marked seasonality (Oct to Apr) Resistance: Documented resistance to chloroquine ranged from 35% in Burkina Faso to 81% in Uganda Dates: Aug 2005 to Jul 2006 Funding: Medicine for Malaria Venture and Sigma‐Tau I.F.R. SpA (Rome). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomisation list stratified by country was generated by an independent off site contract research organisation". |
| Allocation concealment (selection bias) | Low risk | "Each treatment allocation concealed in opaque sealed envelopes that were opened only after the patient’s recruitment". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "assessment of the primary end‐point were made by staff blinded to the treatment assignment and before availability of the PCR results". |
| Blinding for adverse events (performance and detection bias) | High risk | Described as 'open label'. ECG assessment were interpreted in a blinded manner. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number excluded from primary analysis similar between groups (7.5% DHA‐P versus 9.7% AL6). |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the trial protocol were assessed. |
| Other bias | Unclear risk | "Employees of Sigma‐Tau participated in study design, data entry, collection and analysis of data". "An author independent of the sponsor, Umberto D’Alessandro, had access to the primary dataset and takes responsibility for the analyses and manuscript as a whole". |
Grande 2007 PER.
| Methods | Trial design: An open‐label RCT Follow‐up: Days 0, 1, 2, 3, 7, 14, 21, 28, 35, 42, 49, 56, and 63 or any other day they became ill, for a clinical assessment and malaria film. PCV measurement day 0, 7, 14 and 63. P. vivax treated with CQ. Adverse event monitoring: Assessed at each follow‐up visit, an adverse event defined as any unfavourable and unintended sign, symptom or disease temporally associated with the drug administered. Complete blood count, liver, and renal function tests at days 0 and 7. |
|
| Participants | Number of participants: 522 Inclusion criteria: Age 5 to 60 yrs, fever > 37.5 °C or history of fever in the previous 24 hrs, P. falciparum mono‐infection 1000 to 200,000/µL. Exclusion criteria: Pregnancy or lactation, severe malaria, any concomitant illness or underlying disease, contraindication to any of the trial drugs, history of treatment with mefloquine in the previous 60 days or chloroquine, primaquine or quinine in previous 14 days. |
|
| Interventions | 1. DHA‐P, fixed dose combination (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination (Artesunate: Guilin, Lariam: Hoffman La‐Roche)
All doses were supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Peru Setting: Nine rural health posts Transmission: Low malaria transmission Resistance: High CQ and SP resistance Dates: July 2003 to July 2005 Funding: Directorate‐General for Development and Cooperation of the Belgian Government |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized in blocks of 10". No further details given. |
| Allocation concealment (selection bias) | Low risk | "Sealed opaque envelopes were opened only after the final decision to recruit the patient had been made". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | No comment on blinding of laboratory staff. |
| Blinding for adverse events (performance and detection bias) | High risk | An open‐label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar loss to follow‐up in both groups (8.7% DHA‐P versus 5.9% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. |
| Other bias | Low risk | No other sources of bias identified. |
Hasugian 2007 IDN.
| Methods | Trial design: An open label RCT Follow‐up: Daily until fever and parasites cleared then weekly until day 42, for a physical examination, a symptom questionnaire and malaria film. Hb measured on days 0, 7, and 28. Adverse event monitoring: Assessed at each follow‐up visit |
|
| Participants | Number of participants: 340 Inclusion criteria: Age > 1 yr, weight > 5 kg, slide confirmed malaria (P. falciparum, P. vivax or both), fever or history of fever in the preceding 48 hrs Exclusion criteria: Pregnancy or lactation, danger signs or signs of severe malaria, > 4% red blood cells parasitized, concomitant disease that required hospital admission |
|
| Interventions | 1. DHA‐P, fixed dose combination (Artekin: Holley)
2. Artesunate plus amodiaquine, loose combination (Arsumax: Guilin, Flavoquine: Aventis)
All doses supervised |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Indonesia Setting: Rural clinics Transmission: Unstable Resistance: Chloroquine and SP resistance Dates: Jul 2005 to Dec 2005 Funding: Wellcome Trust ‐ National Health and Medical Research Council. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomisation list was generated in blocks of 20 by an independent statistician". |
| Allocation concealment (selection bias) | Low risk | "Treatment allocation concealed in an opaque, sealed envelope that was opened once the patient had been enrolled". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "All slides were read by a certified microscopist who was blinded to treatment allocation". |
| Blinding for adverse events (performance and detection bias) | High risk | An open‐label trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The primary outcome data are unpublished data including only participants with P. falciparum mono or co‐infection at baseline. High losses to follow‐up in both groups at day 42 (21% DHA‐P versus 24.5 % AL6), moderate at day 28 (16.6% DHA‐P versus 18.8 % AL6). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 42 outcomes may underestimate failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Janssens 2007 KHM.
| Methods | Trial design: An open label RCT Follow‐up: Monitored daily until fever and parasites cleared then weekly to day 63. Temperature, symptom questionnaire, malaria film, and haematocrit at each visit. Adverse event monitoring: An adverse event defined as any new sign or symptom appearing after treatment started. At each visit a symptom questionnaire was completed. |
|
| Participants | Number of participants: 464 Inclusion criteria: Age > 1 yr, axillary temp > 37.5 °C or history of fever, signs and symptoms of uncomplicated malaria, P. falciparum mono or mixed infections, written informed consent Exclusion criteria: Pregnancy or lactation, signs or symptoms of severe malaria, > 4% red blood cells parasitized, a history of convulsions or neuropsychiatric disorder, treatment with mefloquine in the past 60 days. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination (Artesunate: Guilin, Mefloquine: Mepha)
All doses supervised. |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Cambodia Setting: Rural health centres and outreach malaria clinics Transmission: Low and seasonal Resistance: Multiple‐drug resistance Dates: Oct 2002 to March 2003 Funding: Médecins sans Frontières |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer generated randomisation (STATA version 8, Statacorp)". |
| Allocation concealment (selection bias) | Unclear risk | "Treatment allocations were concealed in sealed envelopes". No further details. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | No comment on blinding of laboratory staff. |
| Blinding for adverse events (performance and detection bias) | High risk | An open‐label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Losses to follow‐up balanced and low in both groups (9.3% DHA‐P versus 10% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. |
| Other bias | Low risk | No other sources of bias identified. |
Kamya 2007 UGA.
| Methods | Trial design: A single blind (outcome assessors) RCT Follow‐up: Standardized history and examination and malaria film on days 0, 1, 2, 3, 7, 14, 21, 28, 35, 42 and any other day they felt unwell. Hb measured at day 0 and day 42 or day of failure. Anaemia was treated with ferrous sulphate and anthelminthics according to IMCI guidelines. Adverse event monitoring: Assessed for any new or worsening event at each visit. An adverse event defined as any untoward medical occurrence, irrespective of its suspected relationship to the trial medications. |
|
| Participants | Number of participants: 509 Inclusion criteria: Age 6 months to 10 yrs, weight > 5 kg, axillary temp > 37.5 °C or history of fever in the past 24 hrs, P. falciparum mono‐infection 2000 to 200,000/µL, informed consent Exclusion criteria: Danger signs or signs of severe malaria, evidence of concomitant febrile illness, history of serious side effects to trial medication |
|
| Interventions | 1. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Coartem: Novartis)
2. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Duocotexin: HolleyPharm)
All doses supervised. All participants received a glass of milk after each dose |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Uganda Setting: Rural health centre Transmission: Perennial holoendemic malaria with very high transmission intensity Resistance: Not reported Dates: Mar 2006 to July 2006 Funding: US Centres for Disease Control, Malaria Consortium Drugman, DFID, DHA‐P supplied by HolleyPharm |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomisation list was computer generated by an off‐site investigator". |
| Allocation concealment (selection bias) | Low risk | "Sequentially numbered, sealed envelopes containing the treatment group assignments were prepared from the randomisation list". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Study physicians and laboratory personnel involved in assessing outcomes were blinded to treatment assignments". |
| Blinding for adverse events (performance and detection bias) | Low risk | Placebos were used to blind participants to treatment allocation. Trial physicians were also blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in both groups (0.9% AL6 versus 0.9% DHA‐P). A large number of participants were excluded after randomization for failing to meet the entry criteria. |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 42 outcomes may underestimate failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Karema 2006 RWA.
| Methods | Trial design: A 3‐arm open label RCT Follow‐up: History, clinical signs and symptoms, and malaria film on days 0, 1, 2, 3, 7, 14, 21, and 28 and any other day they felt unwell. PCV measured at days 0 and 14. Adverse event monitoring: An adverse event defined as any unfavourable and unintended sign associated temporally with the use of the drug administered. Differential WBC count (and liver function tests at 1 site only) assessed at days 0 and 14. |
|
| Participants | Number of participants: 762 Inclusion criteria: Age 12 to 59 months, weight > 10 kg, axillary temp > 37.5 °C or history of fever in the preceding 24 hrs, P. falciparum mono‐infection 2000 to 200,000/µL. Exclusion criteria: Severe malaria, any other concomitant illness or underlying disease, known allergy to trial drugs, clear history of adequate antimalarial treatment in the previous 72 hrs, PCV < 15%. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleypharm)
2. Artesunate plus amodiaquine, loose combination (Arsumax: Sanofi)
All doses supervised |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Rwanda Setting: Peri‐urban and rural health centres Transmission: Not reported Resistance: Not reported Dates: Oct 2003 to Apr 2004 Funding: Belgian Development Co‐operation in collaboration with the Prince Leopold Institute of Tropical Medicine. DHA‐P provided by Holleypharm |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomly allocated in blocks of 15", computer generated sequence (information from author). |
| Allocation concealment (selection bias) | Unclear risk | "Allocation of treatment was concealed until final recruitment'. No further details. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Laboratory technicians reading malaria slides did not know the treatment received". |
| Blinding for adverse events (performance and detection bias) | High risk | An open‐label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Very low losses to follow‐up in all groups (0.8% DHA‐P versus 0.4% AS+AQ). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 28 outcomes may underestimate failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Karunajeewa 2008 PNG.
| Methods | Trial design: A 4‐arm open label RCT Follow‐up: Standardized follow‐up including temperature and malaria film on days 0, 1, 2, 3, 7, 14, 28, and 42. Drug levels assayed on day 7. Adverse event monitoring: None described |
|
| Participants | Number of participants: 372 Inclusion criteria: Age 0.5 to 5 yrs, axillary temp > 37.5 °C or history of fever in the preceding 24 hrs, > 1000/µL asexual P. falciparum or > 250/µL asexual P. vivax,P. ovale, or P. malariae, informed consent Exclusion criteria: Features of severe malaria, evidence of another infection or coexisting condition including malnutrition, intake of trial drug in previous 14 days. |
|
| Interventions | 1. Artesunate plus sulfadoxine‐pyrimethamine, loose combination (Sanofi‐Aventis, Roche)
2. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Beijing Holley‐Cotec)
3. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Novartis), given with milk
All doses supervised except the evening dose of AL6 |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Papua New Guinea Setting: Health centres Transmission: Holoendemic Resistance: CQ and SP Dates: Apr 2005 to Jul 2007 Funding: WHO Western Pacific Region, Rotary against Malaria in Papua New Guinea, National Health and Medical Research Council of Australia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated randomised assignment with blocks of 24 for each site". |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | Microscopists were unaware of treatment assignments. |
| Blinding for adverse events (performance and detection bias) | High risk | An open label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Moderate losses to follow‐up in all groups (11.5% AS+SP versus 13.0% DHA‐P versus 14.2% AL6). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 42 outcomes may underestimate failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Krudsood 2007 THA.
| Methods | Trial design: An open label RCT Follow‐up: Blood smears every 12 hrs until found to be negative and daily for 28 days. Haematological and biochemical samples, and urine examined on day 0, 1 and 3 and weekly for the 4 weeks trial period. Adverse event monitoring: Regular physical examinations were conducted and assessment was done using non‐suggestive questioning by investigators |
|
| Participants | Number of participants: 191 Inclusion criteria: Male and female patients with uncomplicated malaria confirmed by positive falciparum asexual blood smear, age ≥ 13 years, body weight ≥ 35kg, ability to take oral medication, agreement to stay in hospital for at least 28 days, informed consent Exclusion criteria: Pregnant or lactating women, severe malaria per WHO criteria, vomiting not allowing oral medication, concomitant systemic disease or disorder other than malaria requiring therapy, history of ingestion of antimalarials in preceding 14 days or with sulphonamides or 4‐aminoquinolones in urine. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleypharm)
2. Artemether‐lumefantrine, fixed dose combination (Coartem: Novartis)
All doses supervised |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Thailand Setting: Hospital for tropical diseases Transmission: "No known malaria transmission" Resistance: Some resistance to P. falciparum reported in Southeast Asia reported Dates: Nov 2005 to June 2006 Funding: Mahidol University Research Grant |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported in the trial. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | Blinding of microscopists not reported. |
| Blinding for adverse events (performance and detection bias) | High risk | Trial is 'open‐label'. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Loss to follow‐up in one treatment group was high (15.5% DHA‐P versus 13.8% AL6). |
| Selective reporting (reporting bias) | Low risk | All listed outcomes are reported. |
| Other bias | Low risk | No other sources of bias identified. |
Mayxay 2006 LAO.
| Methods | Trial design: An open label RCT Follow‐up: Temperature was measured every 6 hrs and patient reviewed daily until fever and parasites cleared then weekly until day 42 or anytime they felt unwell. At each visit a malaria film and haematocrit measurement was taken. Adverse event monitoring: Potential adverse events were recorded at each visit |
|
| Participants | Number of participants: 220 Inclusion criteria: Age > 1 year, axillary temp > 37.5 °C or history of fever in the previous 3 days, P. falciparum mono‐infection 1000 to 200,00/µL, were likely to stay in hospital until parasite clearance and complete 42 days follow‐up, informed consent Exclusion criteria: Pregnancy or lactation, signs of severe malaria, antimalarials in the previous 3 days, contraindications to the trial drugs |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination (Artesunate: Guilin, Lariam: Roche)
All doses supervised. |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Lao People's Democratic Republic (Laos) Setting: District clinic Transmission: Not reported Resistance: Not reported Dates: May 2004 to Sept 2004 Funding: Western Pacific Regional office of WHO, Wellcome Trust of Great Britain, Artekin provided by Holleykin Pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized in blocks of 10". No further details given. |
| Allocation concealment (selection bias) | Low risk | "The treatment choice was kept in a sealed opaque envelope, which was opened only after the decision to recruit". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | No comment on blinding of laboratory staff. |
| Blinding for adverse events (performance and detection bias) | High risk | An open‐label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in both groups (3.6% DHA‐P versus 1.8% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | The WHO recommends 63 days follow‐up in studies of AS+MQ. Day 42 outcomes are likely to overestimate the efficacy of the two drugs. |
| Other bias | Low risk | No other sources of bias identified. |
Mens 2008 KEN.
| Methods | Trial design: An open label RCT Follow‐up: Malaria film and Hb level on days 0, 1, 2, 3, 7, 14, and 28, plus QT‐NASBA for detection of sub‐microscopic gametocytaemia Adverse event monitoring: Adverse events were recorded at each visit in the case record form. An adverse event defined as any unfavourable and unintended sign. |
|
| Participants | Number of participants: 146 Inclusion criteria: Age 6 months to 12 years, axillary temp > 37.5 °C or history of fever, P. falciparum mono‐infection 1000 to 200,000/µL, informed consent Exclusion criteria: Severe malaria, any other underlying illness |
|
| Interventions | 1. DHA‐P, fixed dose combination, 20 mg/160 mg tablets (Sigma‐Tau)
2. Artemether‐lumefantrine, fixed dose combination, 20/120 mg tablets (Novartis)
All doses supervised and given with a glass of milk. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Kenya Setting: Health centre Transmission: High transmission Resistance: Not reported Dates: Apr 2007 to Jul 2007 Funding: The Knowledge and Innovation Fund, Koninklijk Instituut voor de Tropen/Royal Tropical Institute. DHA‐P provided free of charge by Sigma‐Tau. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A computer generated randomisation list". |
| Allocation concealment (selection bias) | Unclear risk | None described. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | Microscopists were blinded to treatment allocation. |
| Blinding for adverse events (performance and detection bias) | Unclear risk | No other blinding described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in both groups (8.2% DHA‐P versus 8.2% AL6). |
| Selective reporting (reporting bias) | Low risk | The WHO recommends 42 days follow‐up in studies of AL6. Day 28 outcomes may underestimate treatment failure with AL6 and DHA‐P. |
| Other bias | Low risk | No other sources of bias identified. |
Ratcliff 2007 IDN.
| Methods | Trial design: An open‐label RCT Follow‐up: A symptom questionnaire, physical examination, malaria film and Hb measurement daily until fever and parasites cleared then weekly to day 42 Adverse event monitoring: A symptom questionnaire at each visit |
|
| Participants | Number of participants: 774 Inclusion criteria: Weight > 10 kg, fever or a history of fever in the preceding 48 hrs, slide confirmed malaria (P. falciparum, P. vivax or mixed infections) Exclusion criteria: Pregnancy or lactation, danger signs or signs of severity, parasitaemia > 4%, concomitant disease requiring hospital admission |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Coartem: Novartis)
Only the first dose of each day was supervised. All participants advised to take each dose with a biscuit or milk. |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Indonesia Setting: Rural outpatient clinics Transmission: Unstable Resistance: Multiple‐drug resistance Dates: Jul 2004 to Jun 2005 Funding: Wellcome Trust UK and National Health and Medical Research Council Australia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomisation list was generated in blocks of 20 patients by an independent statistician". |
| Allocation concealment (selection bias) | Low risk | "With each treatment allocation concealed in an opaque sealed envelope". No further details given. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | The microscopists were blinded to treatment allocation. |
| Blinding for adverse events (performance and detection bias) | High risk | An open label trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The primary outcome data are unpublished data including only participants with P. falciparum mono or co‐infection at baseline. Losses to follow‐up were high in both groups at day 42 (28.4 % DHA‐P versus 25.6 % AL6) and moderate at day 28 (19% DHA‐P versus 17.6% AL6). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 42 outcomes may underestimate failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Sawa 2013 KEN.
| Methods | Trial design: RCT Follow‐up: Clinical assessment on days 1, 2, 3, 7, 14, 28, and 42 and any other time when child became ill. Blood smears taken on all follow‐up days except day 1. Adverse event monitoring: Not reported |
|
| Participants | Number of participants: 298 Inclusion criteria: Microscopically confirmed P. falciparum infection with asexual parasite density of 1,000 to 200,000 parasites/µL, tympanic temperature of ≥ 37.5 °C or history of fever in preceding 24 hrs, age 6 months to 10 years, informed consent Exclusion criteria: Hb level of < 5 g/dL, presence of any other Plasmodium species, presence of other febrile disease, severe malaria, history of adverse events with trial drugs |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Duocotexin: Holley Pharm)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Coartem: Novartis)
All doses were supervised. All participants advised to take each dose with fatty food to facilitate absorption. |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Kenya Setting: Community setting Transmission: Moderate transmission intensity Resistance: None reported Dates: Apr to Jun 2009 Funding: Grants from the European Community’s Seventh Framework Programme and the Bill and Melinda Gates Foundation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomization list was generated for different age strata (<2 years, 2–5 years, and 5–10 years), using MS Excel". |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not reported. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Except for those involved in administering medication, all staff members engaged in the trial were blinded to the treatment arm to which each child was assigned". |
| Blinding for adverse events (performance and detection bias) | Low risk | All staff blinded were except those administering drugs. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low number of patients without outcomes in both groups (DHA‐P 7.6%, AL6 5.2%). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes are reported. |
| Other bias | Low risk | No other sources of bias identified. |
Smithuis 2006 MMR.
| Methods | Trial design: A 4‐arm open‐label RCT Follow‐up: A symptom questionnaire, malaria film, and gametocyte count on days 0, 1, 2, 3, 7, 14, 21, 28, 35, and 42. Hb was measured on days 0 and 28. Adverse event monitoring: A symptom questionnaire at each visit |
|
| Participants | Number of participants: 652 Inclusion criteria: Age > 1 year, axillary temperature > 37.5 °C or history of fever in the previous 48 hrs, P. falciparum mono‐infection 500 to 100,000 parasites/µL or co‐infection with P. vivax, informed consent. Exclusion criteria: Pregnancy, signs of severe malaria, signs or symptoms of other diseases, history of taking mefloquine in the previous 2 months or any other antimalarial in the previous 48 hrs, history of psychiatric disease. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
2. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
3. Artesunate plus mefloquine, loose combination (artesunate: Guilin, Lariam: Hoffman‐La Roche)
4. Artesunate plus mefloquine, loose combination (artesunate: Guilin, Lariam: Hoffman‐La Roche)
|
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Myanmar Setting: Rural village tracts Transmission: Seasonal with peaks in the monsoon season Nov to Jan and sometimes in the early monsoon, May to June Resistance: Very high rates of CQ and SP resistance Dates: Nov 2003 to Feb 2004 Funding: Médecins sans Frontières (Holland) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Unmarked and sealed envelopes, containing the treatment allocation were drawn from a box. |
| Allocation concealment (selection bias) | Unclear risk | "Unmarked and sealed envelopes". No further details given. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | No comment on blinding of laboratory staff. |
| Blinding for adverse events (performance and detection bias) | High risk | An open label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Very low losses to follow‐up in both groups. |
| Selective reporting (reporting bias) | Low risk | The WHO recommends 63 days follow‐up in studies of AS+MQ. Day 42 outcomes are likely to overestimate the efficacy of the two drugs. |
| Other bias | Low risk | No other sources of bias identified. |
Smithuis 2010 MMR.
| Methods | Trial design: A 5‐arm open‐label RCT Follow‐up: Assessment done weekly for 9 weeks and at any other time they became ill. Hb was only measured on day 63. Adverse event monitoring: Specific procedures not reported. |
|
| Participants | Number of participants: 811 Inclusion criteria: Acute uncomplicated malaria (parasite density 500 – 200,000 parasites/µL) or mixed infection, weight > 5 kg, age > 6 months, informed consent Exclusion criteria: Pregnancy, severe malaria, severe acute malnutrition (weight‐for‐height below 70% of median with or without symmetrical peripheral oedema), history of antimalarial treatment within preceding 48 hrs, history of taking mefloquine in past 8 weeks, known history of hypersensitivity to trial drugs. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg adult tablets, 20 mg/160mg children's tablets
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120mg tablets
3. Artesunate amodiaquine, fixed dose combination, 25 mg/67.5mg tablets, 50 mg/135 mg tablets, 100 mg/270mg tablets
4. Artesunate plus mefloquine, fixed dose combination, 25 mg/55 mg tablets, 100mg/220mg tablets
5. Artesunate plus mefloquine, loose combination
For children, tablets were crushed and syrup added. |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Myanmar Setting: Three clinics in Rakhine state Transmission: Seasonal and generally low Resistance: No resistance reported Dates: Dec 2008 to Mar 2009 Funding: Médecins sans Frontières (Holland) and the Wellcome Trust Mahidol University Oxford Tropical Medicine Research Programme |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "random assignment was achieved by patients drawing an envelope from a box after enrolment". |
| Allocation concealment (selection bias) | Unclear risk | "Treatment allocations were put in sealed envelopes in blocks of 50 for each age‐group". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Microscopists examining blood films were unaware of treatment allocation". |
| Blinding for adverse events (performance and detection bias) | High risk | Described as "open‐label". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in each treatment group (AA 3.2%, AL6 5.6%, AM‐F 4.1%, AM‐L 7.5%, DP 3.7%). |
| Selective reporting (reporting bias) | Unclear risk | Some WHO outcomes are not reported (for example, LTF, ETF). |
| Other bias | Low risk | No other sources of bias identified. |
Tangpukdee 2005 THA.
| Methods | Trial design: An open label RCT Follow‐up: The patients were admitted to hospital for 28 days. Clinical evaluation and parasite counts were performed 12‐hourly until parasites cleared then daily for 28 days. Adverse event monitoring: Assessed daily using non‐suggestive questioning. Side effects were defined as signs and symptoms which occurred or became more severe after treatment started. Routine haematology, biochemistry, and urinalysis were conducted and baseline and weekly during follow‐up. |
|
| Participants | Number of participants: 180 Inclusion criteria: Age >14 years, weight > 40 kg, P. falciparum on blood smear, ability to take oral medicines, agree to stay in hospital for 28 days, informed consent Exclusion criteria: Pregnancy or lactation, severe malaria, severe vomiting, concomitant systemic diseases, other antimalarials in the previous 14 days or the presence of sulphonamides or 4‐aminoquinolones in the urine |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination
All doses supervised |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Thailand Setting: Bangkok Hospital for Tropical Diseases Transmission: Low Resistance: Multiple‐drug resistance Dates: Not given Funding: Mahidol University Research Grant, Artekin supplied by Holleykin Pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly treated at a ratio of 1:2". No further details given. |
| Allocation concealment (selection bias) | Unclear risk | None described. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | No comment on blinding of laboratory staff. |
| Blinding for adverse events (performance and detection bias) | High risk | An open label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Losses to follow‐up were low and similar between groups (10.8% DHA‐P versus 10% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | Day 28 outcomes may overestimate the efficacy of drugs with long half‐lives such as AS+MQ and DHA‐P. |
| Other bias | Low risk | No other sources of bias identified. |
The 4ABC Study 2011 AF.
| Methods | Trial design: An open label multicenter RCT Follow‐up: The patients were admitted to hospital for 3 days in some facilities while in some, they were kept for only 1h to check for vomiting. They were asked to return on days 3, 7, 14, 21, and 28. Clinical assessment and blood smears taken at each visit and when clinically judged to be necessary. Haematological samples were taken at enrolment and on days 3, 7, 14, and 28. Samples for biochemistry tests (liver and renal function) were taken at enrolment and on days 7 and 28. Adverse event monitoring: Monitored throughout the trial. Method of monitoring not specified. |
|
| Participants | Number of participants: 4116 Inclusion criteria: Suspected uncomplicated malaria, age 6 to 59 months, body weight > 5 kg, microscopically confirmed P. falciparum mono‐infection with asexual parasite density between 2,000 and 200,000/µL, fever (axillary temperature ≥ 37.5 °C) or history of fever in preceding 24 hrs, Hb ≥ 7.0 g/dL, informed consent. Exclusion criteria: Participation in another investigational drug trial in previous 30 days, known hypersensitivity to study drugs, severe malaria or other danger signs for example not able to breast‐feed or drunk, vomiting (more than twice in 24 hrs), recent history of convulsions (more than once in 24 hrs), unconscious state, unable to stand or sit, severe malnutrition (weight for height < 70% of median National Center for Health Statistics/WHO reference) or other concomitant illness or underlying disease, contra‐indication to receive trial drugs, or ongoing prophylaxis with drugs having antimalarial activity. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg adult tablets, 20 mg/160 mg paediatric tablets (Eurartesim®: Sigma Tau)
2. Artemether‐lumefantrine, fixed dose combination (Coartem: Novartis)
3. Artesunate‐amodiaquine, fixed dose combination, 25 mg/67.5 mg tablets, 50 mg/135 mg tablets, 100 mg/270 mg tablets (Coarsucam: Sanofi Aventis)
All doses supervised. |
|
| Outcomes |
Not included in the review:
|
|
| Notes | Country: Seven sub‐Saharan African countries (Burkina Faso, Gabon, Nigeria, Rwanda, Uganda, Zambia, and Mozambique) Setting: We were unable to identify the trial sites as rural, urban or health facilities Transmission: Varied across trial regions. Trial regions in Gabon and Nigeria had perennial and high malaria transmission; trial regions in Burkina Faso and Rwanda had seasonal but high transmission; trial regions in Zambia included areas with seasonal, mesoendemic transmission; trial regions in Mozambique had perennial, mesoendemic transmission; Jinja and Tororo trial regions in Uganda had perennial, low transmission while Mbarara in Uganda had mesoendemic transmission. . Resistance: All sites had notable CQ and SP resistance. CQ resistance ranged from 24% in Burkina Faso to 100% in Gabon while SP resistance ranged from 4% in Burkina Faso to 49% in Jinja, Uganda. Dates: Jul 2007 to Jun 2009 Funding: European Developing Countries Clinical Trials Partnership (EDCTP) and the Medicine for Malaria Venture (MMV). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomization list was produced for each recruiting site by the National Institute for Health Research Medicines for Children Research Network Clinical Trials Unit, University of Liverpool, UK". |
| Allocation concealment (selection bias) | Low risk | "Each treatment allocation concealed in opaque sealed envelopes that were opened only after the patient’s recruitment". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | Trial is described as "open‐label" but the clinician or other staff following up the patient and assessing the end points blinded to the treatment assignment whenever possible. |
| Blinding for adverse events (performance and detection bias) | Low risk | "the clinician or other staff following up the patient and assessing the end points blinded to the treatment assignment whenever possible". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low loss to follow‐up in each treatment group (3.5% ASAQ, 2.5% DHA‐P, 2.4% AL6). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes are reported. |
| Other bias | Low risk | No other sources of bias identified. |
Tran 2004 VNM.
| Methods | Trial design: An open label RCT Follow‐up: Malaria film on days 0, 2, and 7. Participants followed up to day 56 but further details not described Adverse event monitoring: Not described |
|
| Participants | Number of participants: 243 Inclusion criteria: Age > 2 yrs, microscopically confirmed uncomplicated P. falciparum malaria Exclusion criteria: Pregnancy, evidence of organ dysfunction, unable to tolerate oral medication, unable to return for follow‐up, resident in Dac O for > 2 years |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Artekin: Holleykin)
2. Artesunate plus mefloquine, loose combination (artesunate: Guilin, Lariam: Hoffman‐La Roche)
|
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Vietnam Setting: Health station Transmission: Low and seasonal Resistance: Multiple‐drug resistance Dates: Nov 2001 to Mar 2002 Funding: Wellcome Trust of Great Britain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly allocated one of three treatments in a ratio of 2:2:1". No further details given. |
| Allocation concealment (selection bias) | Unclear risk | "Drugs were kept in identically numbered opaque envelopes". No further details. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | No comment on blinding of laboratory staff. |
| Blinding for adverse events (performance and detection bias) | High risk | An open label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "There were no losses to follow‐up". |
| Selective reporting (reporting bias) | Unclear risk | It is unclear from the paper whether it is only clinical failure that is being reported. |
| Other bias | Low risk | No other sources of bias identified. |
Valecha 2010 AS.
| Methods | Trial design: An open‐label (non‐inferiority) RCT Follow‐up: Particiopants were managed as outpatients unless local practice dictated otherwise (some centres used hospital stays of between 3 and 28 days). Outpatients were asked to return on days 1, 2, 3, 7, 14, 21, 28, 35, 42, 49, 56, and 63, and any time they felt unwell. Blood smears were performed at each visit. Adverse event monitoring: Blood and urine samples were taken for analysis on days 0, 28, 63 (if abnormal on day 28) and on the day of any recurrent parasitaemia. Twelve‐lead ECGs were performed at days 0, 2, 7, 28 (if abnormal on day 7), 63 and on the day of any recurrent parasitaemia. |
|
| Participants | Number of participants: 1150 Inclusion criteria: Age 3 months to 65 years (≥18 years in India), P. falciparum mono‐infection (80 to 200,000 parasites/µL) or mixed infection, weight ≥ 5 kg, fever (≥ 37.5 °C) or history of fever, informed consent. Exclusion criteria: Severe malaria, treatment with MQ in the 60 days before screening, treatment with DHA‐P in the 3 months before screening, > 4% parasitised red blood cells, pregnancy or lactation. |
|
| Interventions | 1. DHA‐P, fixed dose combination, adult tablets 40 mg/320 mg, child tablets 20 mg/160 mg (Eurartesim®: Sigma Tau)
2. Artesunate plus mefloquine, loose dose combination, AS 50mg tablets, MQ 250 mg tablets (Mepha Ltd)
All doses supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Thailand (six sites), Laos (two centres), and India (three centres). Setting: Hospitals and research units. Transmission: Varied across trial regions. Trial regions in Thailand had unstable, low and seasonal malaria transmission; trial regions in Laos had seasonal transmission with a peak just after the heavy rainy months of July to August; trial regions in India included areas with perennial transmission, perennial transmission with a seasonal peak from June to September, and transmission active in post monsoon months. Resistance: All sites had notable CQ resistance (estimates of 28 day treatment failure at the Indian sites ranged from 32% to 67% between 2002 and 2007). The Thai sites also had multi‐drug resistant P. falciparum. Dates: Jun 2005 to Feb 2007. Funding: Medicines for Malaria Venture, Sigma Tau, and Oxford University. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation list was generated by an external contract research organisation (MDS Pharma Services) using the plan procedure of SAS (Cary, NC, USA)". |
| Allocation concealment (selection bias) | Unclear risk | "Randomisation was conducted under blinded conditions: the blind to the investigator and patient in the randomisation process was maintained by the use of sealed envelopes". Envelopes are not described as opaque. |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Evaluation of the PCR test results was blinded". |
| Blinding for adverse events (performance and detection bias) | High risk | Trial is described as "open label". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Proportion of participants not completing the trial was low in both groups (6.6% DHA‐P versus 6.0% AS+MQ). |
| Selective reporting (reporting bias) | Low risk | All listed outcomes reported. |
| Other bias | High risk | "A committee representing members of the DHA‐PQP Joint Development Team and trial site Principal Investigators developed the protocol. Sigma Tau conducted the study, collected and analysed data. All authors had access to the primary data and take responsibility for data reporting accuracy and completeness. The corresponding author had responsibility for the final decision to submit for publication". |
Yavo 2011 AF.
| Methods | Trial design: An open‐label (non‐inferiority) RCT Follow‐up: Clinical and physical assessment and blood smears taken on follow‐up days 1, 2, 3, 4, 7, 14, 21, and 28 after swallowing first antimalarial. Haematological and biochemical assessment conducted at baseline and on day 4. Adverse event monitoring: Recorded on the case report forms and graded as mild, moderate or severe. |
|
| Participants | Number of participants: 384 Inclusion criteria: At least 2 years of age, P. falciparum mono‐infection with parasitaemia from 2,000 to 200,000/µL of blood in Cameroon and Côte d’Ivoire and 1,000 to 100,000/µL of blood in Sénégal, fever with axillary temperature ≥ 37.5 °C, written informed consent Exclusion criteria: History of side‐effects to trial drugs, evidence of concomitant febrile illness, severe malaria or danger signs, treatment with 4‐amino quinolones, SP, MG or halofantrine in previous 7 days or quinine, artemisinin or cyclins in previous 3 days, pregnancy or nursing, ongoing antimalarial treatment |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Duocotecxin: Beijing Holley‐Cotect Pharmaceutical Co.)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg (Coartem: Novartis)
All doses supervised. |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Senegal, Côte d’Ivoire, Cameroon Setting: health facilities Transmission: Not reported Resistance: "CQ and SP resistance reported in most parts of the continent" Dates: November 2006 to May 2008 Funding: Beijing Holley‐Cotec Pharmaceutical Co. Ltd which also supplied DHA‐P free of charge |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "In each study site computer generated randomization codes were prepared by an independent individual". |
| Allocation concealment (selection bias) | Low risk | "These codes were enclosed in sequentially numbered opaque sealed envelopes, each of which contained the treatment allocation". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Unclear risk | Blinding of microscopists not described. |
| Blinding for adverse events (performance and detection bias) | High risk | Trial described as "open label". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low percentage of patients without outcomes (3.0% in DHA‐P and 2.1% in AL6). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes are reported. Day 28 outcomes may underestimate effect of DHA‐P. |
| Other bias | Low risk | No other sources of bias identified. |
Yeka 2008 UGA.
| Methods | Trial design: A single blind RCT Follow‐up: Standardized history, physical exam, and malaria film on days 0, 1, 2, 3, 7, 14, 21, 28, 35, and 42 and any other day they were unwell. Hb on days 0 and 42 or the day of failure. Anaemia was treated with ferrous sulphate and anthelmintics according to IMCI guidelines. Adverse event monitoring: Assessed at each visit including neurological examination. Adverse events described as any untoward medical occurrence. |
|
| Participants | Number of participants: 461 Inclusion criteria: Age 6 months to 10 yrs, weight > 5 kg, axillary temp > 37.5 °C or history of fever in the previous 24 hrs, P. falciparum mono‐infection 2000 to 200,000/µL, informed consent Exclusion criteria: Danger signs or evidence of severe malaria, concomitant febrile illness, history of serious side effects to the trial medications |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Duocotexin: HolleyPharm)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Coartem: Novartis)
All doses supervised and given with a glass of milk |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Uganda Setting: Health centre Transmission: Moderate transmission Resistance: Not stated Dates: Aug 2006 to Apr 2007 Funding: CDC, DfID, DHA‐P supplied by Holleypharm, AL6 supplied by Uganda Ministry of Health |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomisation list was computer generated by an off‐site investigator". |
| Allocation concealment (selection bias) | Low risk | "Sealed opaque envelopes containing the study number and assigned treatment were secured in a locked cabinet". |
| Blinding for microscopy outcomes (performance bias and detection bias) | Low risk | "Only the study nurse was aware of assignments. All other study personnel were blinded". |
| Blinding for adverse events (performance and detection bias) | Low risk | "Patients were not informed of their treatment regimen". "Only the study nurse was aware of assignments and adverse events assessed by clinicians. All other study personnel were blinded". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in both groups (1.4% DHA‐P versus 1.5% AL6). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 42 outcomes may underestimate treatment failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Zongo 2007 BFA.
| Methods | Trial design: A 3‐arm RCT Follow‐up: A standardized history, examination, and malaria film on days 0, 1, 2, 3, 7, 14, 21, 28, 35, and 42. Hb measured on days 0 and 42 or day of clinical failure. Children with Hb < 10 g/dL were treated with ferrous sulphate and antihelminthic treatment. Adverse event monitoring: Assessed at each visit. Adverse events defined as untoward medical occurrences. |
|
| Participants | Number of participants: 580 Inclusion criteria: Age > 6 months, weight > 5 kg, axillary temp > 37.5 °C or history of fever in the last 24 hrs, P. falciparum mono‐infection 2000 to 200,000/µL, the ability to participate in 42 days follow‐up, informed consent. Exclusion criteria: Danger signs or signs of severe malaria, history of serious adverse effects related to trial medications, evidence of concomitant febrile illness, antimalarial use other than chloroquine in previous two weeks, Hb < 5 g/dL. |
|
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets (Duocotexin: HolleyPharm)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Coartem: Novartis)
3. Amodiaquine plus sulfadoxine‐pyrimethamine, loose combination (Flavoquine: Aventis, Fansidar: Roche)
All doses supervised |
|
| Outcomes |
Not included in this review:
|
|
| Notes | Country: Burkino Faso Setting: Health dispensaries Transmission: Holoendemic, transmission principally in the rainy season May to Oct Resistance: Not reported Dates: Not reported Funding: Doris Duke Charitable Foundation, Holley Cotec Pharmaceuticals, International Atomic Energy Agency, National Budget of the Institut de Recherche en Sciences de la Sante |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomly assigned on the basis of a computer‐generated code provided by an off‐site investigator". |
| Allocation concealment (selection bias) | Low risk | "Referred for treatment allocation by a study nurse not involved in enrolment or assessment of treatment outcomes". |
| Blinding for microscopy outcomes (performance bias and detection bias) | High risk | "The study was not blinded". |
| Blinding for adverse events (performance and detection bias) | High risk | As above. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low losses to follow‐up in all groups (8% DHA‐P versus 6.4% AL6 versus 8.2% AQ+SP). |
| Selective reporting (reporting bias) | Low risk | All WHO outcomes reported. Day 42 outcomes may underestimate treatment failure with DHA‐P due to its long half‐life. |
| Other bias | Low risk | No other sources of bias identified. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Chinh 2009 | Comparison not relevant to this review: two fixed‐dose combinations of DHA‐P were compared. |
| Guo 1990 | Conference presentation. Comparison not relevant to this review: artesunate versus piperaquine. |
| Gupta 2010 | Molecular genotyping analysis based on Kamya 2007 UGA and Yeka 2008 UGA. Contains no new efficacy data. |
| Karema 2005 | Conference presentation of Karema 2006 RWA. |
| Somé 2010 | Polymorphism selection analysis based on Zongo 2007a BFA. Contains no new efficacy data. |
| Song 2011 KHM | Comparison not relevant to this review: DHA‐P‐phosphate (2 day dosage) versus artemether‐lumefantrine. |
| Sutanto 2013 | Comparison not relevant to this review: DHA‐P + primaquine versus DHA‐P. |
| Tarning 2008 | Pharmacokinetic analysis based on Ashley 2005 THA. Contains no new efficacy data. |
| Thanh 2009 | Quasi‐RCT. |
| Tjitra 2012 | Comparison not relevant to this review: Artemisinin‐naphthoquine versus DHA‐P. |
| Tran 2012 | Comparison not relevant to this review: AS monotherapy versus DHA‐P. |
| Verret 2011 | Nutritional status analysis based on Arinaitwe 2009 UGA. Contains no new efficacy data. |
| Wang 2008 | Quasi‐RCT. |
| Yeka 2013 | A comparison of DHA‐P versus artemether‐lumefantrine as rescue treatments based on The 4ABC Study 2011 AF. Contains no new efficacy data. |
Characteristics of studies awaiting assessment [ordered by study ID]
Borrmann 2011 KEN (a).
| Methods | Trial design: A non‐inferiority RCT Follow‐up: Clinical assessment and blood smears taken on days 0, 1, 2, and 3 then weekly until day 63 and finally on day 84. Adverse event monitoring: Monitoring done on days 0, 1, 2, and 3 then weekly until day 63 and finally on day 84. |
| Participants | Number of participants: 474 Inclusion criteria: Uncomplicated malaria, age 6 to 59 months, body weight ≥ 5kg, microscopically confirmed falciparum mono‐infection with asexual parasite density of 2,000 to 200,000 µL, reported or documented axillary temperature ≥ 37.5 °C, informed consent Exclusion criteria: Known allergies, severe malaria or danger signs, ECG abnormalities that require urgent management, participation in another investigational drug study within previous 30 days. |
| Interventions | 1. DHA‐P, fixed dose combination, 40 mg/320 mg tablets and 20 mg/160 mg tablets (Eurartesim®, Sigma‐Tau)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg (Coartem: Novartis)
All doses were supervised. |
| Outcomes |
Not included in this review:
|
| Notes | Country: Kenya Setting: One hospital site. Transmission: Perennial with peaks trailing two typical annual rainy seasons Resistance: None reported Dates: Sep 2005 to Apr 2008 Funding: DFG and MMV grants, European Developing Countries Clinical Trials Partnership (EDCTP) and the Wellcome Trust |
Characteristics of ongoing studies [ordered by study ID]
Tekete 2012 AF.
| Trial name or title | EDCTP Longitudinal study |
| Methods | Trial design: An open‐label RCT Follow‐up: Patients will receive same study drug for subsequent episodes of uncomplicated malaria for up to 2 years after first randomization. Haematology, biochemistry and clinical safety will be assessed over this two year period. |
| Participants | Number of participants: 4032 Inclusion criteria: Acute uncomplicated malaria, age > 6 months, weight ≥ 5 kg with no clinical sign of severe malnutrition, axillary temp > 37.5 °C, oral/rectal/tympanic temperature > 38 °C or history of fever in the last 24 hrs, microscopically confirmed P. falciparum with parasite density less than 200,000/µL, ability to swallow oral medication, no documented malaria treatment in preceding 2 weeks or 4 weeks for re‐inclusion, the ability to participate in the scheduled follow‐up visits, written informed consent Exclusion criteria: Signs and symptoms of severe/complicated malaria, severe vomiting (more than 3 times in preceding 24hrs) or inability to tolerate oral medication, severe diarrhoea (3 or more watery stools per day), known history of clinically significant disorders such as cardiovascular (QTc interval > 450 ms), respiratory (including active tuberculosis), history of jaundice, renal, hepatic, gastrointestinal, immunological (including active HIV), neurological, endocrine, or other major psychiatric disorders, history of convulsions or other abnormality (including recent head trauma), Hb < 7 g/dL, concomitant febrile illness, hypersensitivity to study drugs, use of any other antimalarial in preceding 2 weeks before enrolment, pregnant or lactating women, known or suspected chronic alcohol abuse, active Hepatitis A, B or C, liver function tests > 2 times upper limit of normal, known significant renal impairment indicated by serum creatinine more than 1.5 x ULN |
| Interventions | 1. DHA‐P, fixed dose combination, adult tablets 40 mg/320 mg, child tablets 20 mg/160 mg (Eurartesim®: Sigma Tau)
2. Artemether‐lumefantrine, fixed dose combination, 20 mg/120 mg tablets (Coartem: Novartis)
3. Artesunate‐Amodiaquine, fixed dose combination, 3 strengths: 25 mg/67.5 mg, 50 mg/135 mg, 100 mg/270 mg (Winthrop: Sanofi‐aventis)
4, Artesunate‐Amodiaquine, fixed dose combination (Pyramax: Shin Poong)
|
| Outcomes |
|
| Starting date | 01 June 2011 |
| Contact information | Principal investigator ‐ Dr Abdoulaye Djimde ‐ adjimde@mrtcbko.org |
| Notes | Country: Burkino Faso (2 centres), Mali (1 centre), Guinea (1 centre) Dates: 01 June 2011 to 29 June 2014 Funding: Medicines for Malaria Venture and European & Developing Countries Clinical Trials Partnership (EDCTP) |
Differences between protocol and review
This review was originally incorporated in a larger review of ACTs (Sinclair 2009). In this review we have included additional appraisal and GRADE assessments of adverse effects.
Contributions of authors
DS, BZ, SD, and PO developed the protocol as used in Sinclair 2009. For this update, BZ and MG reviewed the reference list, extracted data, and entered it into Review Manager (RevMan). BZ, MG, and DS conducted the analyses, constructed summary of findings tables, and evaluated the quality of evidence using the GRADE approach. All authors reviewed and edited the final draft.
Declarations of interest
None known.
Unchanged
References
References to studies included in this review
Adam 2010 SDN {published data only}
- Adam I, Salah MT, Eltahir HG, Elhassan AH, Elmardi KA, Malik EM. Dihydroartemisinin‐piperaquine versus artemether‐lumefantrine, in the treatment of uncomplicated Plasmodium falciparum malaria in central Sudan. Annals of Tropical Medicine and Parasitology 2010;104(4):319‐26. [DOI] [PubMed] [Google Scholar]
Agarwal 2013 KEN {published data only}
- Agarwal A, McMorrow M, Onyango P, Otieno K, Odero C, Williamson J, et al. A randomized trial of artemether‐lumefantrine and dihydroartemisinin‐piperaquine in the treatment of uncomplicated malaria among children in western Kenya. Malaria Journal 2013;12(254):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Arinaitwe 2009 UGA {published data only}
- Arinaitwe E, Sandison TG, Wanzira H, Kakuru A, Homsy J, Kalamya J, et al. Artemether‐lumefantrine versus dihydroartemisinin‐piperaquine for falciparum malaria: a longitudinal, randomized trial in young Ugandan children. Clinical Infectious Diseases 2009;49(11):1629‐37. [DOI] [PubMed] [Google Scholar]
- Creek D, Bigira V, Arinaitwe E, Wanzira H, Kakuru A, Tappero J, et al. Increased risk of early vomiting among infants and young children treated with dihydroartemisinin‐piperaquine compared with artemether‐lumefantrine for uncomplicated malaria. American Journal of Tropical Medicine and Hygiene 2010;83(4):873‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuru A, Jagannathan P, Arinaitwe E, Wanzira H, Muhindo M, Bigira V, et al. The effects of ACT treatment and TS prophylaxis on Plasmodium falciparum gametocytemia in a cohort of young Ugandan children. American Journal of Tropical Medicine and Hygiene 2013;88(4):736‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrak S, Gasasira A, Arinaitwe E, Kakuru A, Wanzira H, Bigira V, et al. Safety and tolerability of artemether‐lumefantrine versus dihydroartemisinin‐piperaquine for malaria in young HIV‐infected and uninfected children. Malaria Journal 2009;8(272):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ashley 2004a THA {published data only}
- Ashley EA, Krudsood S, Phaiphun L, Srivilairit S, McGready R, Leowattana W, et al. Randomized, controlled dose‐optimization studies of dihydroartemisinin‐piperaquine for the treatment of uncomplicated multidrug‐resistant falciparum malaria in Thailand. Journal of Infectious Diseases 2004;190(10):1773‐82. [DOI] [PubMed] [Google Scholar]
Ashley 2004b THA {published data only}
- Ashley EA, Krudsood S, Phaiphun L, Srivilairit S, McGready R, Leowattana W, et al. Randomized, controlled dose‐optimization studies of dihydroartemisinin‐piperaquine for the treatment of uncomplicated multidrug‐resistant falciparum malaria in Thailand. Journal of Infectious Diseases 2004;190(10):1773‐82. [DOI] [PubMed] [Google Scholar]
Ashley 2005 THA {published data only}
- Ashley EA, McGready R, Hutagalung R, Phaiphun L, Slight T, Proux S, et al. A randomized, controlled study of a simple, once‐daily regimen of dihydroartemisinin‐piperaquine for the treatment of uncomplicated, multidrug‐resistant falciparum malaria. Clinical Infectious Diseases 2005; Vol. 41, issue 4:425‐32. [DOI] [PubMed]
Bassat 2009 AF {published data only}
- Bassat Q, Mulenga M, Tinto H, Piola P, Borrmann S, Menéndez C, et al. Dihydroartemisinin‐piperaquine and artemether‐lumefantrine for treating uncomplicated malaria in African children: a randomised, non‐inferiority trial. PLoS One 2009;4(11):e7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann S, Sasi P, Mwai L, Bashraheil M, Abdallah A, Muriithi S, et al. Declining responsiveness of Plasmodium falciparum infections to artemisinin‐based combination treatments on the Kenyan coast. PloS One 2011;6(11):e26005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambozi M, Geertruyden JP, Hachizovu S, Chaponda M, Mukwamataba D, Mulenga M, et al. Safety and efficacy of dihydroartemisininpiperaquine versus artemether‐lumefantrine in the treatment of uncomplicated Plasmodium falciparum malaria in Zambian children. Malaria Journal 2011;10(50):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grande 2007 PER {published data only}
- Grande T, Bernasconi A, Erhart A, Gamboa D, Casapia M, Delgado C, et al. A randomised controlled trial to assess the efficacy of dihydroartemisinin‐piperaquine for the treatment of uncomplicated falciparum malaria in Peru. PLoS One 2007;2(10):e1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hasugian 2007 IDN {published data only}
- Hasugian AR, Purba HL, Kenangalem E, Wuwung RM, Ebsworth EP, Maristela R, et al. Dihydroartemisinin‐piperaquine versus artesunate‐amodiaquine: superior efficacy and posttreatment prophylaxis against multidrug‐resistant Plasmodium falciparum and Plasmodium vivax malaria. Clinical Infectious Diseases 2007;44(8):1067‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Janssens 2007 KHM {published data only}
- Janssens B, Herp M, Goubert L, Chan S, Uong S, Nong S, et al. A randomized open study to assess the efficacy and tolerability of dihydroartemisinin‐piperaquine for the treatment of uncomplicated falciparum malaria in Cambodia. Tropical Medicine and International Health 2007;12(2):251‐9. [DOI] [PubMed] [Google Scholar]
Kamya 2007 UGA {published data only}
- Kamya MR, Yeka A, Bukirwa H, Lugemwa M, Rwakimari JB, Staedke SG, et al. Artemether‐lumefantrine versus dihydroartemisinin‐piperaquine for treatment of malaria: a randomized trial. PLoS Clinical Trials 2007;2(5):e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karema 2006 RWA {published data only}
- Karema C, Fanello CI, Overmeir C, Geertruyden JP, Doren W, Ngamije D, et al. Safety and efficacy of dihydroartemisinin/piperaquine (Artekin) for the treatment of uncomplicated Plasmodium falciparum malaria in Rwandan children. Transactions of the Royal Society of Tropical Medicine and Hygiene 2006; Vol. 100, issue 12:1105‐11. [DOI] [PubMed]
Karunajeewa 2008 PNG {published data only}
- Davis WA, Clarke PM, Siba PM, Karunajeewa HA, Davy C, Mueller I, et al. Cost‐effectiveness of artemisinin combination therapy for uncomplicated malaria in children: data from Papua New Guinea. Bulletin of the World Health Organization 2011;89(3):211‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunajeewa HA, Mueller I, Senn M, Lin E, Law I, Gomorrai PS, et al. A trial of combination antimalarial therapies in children from Papua New Guinea. New England Journal of Medicine 2008;359(24):2545‐57. [DOI] [PubMed] [Google Scholar]
Krudsood 2007 THA {published data only}
- Krudsood S, Tangpukdee N, Thanchatwet V, Wilairatana P, Srivilairit S, Pothipak N, et al. Dose ranging studies of new artemisinin‐piperaquine fixed combinations compared to standard regimens of artemisinin combination therapies for acute uncomplicated falciparum malaria. Southeast Asian Journal of Tropical Medicine and Public Health 2007;38(6):971‐8. [PMC free article] [PubMed] [Google Scholar]
Mayxay 2006 LAO {published data only}
- Mayxay M, Thongpraseuth V, Khanthavong M, Lindegårdh N, Barends M, Keola S, et al. An open, randomized comparison of artesunate plus mefloquine vs. dihydroartemisinin‐piperaquine for the treatment of uncomplicated Plasmodium falciparum malaria in the Lao People's Democratic Republic (Laos). Tropical Medicine and International Health 2006;11(8):1157‐65. [DOI] [PubMed] [Google Scholar]
Mens 2008 KEN {published data only}
- Mens PF, Sawa P, Amsterdam SM, Versteeg I, Omar SA, Schallig HD, et al. A randomized trial to monitor the efficacy and effectiveness by QT‐NASBA of artemether‐lumefantrine versus dihydroartemisinin‐piperaquine for treatment and transmission control of uncomplicated Plasmodium falciparum malaria in western Kenya. Malaria Journal 2008;7(237):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ratcliff 2007 IDN {published data only}
- Ratcliff A, Siswantoro H, Kenangalem E, Maristela R, Wuwung RM, Laihad F, et al. Two fixed‐dose artemisinin combinations for drug‐resistant falciparum and vivax malaria in Papua, Indonesia: an open‐label randomised comparison. The Lancet 2007;369(9563):757‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sawa 2013 KEN {published data only}
- Sawa P, Shekalaghe SA, Drakeley CJ, Sutherland CJ, Mweresa CK, Baidjoe AY, et al. Malaria transmission after artemether‐lumefantrine and dihydroartemisinin‐piperaquine: a randomized trial. Journal of Infectious Diseases 2013;207(11):1637‐45. [DOI] [PubMed] [Google Scholar]
Smithuis 2006 MMR {published data only}
- Smithuis F, Kyaw MK, Phe O, Aye KZ, Htet L, Barends M, et al. Efficacy and effectiveness of dihydroartemisinin‐piperaquine versus artesunate‐mefloquine in falciparum malaria: an open‐label randomised comparison. The Lancet 2006;367(9528):2075‐85. [DOI] [PubMed] [Google Scholar]
Smithuis 2010 MMR {published data only}
- Smithuis F, Kyaw MK, Phe O, Win T, Aung PP, Oo AP, et al. Effectiveness of five artemisinin combination regimens with or without primaquine in uncomplicated falciparum malaria: an open‐label randomised trial. Lancet Infectious Diseases 2010;10(10):673‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tangpukdee 2005 THA {published data only}
- Tangpukdee N, Krudsood S, Thanachartwet W, Chalermrut K, Pengruksa C, Srivilairit S, et al. An open randomized clinical trial of Artekin vs artesunate‐mefloquine in the treatment of acute uncomplicated falciparum malaria. Southeast Asian Journal of Tropical Medicine and Public Health 2005;36(5):1085‐91. [PubMed] [Google Scholar]
The 4ABC Study 2011 AF {published data only}
- The Four Artemisinin‐Based Combinations (4ABC) Study Group. A head‐to‐head comparison of four artemisinin‐based combinations for treating uncomplicated malaria in African children: a randomized trial. PLoS Medicine 2011;8(11):e1001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tran 2004 VNM {published data only}
Valecha 2010 AS {published data only}
- Gargano N, Ubben D, Tommasini S, Bacchieri A, Corsi M, Bhattacharyya PC, et al. Therapeutic efficacy and safety of dihydroartemisinin‐piperaquine versus artesunate‐mefloquine in uncomplicated Plasmodium falciparum malaria in India. Malaria Journal 2012;11(233):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayxay M, Keomany S, Khanthavong M, Souvannasing P, Stepniewska K, Khomthilath T, et al. A phase III, randomized, non‐inferiority trial to assess the efficacy and safety of dihydroartemisinin‐piperaquine in comparison with artesunate‐mefloquine in patients with uncomplicated Plasmodium falciparum malaria in southern Laos. American Journal of Tropical Medicine and Hygiene 2010;83(6):1221‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valecha N, Phyo A P, Mayxay M, Newton P N, Krudsood S, Keomany S, et al. An open‐label, randomised study of dihydroartemisinin‐piperaquine versus artesunate‐mefloquine for falciparum malaria in Asia. PLoS One 2010;5(7):e11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yavo 2011 AF {published data only}
- Yavo W, Faye B, Kuete T, Djohan V, Oga SA, Kassi RR, et al. Multicentric assessment of the efficacy and tolerability of dihydroartemisinin‐piperaquine compared to artemether‐lumefantrine in the treatment of uncomplicated Plasmodium falciparum malaria in sub‐Saharan Africa. Malaria Journal 2011;10(198):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yeka 2008 UGA {published data only}
- Yeka A, Dorsey G, Kamya MR, Talisuna A, Lugemwa M, Rwakimari JB, et al. Artemether‐lumefantrine versus dihydroartemisinin‐piperaquine for treating uncomplicated malaria: a randomized trial to guide policy in Uganda. PLoS One 2008;3(6):e2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zongo 2007 BFA {published data only}
- Zongo I, Dorsey G, Rouamba N, Dokomajilar C, Séré Y, Rosenthal PJ, et al. Randomized comparison of amodiaquine plus sulfadoxine‐pyrimethamine, artemether‐lumefantrine, and dihydroartemisinin‐piperaquine for the treatment of uncomplicated Plasmodium falciparum malaria in Burkina Faso. Clinical Infectious Diseases 2007; Vol. 45, issue 11:1453‐61. [DOI] [PubMed]
References to studies excluded from this review
Chinh 2009 {published data only}
- Chinh NT, Quang NN, Thanh NX, Dai B, Geue JP, Addison RS, et al. Pharmacokinetics and bioequivalence evaluation of two fixed‐dose tablet formulations of dihydroartemisinin and piperaquine in Vietnamese subjects. Antimicrobial Agents and Chemotherapy 2009;53(2):828‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guo 1990 {published data only}
- Guo XB, Fu YX, Chen PJ. A randomised comparative study on the treatment of falciparum malaria with artesunate tablets and piperaquine [Chinese]. Clinical Trials on Qinghaosu and its Derivatives. Guangzhou, China, 1990:39‐42.
Gupta 2010 {published data only}
- Gupta V, Dorsey G, Hubbard AE, Rosenthal PJ, Greenhouse B. Gel versus capillary electrophoresis genotyping for categorizing treatment outcomes in two anti‐malarial trials in Uganda. Malaria Journal 2010;9(19):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karema 2005 {published data only}
- Karema C, Fanello C, Doren W, Rwagacondo C, Overmeir C, Dujardin J, et al. Safety and efficacy of dihydroartemisinin‐piperaquine in Rwandan children with uncomplicated P. falciparum malaria. 4th MIM Malaria Conference: November, 2005. Yaounde, Cameroon, 2005.
Somé 2010 {published data only}
- Somé A F, Séré YY, Dokomajilar C, Zongo I, Rouamba N, Greenhouse B, et al. Selection of known Plasmodium falciparum resistance‐mediating polymorphisms by artemether‐lumefantrine and amodiaquine‐sulfadoxine‐pyrimethamine but not dihydroartemisinin‐piperaquine in Burkina Faso. Antimicrobial Agents and Chemotherapy 2010;54(5):1949‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Song 2011 KHM {published data only}
- Song J, Socheat D, Tan B, Seila S, Xu Y, Ou F, et al. Randomized trials of artemisinin‐piperaquine, dihydroartemisinin‐piperaquine phosphate and artemether‐lumefantrine for the treatment of multi‐drug resistant falciparum malaria in Cambodia‐Thailand border area. Malaria Journal 2011;10(231):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sutanto 2013 {published data only}
- Sutanto I, Suprijanto S, Kosasih A, Dahlan MS, Syafruddin D, Kusriastuti R, et al. The effect of primaquine on gametocyte development and clearance in the treatment of uncomplicated falciparum malaria with dihydroartemisinin‐piperaquine in South Sumatra, Western Indonesia: an open‐label, randomized, controlled trial. Clinical Infectious Diseases 2013;56(5):685‐93. [DOI] [PubMed] [Google Scholar]
Tarning 2008 {published data only}
- Tarning J, Ashley EA, Lindegardh N, Stepniewska K, Phaiphun L, Day NP, et al. Population pharmacokinetics of piperaquine after two different treatment regimens with dihydroartemisinin‐piperaquine in patients with Plasmodium falciparum malaria in Thailand. Antimicrobial Agents and Chemotherapy 2008;52(3):1052‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thanh 2009 {published data only}
- Thanh NX, Trung TN, Phong NC, Thien NX, Dai B, Shanks GD, et al. Open label randomized comparison of dihydroartemisinin‐piperaquine and artesunate‐amodiaquine for the treatment of uncomplicated Plasmodium falciparum malaria in central Vietnam. Tropical Medicine and International Health 2009;14(5):504‐11. [DOI] [PubMed] [Google Scholar]
Tjitra 2012 {published data only}
- Tjitra E, Hasugian AR, Siswantoro H, Prasetyorini B, Ekowatiningsih R, Yusnita EA, et al. Efficacy and safety of artemisinin‐naphthoquine versus dihydroartemisinin‐piperaquine in adult patients with uncomplicated malaria: a multi‐centre study in Indonesia. Malaria Journal 2012;11(153):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tran 2012 {published data only}
- Tran TH, Nguyen TT, Nguyen HP, Boni MF, Ngo VT, Nguyen TN, et al. In vivo susceptibility of Plasmodium falciparum to artesunate in Binh Phuoc Province, Vietnam. Malaria Journal 2012;11(355):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Verret 2011 {published data only}
- Verret WJ, Arinaitwe E, Wanzira H, Bigira V, Kakuru A, Kamya M, et al. Effect of nutritional status on response to treatment with artemisinin‐based combination therapy in young Ugandan children with malaria. Antimicrobial Agents and Chemotherapy 2011;55(6):2629‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2008 {published data only}
- Wang SQ, Christophel E, Lin SG, Meng F, Hu XM, Wang GZ, et al. [Efficacy of dihydroartemisinin‐piperaquine and artemether‐lumefantrine in the treatment of uncomplicated falciparum malaria in Hainan, China]. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 2008;26(1):50‐2. [PubMed] [Google Scholar]
Yeka 2013 {published data only}
- Yeka A, Tibenderana J, Achan J, D'Alessandro U, Talisuna AO. Efficacy of quinine, artemether‐lumefantrine and dihydroartemisinin‐piperaquine as rescue treatment for uncomplicated malaria in Ugandan children. PloS One 2013;8(1):e53772. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Borrmann 2011 KEN (a) {published data only}
- Borrmann S, Sasi P, Mwai L, Bashraheil M, Abdallah A, Muriithi S, et al. Declining responsiveness of Plasmodium falciparum infections to artemisinin‐based combination treatments on the Kenyan coast. PloS One 2011;6(11):e26005. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
Tekete 2012 AF {published data only}
- Tekete M, Djimde A, Borrmann S. A phase IIIb/IV comparative, randomised, multi‐centre, open label, parallel 3‐arm clinical study to assess the safety and efficacy of repeated administration of four artemisinin‐based combination therapy (ACT) over a two‐year period in children and adult patients with acute uncomplicated Plasmodium sp. malaria. Chemotherapie Journal. 2012; Vol. Conference: 9.
Additional references
Abba 2011
- Abba K, Deeks JJ, Olliaro P, Naing CM, Jackson SM, Takwoingi Y, et al. Rapid diagnostic tests for diagnosing uncomplicated P. falciparum malaria in endemic countries. Cochrane Database of Systematic Reviews 2011, Issue 7. [DOI: 10.1002/14651858.CD008122.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adjuik 2004
- Adjuik M, Babiker A, Garner P, Olliaro P, Taylor W, White N, et al. Artesunate combinations for treatment of malaria: meta‐analysis. The Lancet 2004;363(9402):9‐17. [DOI] [PubMed] [Google Scholar]
Bloland 2003
- Bloland PB. Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria [WHO/HTM/RBM/2003.50]. Geneva: World Health Organization, 2003. [Google Scholar]
Cattamanchi 2003
- Cattamanchi A, Kyabayinze D, Hubbard A, Rosenthal PJ, Dorsey G. Distinguishing recrudescence from reinfection in a longitudinal antimalarial drug efficacy study: comparison of results based on genotyping of msp‐1, msp‐2, and glurp. American Journal of Tropical Medicine and Hygiene 2003;68(2):133‐9. [PubMed] [Google Scholar]
Davis 2005
- Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. Piperaquine: a resurgent antimalarial drug. Drugs 2005;65(1):75‐87. [DOI] [PubMed] [Google Scholar]
European Medicines Agency 2011
- European Medicines Agency. Eurartesim: Assessment report. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/001199/WC500118116.pdf 2011:1‐129.
Guyatt 2008
- Guyatt GH, Oxman AD, Kunz R, Vist GE, Falck‐Ytter Y, Schünemann HJ, et al. What is "quality of evidence" and why is it important to clinicians?. BMJ 2008;336(7651):995‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors), Cochrane Handbook of Systematic Reviews of Interventions. Version 5.0.0 [updated February 2008]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org. Chichester: John Wiley & Sons Ltd.
Jansen 2010
- Jansen FH. The pharmaceutical death‐ride of dihydroartemisinin. Malaria Journal 2010;9(212):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karunajeewa 2004
- Karunajeewa H, Lim C, Hung TY, Ilett KF, Denis MB, Socheat D, et al. Safety evaluation of fixed combination piperaquine plus dihydroartemisinin (Artekin) in Cambodian children and adults with malaria. British Journal of Clinical Pharmacology 2004;57(1):93‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Keating 2012
- Keating GM. Dihydroartemisinin/Piperaquine: a review of its use in the treatment of uncomplicated Plasmodium falciparum malaria. Drugs 2012;72(7):937‐61. [DOI] [PubMed] [Google Scholar]
Lefebvre 2008
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Review of Interventions. Version 5.0.1 [updated September 2008]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
McIntosh 2000
- McIntosh HM, Olliaro P. Artemisinin derivatives for treating uncomplicated malaria. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000256] [DOI] [PMC free article] [PubMed] [Google Scholar]
Meshnick 1996
- Meshnick SR, Taylor TE, Kamchonwongpaisan S. Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiological Reviews 1996;60(2):301‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Myint 2007
- Myint HY, Ashley EA, Day NP, Nosten F, White NJ. Efficacy and safety of dihydroartemisinin‐piperaquine. Transactions of the Royal Society of Tropical Medicine and Hygiene 2007;101(9):858‐66. [DOI] [PubMed] [Google Scholar]
Naing 2013
- Naing C, Mak JW, Aung K, Wong JY. Efficacy and safety of dihydroartemisinin‐piperaquine for treatment of uncomplicated Plasmodium falciparum malaria in endemic countries: meta‐analysis of randomised controlled studies. Transactions of the Royal Society of Tropical Medicine and Hygiene 2013;107(2):65‐73. [DOI] [PubMed] [Google Scholar]
Nosten 2007
- Nosten F, White NJ. Artemisinin‐based combination treatment of falciparum malaria. American Journal of Tropical Medicine and Hygiene 2007;77(6 Suppl):181‐92. [PubMed] [Google Scholar]
Price 1996
- Price RN, Nosten F, Luxemburger C, ter Kuile FO, Paiphun L, Chongsuphajaisiddhi T, et al. Effects of artemisinin derivatives on malaria transmissibility. The Lancet 1996;347(9016):1654‐8. [DOI] [PubMed] [Google Scholar]
Price 1999
- Price R, Vugt M, Phaipun L, Luxemburger C, Simpson J, McGready R, et al. Adverse effects in patients with acute falciparum malaria treated with artemisinin derivatives. American Journal of Tropical Medicine and Hygiene 1999;60(4):547‐55. [DOI] [PubMed] [Google Scholar]
Review Manager (RevMan) [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2013.
Schmatz 2010
- Schmatz D. Dihydroartemisinin: more life‐boat than death‐ride. Malaria Journal 2010;9(212):1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sinclair 2009
- Sinclair D, Zani B, Donegan S, Olliaro PL, Garner P. Artemisinin‐based combination therapy for treating uncomplicated malaria. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007483.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sinclair 2012
- Sinclair D, Donegan S, Isba R, Lalloo DG. Artesunate versus quinine for treating severe malaria. Cochrane Database of Systematic Reviews 2012, Issue 6. [DOI: 10.1002/14651858.CD005967.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Targett 2001
- Targett G, Drakeley C, Jawara M, Seidlein L, Coleman R, Deen J, et al. Artesunate reduces but does not prevent posttreatment transmission of Plasmodium falciparum to Anopheles gambiae. Journal of Infectious Diseases 2001;183(8):1254‐9. [DOI] [PubMed] [Google Scholar]
White 1996
- White NJ, Olliaro PL. Strategies for the prevention of antimalarial drug resistance: rationale for combination chemotherapy for malaria. Parasitology Today 1996;12(10):399‐401. [DOI] [PubMed] [Google Scholar]
White 1999
- White NJ, Nosten F, Looareesuwan S, Watkins WM, Marsh K, Snow RW, et al. Averting a malaria disaster. The Lancet 1999;353(9168):1965‐7. [DOI] [PubMed] [Google Scholar]
White 2002
- White NJ. The assessment of antimalarial drug efficacy. Trends in Parasitology 2002;18(10):458‐64. [DOI] [PubMed] [Google Scholar]
WHO 2010
- WHO Global Malaria Programme. Guidelines for the treatment of malaria. Geneva: World Health Organization, 2010. [Google Scholar]
WHO 2012
- WHO Global Malaria Programme. World Malaria Report 2012. World Malaria Report 2012. Geneva: World Health Organization, 2012. [Google Scholar]
WWARN 2013
- WorldWide Antimalarial Resistance Network (WWARN). WWARN. http://www.wwarn.org/about‐us/our‐work 2013.
WWARN 2013b
- WWARN DP Study Group. The effect of Dosing Regimens on the Antimalarial Efficacy of Dihydroartemisinin‐Piperaquine: A pooled analysis of Individual Patient Data. PLoS Medicine 2013;10(12):e1001564. [DOI] [PMC free article] [PubMed] [Google Scholar]