

Abstract
Staphylococcus aureus infections exert a tremendous burden on the health-care system, and the threat of drug-resistant strains continues to grow. The bacteriolytic enzyme lysostaphin is a potent antistaphylococcal agent with proven efficacy against both drug-sensitive and drug-resistant strains; however, the enzyme’s own bacterial origins cause undesirable immunogenicity and pose a barrier to clinical translation. Here, we deimmunized lysostaphin using a computationally guided process that optimizes sets of mutations to delete immunogenic T cell epitopes without disrupting protein function. In vitro analyses showed the methods to be both efficient and effective, producing seven different deimmunized designs exhibiting high function and reduced immunogenic potential. Two deimmunized candidates elicited greatly suppressed proliferative responses in splenocytes from humanized mice, while at the same time the variants maintained wild-type efficacy in a staphylococcal pneumonia model. Overall, the deimmunized enzymes represent promising leads in the battle against S. aureus.
Introduction
Staphylococcus aureus causes a range of potentially life-threatening infections including skin lesions,1 lung infections,2 bacteremia,3 ocular infections, and endocarditis.4 In 2009 alone, there were 697,248 S. aureus associated hospitalizations, a full two-third of which were caused by methicillin-resistant S. aureus (MRSA).5 MRSA causes more than 11,000 deaths in the United States each year,6 and it is now common among both hospital-associated and community-associated infections. A 2012 estimate puts the cost of community associated MRSA alone at $1–13 billion annually.7 In addition to the high clinical incidence of MRSA,8 S. aureus resistance to other antibiotics is on the rise.9 The enormous financial and human costs of drug-resistant S. aureus infections has prompted both the CDC and WHO to highlight this threat and call for new treatment options.6,10 However, technical constraints and economic concerns have stymied antibiotic development pipelines. Moreover, seven decades of experience has shown that bacterial pathogens begin developing resistance to new chemotherapies within a few years of their initial use.11 Therefore, addressing this medical need in a comprehensive fashion will require more careful antibiotic stewardship combined with a search for novel and more robust therapeutic agents.
As opposed to further elaboration of small molecule chemotherapies, there is mounting evidence that bacteriolytic enzymes represent a powerful new class of antibacterial agents.12–14 Staphylococcus simulans lysostaphin (LST) is a highly effective, two-domain antistaphylococcal bacteriocin that efficiently kills both drug-sensitive and drug-resistant S. aureus. LST efficacy has been demonstrated in a wide range of in vitro studies, in vivo studies, and even human trials.15 Because of its own bacterial origins, however, LST drives a potent immune response in animals and humans. LST-antibody immune complexes are thought to cause toxicity in numerous models, including nonhuman primates.15 Thus, immunogenicity represents a serious barrier to LST clinical translation, and developing deimmunized variants would reduce associated health risks, facilitate clinical testing and approval, and ultimately enable repeated use in individuals requiring multiple treatment cycles.
To address the issue of LST immunogenicity, we sought to capitalize on a detailed understanding of cellular and molecular processes that underlie development of antidrug antibodies. Immune surveillance of biotherapeutics begins with internalization of a protein drug by antigen-presenting cells. These cells process the protein into short peptide fragments, putative immunogenic sequences are bound by class II major histocompatibility complex proteins (MHC II, also known as HLA in humans), and the complexes are trafficked to the cell surface where they are interrogated by CD4+ helper T cells.16 Bona fide immunogenic peptides, termed T cell epitopes, mediate formation of ternary MHC-peptide-T cell receptor complexes. The result of this cell–cell interaction is a complex signaling cascade that drives T cell stimulation, B cell maturation, and ultimately production of class-switched IgG antibodies that bind the offending biotherapeutic with high specificity and affinity.17 Formation of antidrug antibodies may therefore be circumvented by engineering proteins so as to prevent molecular recognition of constituent peptides, a process termed T cell epitope deletion.18,19
LST is a two domain protein, with an N-terminal glycylglycine zinc endopeptidase domain (LSTCAT) and a C-terminal SH3-like cell wall binding domain (LSTBD). This modular architecture, which is a common feature of many lytic enzymes,13,20 enables mix and match construction of chimeric cell wall hydrolases from natural catalytic and cell wall binding domains.21–26 Prompted by the modular nature of staphylolytic enzymes, we elected to pursue aggressive deimmunization of the LSTCAT domain only, leaving the native LSTBD domain intact. We anticipated that a globally deimmunized catalytic domain would have broad utility as a fusion partner to any number of different bacterial targeting moieties, including both native and engineered cell wall binding domains.
In order to deimmunize LSTCAT, we applied the EpiSweep protein design algorithm27 to simultaneously optimize variants for two competing objective functions: low immunogenic potential (assessed via predicted T cell epitope content) and retention of molecular fitness (assessed via molecular mechanics energies). Variants whose predicted immunogenicities and molecular energies are not simultaneously dominated by any other single enzyme represent the Pareto optimal frontier of the protein design space.28 EpiSweep generates all such Pareto optimal variants along with successively sub-optimal designs,27 thereby enabling the selection of diverse candidates meriting experimental evaluation. A small panel of engineered LSTCAT designs was selected, produced, and evaluated using both in vitro and in vivo assays of molecular function and immunogenicity. The results demonstrate efficient and effective production of deimmunized yet highly functional LST variants, some of which may ultimately prove useful in combatting MRSA and other dangerous S. aureus infections.
Results
A predictive analysis identified putative T cell epitopes clustered throughout the LSTCAT sequence (Figure 1). To address this immunogenicity risk, deimmunized LST variants were rapidly generated in a closed-loop engineering process that tightly coupled computational design and experimental analysis. Stage one narrowed the pool of prospective deimmunizing mutations to a functionally validated subset (Figure 2, left), and stage two integrated these individual mutations into globally deimmunized LSTCAT designs (Figure 2, right). In a subsequent third stage, the most promising lead candidates were more rigorously characterized, including assessment of in vivo efficacy and immunogenicity in humanized mice. Detailed results from each stage of the process follow.
Figure 1.

Epitope map of wild-type LSTCAT catalytic domain and positions of deimmunizing mutations. A map of predicted epitopes in wild type LSTCAT is shown as a bar plot, where amino acid positions are indicated on the x-axis and the number of MHC II alleles predicted to bind a given nonamer peptide are shown on the y-axis. Epitopes are indicated by bars at the first residue of the nonamer peptide. Epitopes cluster into five distinct regions shown as purple, blue, green, orange, and red bars, respectively. The positions of deimmunizing mutations are indicated with black triangles on the x-axis, and wild type residues are noted in red text. The mutational compositions of the seven characterized variants are shown below in black text, and the fraction of putative epitopes deleted by each mutation is indicated in parenthesis next to the mutant residue (i.e., “3/10” indicates deletion of 3 out of 10 putative epitopes in that cluster).
Figure 2.

Two stage computationally-driven development of LSTCAT variants: Stage 1-left; stage 2-right. A structural map of putative LSTCAT epitope content bridges the Design Priors for both stages. Thick red tubes indicate dense, overlapping MHC II binding peptides, and thin white tubes indicate no predicted MHC II binding. (a) Priors for the initial design calculations identified allowed mutations based on their frequencies in a multiple sequence alignment of LSTCAT homologs, represented here as a logo plot. (b) Initial EpiSweep calculations generated an undominated Pareto optimal frontier, and near-optimal frontiers, at each mutational load. Mutational loads are: red = 8, blue = 7, purple = 6, green = 5, orange = 4, dark blue = 3, yellow = 2, black = wild type. X-axis is reduction in predicted epitope content and y-axis is computed molecular energy in arbitrary units, with wild type reference at 0. (c, top) Characterization of 15 individual point mutations that appeared frequently in designs from b. Expression level is shown in blue on left y-axis and specific lytic activity towards S. aureus in green on the right y-axis. (c, bottom) Predicted number of epitopes deleted by each point mutation. Validated substitutions selected for stage 2 protein design are highlighted in green boxes. (d) Priors for stage 2 design calculations were constrained to twelve specific mutations validated in c. Mutation S122D was included in all designs. (e) Final EpiSweep calculations generated a new set of Pareto optimal and near-optimal designs at mutational loads of 2–8 per enzyme, and fourteen candidates were selected for experimental analysis (indicated as filled diamonds). (f, top) Functional characterization of seven high expressing constructs. Apparent melting temperature is shown in purple on the left y-axis, and specific lytic activity in green on the right y-axis. (f, bottom) Measured MHC II binding for the seven engineered variants. Five corresponding constituent peptides from each LST design were evaluated for binding to eight different human MHC II proteins. Peptides were categorized as strong – red (IC50 < 0.1 µmol/l), moderate – orange (0.1 µmol/l ≤ IC50 < 1 µmol/l), weak – yellow (1 µmol/l ≤ IC50 < 10 µmol/l), or nonbinders (IC50 ≥ 10 µmol/l), and total counts of binders are shown for each variant. Variants F4a and F8a, highlighted in green boxes, were chosen for further analysis.
Stage 1: design and deimmunization priors
At the time of these studies, there was no available LSTCAT crystal or NMR structure, and a homology model was therefore constructed using the crystal structure of the homologous enzyme LytM29 (Figure 2a,d inset). Overall, the LSTCAT and LytM sequences aligned closely, and there was a well-matched loop model for the one small region of LSTCAT that lacked a template segment in the LytM structure. As a result, the energy minimized model appeared to be of very high quality, attaining a QMEAN score of 0.63 (within the 0.6–0.7 range typical of high resolution crystal structures).30
Computational methods, such as the EpiMatrix epitope predictor31 used here, enable efficient analysis of epitopes in the wild-type sequence, as well as optimization of possible mutations for epitope deletion. The wild type LSTCAT domain, previously engineered to delete an N-linked glycosylation sequon,32 was predicted to contain 21 nonamer peptides having 50 cumulative MHC II binding interactions with the eight most common MHC II alleles (Figure 1). The majority of putative epitopes clustered into five distinct regions (named C1WT to C5WT), two of which exhibited dense, overlapping, and highly promiscuous MHC II binding (Supplementary Figure S1, clusters C2WT and C5WT). These epitope hotspots were distributed throughout the LSTCAT molecular structure (Figure 2a,d inset).
In stage 1, prospective deimmunizing mutations were limited to a set of 36 amino acid substitutions at 20 different residue positions. The allowed mutations were identified based on frequency in a set of diverse LSTCAT homologs (Figure 2a), constraining that subset to avoid known detrimental mutations, avoid active site residues, and require deletion of at least one putative epitope.
Initial computational designs
Based on the deimmunization priors, EpiSweep generated a total of 1,663 designs having two to eight mutations each. The protein plans mapped a Pareto optimal frontier in the two-dimensional energy score versus epitope score design space, and the design set also included 19 near-optimal frontiers at each mutational load (Figure 2b). As seen in the graphic, increased aggressiveness in deleting epitopes (lower epitope score) requires an increased energetic penalty higher energy score), though many of the designs have better predicted energy than wild type, and no penalty is particularly severe. As is typically observed, higher mutational loads (left most sets of frontiers) leverage combinations of mutations to achieve a desired reduction in epitope content, and relative to aggressive single mutations, these combinations incur a smaller energetic penalty. Conversely, to achieve a given epitope score at lower mutational loads, each mutation must delete a larger relative number of epitopes, and these more aggressive substitutions tend to have a greater energetic cost. Compare, for example, the energies of designs having identical epitope scores but four mutations (in orange) versus eight mutations (in red) (Figure 2b).
Functional analysis of point mutants
While the ultimate goal was production of integrated multi-mutation variants, we first sought to quickly identify and eliminate detrimental mutations that individually undermined expression or activity. The 15 mutations appearing most frequently among the initial designs were individually introduced into the aglycosylated wild-type LST backbone.32 Mutant genes were cloned into Pichia pastoris, expressed on a small scale, and LST activities and expression levels were determined by analysis of crude culture supernatants. Based on densitometry of polyacrylamide gels (Supplementary Figure S2), 10 variants had expression yields greater than or equal to 90% wild type, with only 2 variants exhibiting less than 70% wild type expression (Figure 2c, variants R118T = 47% and S124Y = 35%). Lytic activities towards S. aureus strain SA113 were quantified using a fluorometric kinetic assay, and rates were normalized to estimated expression levels. Thirteen of 15 variants had estimated specific activities equal to or greater than wild type (Figure 2c). The two exceptions, variants F38G and S124Y, experienced 30 and 84% rate reductions, respectively.
Stage 2: deimmunization priors
According to the stage 1 functional analysis (Figure 2c), F38G and S124Y were deemed detrimental and excluded from future designs. Mutation S122G, although fully functional, was also excluded, as it targeted the same epitope cluster as S122D but did so less effectively (deleting only 3 putative epitopes as opposed to a net of 10, respectively, Figure 2c and Supplementary Figure S1). Given the fact that S122D was a particularly efficient epitope silencer (Figure 2c and Supplementary Figure S1), it was specifically incorporated into all stage 2 plans. Thus stage 2 design considered 11 prospective mutations (in addition to S122D): N12G, N13H, I41E, K46H, L83M, Y93H, K95E, I99Q, R118T, N121G, and S124G (Figure 2d).
Lead computational designs
Based on this more restricted set of prospective deimmunizing mutations, EpiSweep was again implemented to generate Pareto optimal frontiers and near-optimal frontiers. It optimized a total of 1,400 variants with mutational loads from two to eight (Figure 2e). With the reduced pool of candidate mutations, there was less distinction between the curves for different mutational loads, but the same trade-off between deimmunizing aggressiveness and energetic penalty was observed.
Prior to candidate selection, each of the designs was subjected to global energy minimization, allowing both side chains and backbone atoms to relax. Fourteen low energy designs at mutational loads of two to eight were selected for experimental analysis (Supplementary Table S1). Among selected designs, predicted epitope scores ranged from 36 to 24, corresponding to a 28–52% reduction in epitope content relative to wild type (epitope score = 50).
Functional analysis of globally deimmunized variants
The 14 chosen designs were cloned, expressed, purified, and subjected to preliminary analysis (Supplementary Table S1). Seven of the 14 variants expressed at reasonable levels (≥30% wild type), and apparent melting temperatures for these enzymes ranged from 89 to 99% that of wild-type LST (Figure 2f). The activity of the seven designs was assessed using a fluorometric kinetic assay of bacterial lysis, and all variants maintained specific lytic rates greater than 50% that of wild type (Figure 2f). Overall, the engineered designs exhibited good thermostability and high levels of antibacterial activity, despite having as many as eight mutations in the catalytic domain. Interestingly, the seven designs that initially failed to express all encoded the R118T mutation, which by itself had resulted in a 50% reduction in expression levels (Figure 2c). Reversion of this single mutation restored expression in all seven associated designs, and the seven reverted variants were all found to have good activity and stability (Supplementary Table S1). Thus, 80% of the prospective deimmunizing mutations selected by EpiSweep in stage 1 produced stable and functional enzymes when combined as integrated designs, although preanalysis as individual point mutations was a key step in rapidly eliminating deleterious substitutions (F38G, S124Y, and in hindsight, R118T).
Immunoreactivity by peptide-MHC II binding assays
To deimmunize a biotherapeutic agent, the EpiSweep algorithm seeks to disrupt class II MHC binding of peptide fragments derived from the protein. As a measure of success, competition binding assays33 were performed with recombinant human MHC II, known peptide antigens, and LSTCAT peptide fragments. Peptides representing wild-type LSTCAT epitope clusters (Supplementary Figure S1) were synthesized, as were cognate peptides derived from each of the seven successfully expressed designs from Supplementary Table S1. Peptide affinities for MHC II proteins were categorized as either strong (IC50 < 0.1 μmol/l), moderate (0.1 μmol/l ≤ IC50 < 1 μmol/l), weak (1 μmol/l ≤ IC50 < 10 μmol/l), or nonbinders (IC50 ≥ 10 μmol/l). High-affinity interaction between peptide antigens and class II MHC is a key determinant of subsequent T cell immunogenicity,34–36 and all five wild-type LST peptides exhibited submicromolar IC50’s for at least one human MHC II protein (Supplementary Table S2). Peptides C3WT, C4WT, and C5WT proved to be promiscuous tight binders, each exhibiting high-affinity interactions with three to four MHC II alleles.
The peptide-MHC II binding analysis included a total 26 peptides and eight MHC II alleles yielding 208 separate affinity determinations. Of the 168 pairs of wild-type and cognate deimmunized affinities, there were 63 instances in which the deimmunizing mutations reduced affinity by greater than 10-fold and/or ablated binding entirely (i.e., IC50 > 250 μmol/l, Supplementary Figure S3). There were another 13 cases in which the mutation reduced binding by 5- to 10-fold. In contrast, there were only 14 examples of >10-fold enhanced binding by a variant peptide, and notably seven of these were instances in which the corresponding wild-type peptide was already a high-affinity binder (Supplementary Table S2, C2WT variants and C5WT variants with allele 0101). To correlate the experimentally measured MHC II affinities with the algorithm’s binary prediction of peptide binding/nonbinding, an experimental binding threshold was set at IC50 ≤ 10 μmol/l, i.e., counting all strong, moderate, and weak binders as defined above. Given this experimental threshold and an EpiMatrix prediction threshold of 5%, the protein design process yielded a 69% positive prediction rate across all eight alleles (Supplementary Table S2). Overall, we observed a 10% false-positive rate and a 21% false-negative rate, similar to those we have reported previously.37–39 At the level of individual peptides, EpiSweep efficiently identified both high affinity MHC-binders and disruptive mutations, realizing a 37.5% success rate in ablating binding and/or reducing affinity by more than 10-fold.
For each of the stage 2 variants that expressed as originally designed, a categorical immunoreactivity score was obtained by summing the number of strong, moderate, and weak MHC binders among each protein’s component peptides (5 peptides • 8 MHC alleles = 40 possible interactions per protein). Wild type LSTCAT possessed 11 strong, 4 moderate, and 4 weak binding interactions. As predicted by the Episcore design parameter (Supplementary Table S1), immunoreactivity generally decreased across the panel of variants from F2 to F8 (Figure 2f). Variant F8a in particular exhibited dramatically reduced immunoreactivity, disrupting 10 of 11 strong binders from wild-type LST. At a more moderate mutational load, variant F4a eliminated three strong binders and manifested an overall reduction of five binding interactions. Given their strong performance during preliminary in vitro characterization, these two deimmunized candidates were selected for more detailed functional and immunogenicity analysis.
Stage 3: stability and antibacterial activity of lead candidates
Variants F4a and F8a were expressed, purified, and characterized in biological duplicate. In addition to our own recombinant wild type LST,32 LST was obtained from a commercial supplier and analyzed in parallel. The apparent melting temperatures of both F4a and F8a were consistent with values obtained during preliminary testing, but their specific rates of bacterial lysis were found to be somewhat higher upon more rigorous analysis (Table 1). Importantly, the deimmunized variants were equivalent to or better than commercially sourced LST in both assays. The enzymes’ antibacterial activities were further quantified by assessing minimal inhibitory concentration (MIC) towards four strains of S. aureus. The F4a MIC for strain SA113 was equivalent to that of wild type and commercial LST, and it was within a single 2-fold serial dilution for three clinical isolates, including MRSA strain 3425-3. Variant F8a also retained good bactericidal/bacteriostatic activity, preventing outgrowth of all four strains at 200 ng/ml (~7 nmol/l) or less. Given the fact that the LSTCAT variants encoded four or eight mutations, respectively, their high levels of anti-staphylococcal activity were striking.
Table 1. Detailed performance analysis of deimmunized LST designsa.
| |
MIC (µg/ml) |
|||||
|---|---|---|---|---|---|---|
| Design ID | %WT lytic rate | Tm (°C) | Strain 1 SA113 | Strain 2 6445 | Strain 3 3425-1 | Strain 4 3425-3 |
| Wild type | 100 ± 30 | 59.0 ± 0.4 | 0.02 ± 0.00 | 0.03 ± 0.01 | 0.04 ± 0.02 | 0.03 ± 0.02 |
| Commercial | 60 ± 20 | 47.3 ± 0.4 | 0.03 ± 0.00 | 0.03 ± 0.00 | 0.04 ± 0.01 | 0.03 ± 0.00 |
| F4a | 70 ± 30 | 55.8 ± 0.2 | 0.02 ± 0.01 | 0.06 ± 0.04 | 0.11 ± 0.09 | 0.05 ± 0.03 |
| F8a | 60 ± 20 | 52.8 ± 0.3 | 0.04 ± 0.01 | 0.13 ± 0.07 | 0.2 ± 0.1 | 0.2 ± 0.1 |
LST, Staphylococcus simulans lysostaphin; MIC, minimal inhibitory concentration.
Errors are standard deviation from a minimum of biological duplicates measured in triplicate.
In vivo efficacy and immunogenicity of lead candidates
To assess antibacterial activities in a more clinically relevant fashion, we employed a murine lung infection model using an S. aureus clinical isolate. Mice were infected with live bacteria via oropharyngeal aspiration, and one hour later they were treated via the same route with a solution containing 2.5 μg of wild type LST, variant F4a, or variant F8a. Twenty-four hours post infection, mice were sacrificed, lungs were harvested, and viable bacterial counts were determined by plating serial dilutions of lung homogenate. All three enzymes yielded a statistically significant 10-fold reduction in bacterial burden relative to a saline buffer control (one-way analysis of variance P = 0.007, Tukey post hoc test), but there was no significant difference between the three treatments (Figure 3a). Thus, the deimmunized candidates retained wild type efficacy in the infected and inflamed lung environment.
Figure 3.
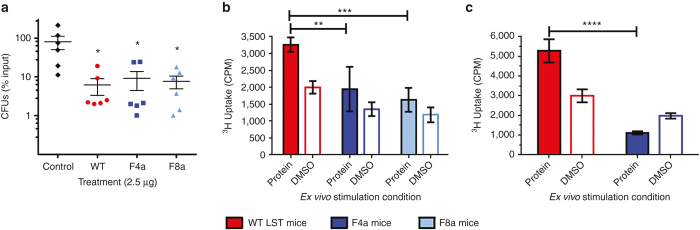
In vivo efficacy and immunogenicity analysis. (a) Bacterial burden in the lungs of C57Bl/6 mice following infection with S. aureus and treatment with wild type LST (red), variant F4a (dark blue), variant F8a (light blue), or a PBS control (black). Shown are mean values and standard deviations (n = 6 per group). (b) BLT mice (all humanized from a single donor) were immunized subcutaneously with either wild type LST, variant F4a, or variant F8a, and splenocytes were harvested and restimulated ex vivo with the same protein or DMSO. Proliferation was measured as uptake of tritiated thymidine. Shown are mean values and standard deviations (n = 4 per group, pooled and measured in triplicate). (c) Transgenic DR4 mice were immunized with multiple subcutaneous injections of wild-type LST. Following the final boost, mice were allowed to recover for 20 weeks, divided into two groups, and rechallenged with either wild type LST or variant F4a. Splenocytes were harvested and restimulated ex vivo with the rechallenge protein or DMSO, and proliferation was measured as uptake of tritiated thymidine. Shown are mean values and standard deviations (n = 5 per group, pooled and measured in triplicate). Statistical significance was assessed by one-way analysis of variance (panel a) or two-way analysis of variance (panels b and c). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In vivo immunogenicity was evaluated using NOD/SCID/γc-/- mice that had been surgically humanized with human immune cells, liver tissue, and thymus tissue (BLT mice). Following transplantation of human tissues at 6 weeks of age, BLT mice were allowed to mature and develop circulating repertoires of human B and T cells. At 14 weeks posttransplantation, mice were divided into three groups of four each and immunized subcutaneously with 100 ug of wild-type LST, F4a, or F8a in adjuvant. Thirteen days post immunization, mice were sacrificed, splenocytes were harvested and pooled for each group, and the pooled cells were subjected to ex vivo restimulation with their cognate proteins. Cell proliferation was measured by tritiated thymidine uptake at 72 hours, and the stimulation index (protein versus DMSO proliferative response) was 1.6-fold for wild-type LST (Figure 3b). While this stimulation index is lower than the twofold to threefold cutoff sometimes used in experiments with peripheral blood mononuclear cells taken directly from human subjects, it bears noting that T cells from humanized mice are widely known to exhibit impaired function. In particular, humanized mouse splenocytes yield poor ex vivo proliferative responses (e.g., twofold stimulation index) even to potent stimulatory agents such as phytohaemagglutinin, ionomycin-PMA, keyhole limpet hemocyanin, and anti-CD3/anti-CD28 antibody cocktails.40,41 Thus, the significant (P = 0.0005, two-way analysis of variance) 1.6-fold stimulation index observed here with splenocytes from mice immunized with wild type LST is a reasonable indicator of an antigen specific immune response, particularly given the fact that the mice of the current study received only a single immunization. Relative to the wild type LST immunized group, pooled splenocytes from F4a and F8a immunized mice exhibited significantly reduced proliferation (Figure 3b). After background subtraction, F4a pooled cells showed a 50% reduced response and F8a pooled cells a 65% reduced response.
In addition to inherent immunogenicity in the context of a naive immune system, we also considered the extent to which a deimmunized protein might evade an established memory response directed against the wild type LST sequence. Due to the long timeframe of such a study and the short lifespan of BLT mice, we elected to assess the memory response in transgenic DR4 mice. This homozygous strain bears a chimeric class II MHC derived from murine signaling domains and the peptide binding domains of human HLA DRA and DRB1*0401.42 This stable transgenic model has a normal, healthy lifespan enabling extended studies, yet it manifests human antigen presentation specificity. To establish an anti-LST immune response, 10 DR4 mice were immunized and repeatedly boosted with sub-cutaneous injections of wild type LST. Nineteen weeks after the final boost, mice were divided into two groups of five each such that each group exhibited similar average antibody titers (Supplementary Figure S4). Mice were then rechallenged with either 100 μg wild-type LST or 100 μg variant F4a. Thirteen days later, splenocytes were harvested, pooled for each group, and subjected to ex vivo restimulation with the cognate protein from the final rechallenge. Similar to the results in the BLT mice, ex vivo restimulation of DR4 splenocytes with wild type LST yielded a 1.8-fold stimulation index (Figure 3c). It bears noting that similarly small stimulation indices in DR2, DR3, and DQ8 transgenic mice have been shown to correlate with antigen specific immune responses.43 Thus, the significant (P = 0.0002, two-way analysis of variance) 1.8-fold stimulation index seen here was a reasonable indicator of an anti-drug immune response. In contrast to mice rechallenged with wild-type LST, pooled splenocytes from the F4a rechallenge group exhibited no detectable proliferation above background levels (Figure 3c). Thus, immune cells primed to recognize wild type LST exhibited reduced activity upon rechallenge with F4a, suggesting that the deimmunized variant effectively evaded the memory response directed against the wild type enzyme.
Discussion
LST is a potent bacteriolytic enzyme and a promising candidate for combating S. aureus infections. However, its inherent immunogenicity poses a barrier to clinical translation. Prior studies sought to shield LST from the immune system via conjugation with polyethylene glycol (PEGylation).44 While PEGylation reduced LST immunoreactivity and elongated serum half-life, it was ultimately found that covalent coupling to PEG inactivated the enzyme; residual activity in PEGylated samples was due to LST that had been noncovalently and reversibly associated with the polymer.15 Here, we have taken an alternative approach to mitigating LST immunogenicity, employing a structure-based deimmunization algorithm to design variants having reduced T cell epitope content yet high levels of antibacterial activity. Importantly, neither an LST x-ray crystal structure nor an NMR structure was available, and we therefore built a model based on the structure of the homologous enzyme LytM.29 Leveraging this molecular model and a Pareto optimal deimmunization methodology, we were able to employ high mutational loads to simultaneously redesign as many as five broadly distributed epitope clusters in the LST catalytic domain.
The most recent advances in lytic enzyme biotherapies include construction of synthetic antibacterial enzymes based on chimeragenesis of modular lysin domains.13,20–26 We anticipated that a deimmunized LST catalytic domain would prove useful as a fusion partner to a wide variety of cell wall targeting entities, and we therefore focused our deimmunization efforts on LSTCAT, leaving the LSTBD cell wall binding domain in its native form. Interestingly, we found that deimmunization of the catalytic domain was sufficient to suppress the cellular immune response against full length fusion proteins that bore the native LSTBD sequence. We therefore speculate that immunodominant LST epitopes are localized predominantly to the catalytic domain, although a rigorous test of this hypothesis awaits more detailed experimental analyses of the phenotype and function of cells that proliferate in response to various constructs. Regardless, mutations targeted to three of five (in the case of F4a) or five of five (in the case of F8a) putative epitope clusters in the catalytic domain produced marked reductions in global immunogenicity, as measured by ex vivo proliferative response of humanized murine splenocytes following in vivo immunization with full length fusion proteins. At the same time, both variants exhibited wild-type efficacy in a murine lung infection model, a highly relevant system given the prevalence of S. aureus in both hospital and community acquired pneumonias.2 Taken as a whole, these results show that the EpiSweep algorithm efficiently identified mutations and combinations of mutations that suppressed LST immunogenic potential while maintaining high level therapeutic activity. Should the enzymes’ ability to evade immune cell surveillance ultimately translate into suppressed antidrug antibody responses, one key impediment to LST clinical development will have been addressed. More generally, we expect that a similar strategy could be employed with other bacteriolytic drug candidates, many of which are known to elicit an undesirable immune response in preclinical models.13
Therapeutic enzymes’ mechanisms of action are fundamentally grounded in reaction catalysis, and the inherent complexity of their sequence-structure-function relationships may render them particularly sensitive to disruption by molecular manipulations. As a result, there are currently only six enzymes that have been successfully engineered by T cell epitope deletion.37,38,45–51 The work described here represents an important contribution to this space. First, the threat of drug-resistant bacterial infections is driving growing interest in antibacterial enzymes, and this report is the first and currently only example of a bacteriolytic biocatalyst deimmunized via T cell epitope deletion. Second, and more generally, this work serves as a preliminary validation of an efficient and effective computationally driven deimmunization process that is broadly applicable and might enable deimmunization of other challenging biotherapeutic targets. To be sure, humanized mouse models have limitations, and the success of protein deimmunization must ultimately be measured in the clinic. Nevertheless, the results of EpiSweep deimmunization efforts with both LST and P99 beta-lactamase52 suggest that the methodology holds great promise as a general tool for accelerating biotherapeutic deimmunization and development.
Materials and Methods
Reagents and media
Primers were ordered with standard desalting from IDT Technologies (Coralville, IA). Commercial lysostaphin, SYPRO Orange 5000x Protein Stain, and crystal violet dye were purchased from Sigma (St Louis, MO). All enzymes were obtained from New England BioLabs (Ipswich, MA), and all reagents from VWR Scientific (Philadelphia, PA), unless otherwise noted. Peptides derived from the LST catalytic domain were ordered from GenScript (Piscataway, NJ), and were greater than 85% pure. MHC-II DR molecules were purchased from Benaroya Research Institute (Seattle, WA), anti-MHC-II-DR antibody from Biolegend (San Diego, CA), and DELFIA Eu-labeled Streptavidin was from PerkinElmer (Boston, MA).
Murine model studies
The protocols for animal infection, immunization, and treatment were approved by the Institutional Animal Care and Use Committee of Dartmouth College (Hanover, NH), in accordance with the Association for the Assessment and Accreditation of Laboratory Animal Care Guidelines. All efforts were made to minimize animal suffering. All in vivo studies used recombinant wild type LST that had been produced and purified in house, and differed from variants F4a and F8a by only the deimmunizing mutations.
Plasmids and strains
P. pastoris strain GS115 and expression vector pPIC9 were from Invitrogen (Grand Island, NY). S. aureus strains SA113 and S. aureus subsp. aureus (ATCC 25923) were obtained from the American Type Culture Collection (Manassas, VA). Other clinical isolates of S. aureus (methicillin sensitive strains 6445 and 3425-1, and MRSA strain 3425-3) were the kind gift of Dr Ambrose Cheung (Dartmouth, Hanover, NH).
Homology modeling
An LSTCAT structural model was constructed using three LytM structures as templates (1QWY:A, 2B0P:A, and 2B44:A). Two hundred and fifty models were constructed using MODELLER (Ver. 9.11; ref. 53) and the best model was selected according to the DOPE potential.54 The templateless region at 24 PLGINGG was separately modeled using FREAD,55 which identified 180 PRGEEGG in 1GMN:A as the best match. The modeled loop structure was grafted on, and the entire model was minimized using TINKER56 against AMBER99sb57 with GB/SA.58 The aglycosylated wild type (125 NPT) was modeled by applying in silico mutations to the original model using Scwrl4,59 followed by energy re-minimization.
Epitope prediction
Epitopes were predicted by the EpiMatrix pocket profile method.31 Considered epitopes were limited to those in the top 5% of predicted binders against MHC II alleles DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301, and 1501, which are broadly representative of global populations.36
Stage 1 allowed mutations
A total of 10,000 homologs to LSTCAT were collected by running PSI-BLAST60 (3 iterations, e-value <0.001). These sequences were filtered to remove those with >50% gaps or <35% sequence identity to the wild-type. A diverse set of 218 representative sequences was subselected so as to have at most 90% sequence identity to each other. Allowed mutations were those predicted to delete at least one putative epitope while appearing as frequently as expected in terms of a background probability distribution.61 Additional filters excluded mutations to/from Pro and Cys, mutations involving active site residues (32H, 36D, 82H, 113H, and 115H), and mutations previously found to be detrimental (T43D, S50D, N121D, and L135S).
Computational deimmunization
EpiSweep integrates epitope prediction and molecular mechanics energy evaluation in order to simultaneously reduce epitope content while maintaining stability and function. It employs an optimization algorithm that identifies all designs making optimal trade-offs between these criteria (the Pareto frontier) at a specified mutational load; it can be iterated to identify successively near-optimal frontiers. Here, EpiSweep was applied as previously described,27 though using the EpiMatrix epitope predictor as discussed above. Mutations were restricted to those specified in the deimmunization prior, though all side-chains were allowed to change rotameric conformation in the search for low energy enzyme designs predicted to maintain the parental enzyme’s antibacterial activity. Molecular mechanics energies (a proxy for stability and thereby maintenance of function) were computed according to AMBER62 as implemented under the open source protein design software OSPREY.63 For each stage and each mutational load from 2 to 8, the Pareto frontier designs were supplemented with designs comprising 19 successive near-optimal frontiers.
Stage 2 lead variants were selected for a combination of factors including mutational load, minimized energy, and epitope score. All designs were energy minimized following the same protocol described in Homology Modeling section (all-atom minimization under AMBER99sb with GB/SA via the Tinker package). Designs were chosen across the epitope score range from 24 to 36 at intervals of 2 (compared to wild-type at 50). A total of 14 designs were selected for experimental analysis (Supplementary Table S1).
Cloning of LST single point mutants
Genes encoding LST single point mutants were synthesized by splice overlap extension PCR with synthetic oligonucleotides (Supplementary Table S3) and an optimized synthetic gene encoding the S126P aglycosylated protein backbone.32 All PCR reactions were performed using Phusion® High-Fidelity DNA polymerase. The lysostaphin gene harboring the desired mutations was digested with EcoRI and XhoI, and ligated into the pPIC9 plasmid using T4 DNA ligase. The end product of ligation was the lysostaphin gene fused to the alpha mating factor secretion signal from Saccharomyces cerevisiae. The plasmid was transformed into E. coli DH5α electrocompetent cells [F- ɸ80lacZΔM15Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 phoA supE44 λ- thi-1 gyrA96 relA1]. Clones were evaluated for the presence of lysostaphin gene using Syn_F and Syn_R primers and sent for DNA sequencing to confirm the presence of mutations.
Cloning of LST integrated designs
Lysostaphin variants were synthesized with standard desalting as 432 bp long gBlocks® gene fragments by IDT Technologies (Coralville, IA). To prepare the pPIC9 plasmid for insertion of synthesized genes, a silent mutation was introduced into the lysostaphin linker (residues 135–136) to accommodate a cutting site for the restriction enzyme AflII. To introduce the necessary mutations, splice overlap extension PCR was used with primers AflII_F and AflII_R (Supplementary Table S3). The synthetic genes were digested with XhoI and AflII restriction enzymes, and ligated in similarly digested pPIC9 plasmid using T4 DNA ligase. The resulting plasmid was transformed into E. coli DH5α electrocompetent cells and the resulting clones were sequenced to confirm the presence of mutations.
LST expression and purification
Following sequence confirmation, purified plasmids were digested with SacI High-Fidelity restriction enzyme prior to electroporation into P. pastoris strain GS115. The resulting transformants were grown on MD plates (1.34% yeast nitrogen base (YNB), 0.000004% biotin, 2% dextrose, and 1% agar). For expression studies, P. pastoris clones were grown in 100 ml BMGY medium (1% yeast extract, 2% peptone, 1.34% YNB, 0.000004% biotin, 1% glycerol, 100 mmol/l phosphate buffer, pH 6) at 30 °C in 500 ml shake flasks covered with four layers of cheese cloth for enhanced oxygen flow to the yeast. After 24 hours, cells were centrifuged at 3,000 rpm in a table top centrifuge for 10 minutes. The cells were then resuspended in 100 ml of induction medium (BMMY: 1% yeast extract, 2% peptone, 1.34% YNB, 0.000004% biotin, 0.5% methanol, 100 mmol/l phosphate buffer, pH 6) and allowed to grow for the next 48 hours at 30 °C. At 12-hour intervals, a 1/100 v/v aliquot of 100% methanol was added to the flasks. After 48 hours of induction, shake flask culture was centrifuged in a table top centrifuge at 3,000 rpm for 15 minutes. The resulting supernatant was sterile filtered to remove any yeast cells and diluted 1:5 with 10 mmol/l KH2PO4 buffer at pH 7.5. Diluted supernatant was poured over a gravity column packed with 500 µl SP-Sepharose Fast Flow resin (GE Healthcare; Cleveland, OH). The column was washed with 5 ml of 50 mmol/l NaCl in 10 mmol/l KH2PO4 at pH 7.5. Protein was eluted with 500 µl aliquots of 200 mmol/l NaCl in 10 mmol/l KH2PO4, pH 7.5. The purity of lysostaphin was determined using SDS–PAGE. The protein concentration was quantified using ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE).
Lytic assay
S. aureus cells were grown either to mid-log or to saturation in tryptic soy broth (TSB) at 37 °C with shaking. The cells were harvested by centrifugation and washed once in phosphate buffered saline (PBS: 2.7 mmol/l KCl, 1.5 mmol/l KH2PO4, 8.9 mmol/l Na2HPO4, 136.9 mmol/l NaCl, pH 7.4). The assay was performed in 96-well, black, clear bottom plates from Greiner Bio-One (Monroe, NC). The final reaction volume (250 µl in PBS) consisted of 10 µl of P. pastoris culture supernatant (or 200 ng of purified protein), S. aureus cells at OD600 = 1.5, and 5 µmol/l SYTOX Green (Thermo Fisher Scientific; Waltham, MA). Data was collected using SpectraMax Gemini Fluorescence Microplate Reader (Molecular Devices, Sunnyvale, CA) using an excitation/emission of 504/523 nm, and rates were determined from the slope of the steepest linear portion of the trace. For the assays performed using P. pastoris culture supernatant, the amount of protein used in each assay was quantified from SDS–PAGE using Quantity Tools from Image Lab 5.1 software (Bio-Rad Laboratories, Hercules, CA) (note that in some cases values were extrapolated from the explicitly measured standard curve).
MIC assay
The MICs of wild type LST and deimmunized variants were determined by adding twofold serial dilutions of enzymes into wells of a polypropylene 96-well plate (Costar 3879) containing ~40,000 S. aureus (strains SA113, 6445, 3425-1, or 3425-3) cells in Muller Hinton II broth (BD) supplemented with 2% NaCl and 0.1% bovine serum albumin, yielding a total volume of 100 µl. Plates were grown overnight at 37 °C with shaking at 900 rpm on an Orbit P4 orbital shaker (Labnet, Edison, NJ). The inhibitory activity of purified lysostaphin was determined by the concentration of enzyme that prevented visible bacterial outgrowth. The assay was performed in triplicate for each enzyme.
Thermostability assay
The relative thermostability of the LST variants was determined by differential scanning fluorimetry, as previously described.64 Briefly, proteins and SYPRO Orange were diluted in PBS (final concentrations of 100 µg/ml and 5× in 20 µl reaction volume, respectively), and fluorescence was quantified at 1-degree increments from 25 to 94 °C using an Applied Biosystems ABI 7500 fast real-time PCR system. The reactions were performed using PCR Plates for Fast Thermocyclers (VWR, Radnor, PA). Fluorescence was quantified using the preset TAMRA parameters. Melting temperatures were determined by data analysis with the “DSF Analysis v3.0.xlsx” Excel sheet (ftp://ftp.sgc.ox.ac.uk/pub/biophysics/) and GraphPad Prism v.6.02 software (GraphPad Software, La Jolla, CA).
MHC binding assays
MHC II competition binding assays were performed using a 384-well high-throughput assay as previously described.34 Binding assays were performed for the eight alleles: DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301, and 1501. Briefly, 100 nmol/l biotinylated control peptides consisting of known peptide antigens for each MHC II allele were incubated in polypropylene 384-well plates with 50 nmol/l purified recombinant MHC II protein and serial dilutions of LST or variant peptide fragments (100 μmol/l to 10 nmol/l). Peptide-MHC II complexes were captured from equilibrated solutions using the conformation specific anti-HLA-DR antibody L243 coated on high binding ELISA plates. Bound control peptide was quantified using the DELFIA streptavidin-Europium conjugate and time resolved fluorescence (SpectraMax Gemini Fluorescence Microplate Reader).
Murine lung infection model
Overnight LB cultures of S. aureus strain ATCC 25923 were pelleted, washed twice with PBS, and resuspended to give 108–109 colony-forming units (CFU) in 40 μl of PBS. The actual inoculum was determined by serial dilution of the input bacterial suspension on LB agar (Difco), followed by incubation at 37 °C for 24 hours. Adult female C57BL/6J mice (age, 8 to 12 weeks; Jackson Laboratories, Detroit, MI) were anesthetized briefly with isoflurane and inoculated with 40 μl of bacterial suspension via oropharyngeal aspiration. At 1 hour postinfection, a second 40 μl PBS inoculation containing either 2.5 μg wild-type LST, 2.5 μg variant F4a, 2.5 μg variant F8a, or a blank control. At 24 hours postinfection, mice were sacrificed and lungs were excised, placed into 1 ml of cold PBS, and homogenized. Viable bacterial counts in the lung homogenate were determined by plating serial dilutions onto LB agar, followed by incubation at 37 °C for 24 hours.
BLT murine immunogenicity studies
BLT mice were constructed by surgical transplantation of human bone marrow, liver, and thymus tissues into NOD/SCID/γc-/- mice (Dartmouth Transgenics & Genetic Constructs Shared Resource) as described in detail elsewhere.65 All animals were humanized using tissue from a single DRB1*0101/DRB1*0701 heterozygous donor. Mice used experimentally had human lymphocytes as a minimum of 25% of their total peripheral blood leukocytes. Fourteen weeks post engraftment, 12 female BLT mice were divided into 3 groups of 4 each and immunized with a single 50 μl subcutaneous injection of 100 μg wild type LST, 100 μg variant F4a, or 100 μg variant F8a in complete Freund’s adjuvant (CFA). Two weeks following the immunization, mice were sacrificed and splenocytes were harvested and pooled for each group. Pooled splenocytes (5 × 105/well) were plated in triplicate into 96-well plates with medium containing 5% fetal calf serum, l-glutamine, antibiotics, and a final concentration of 10 μg/ml LST or variants (or 1% DMSO as a control). After 72 hours of incubation, wells were pulsed with 1 μCi of [3H]thymidine (Dupont NEN, Boston, MA) and harvested 6 hours later onto Unifilter 96-well GF/C plates for assessment of thymidine incorporation by scintillation counting (Packard MicroSant NXT counter). Serum from immunized mice was assayed by ELISA for anti-lysostaphin IgM and IgG antibody titers, but no significant anti-lysostaphin antibody signal could be detected in either wild type or variant immunized groups. This lack of a significant humoral immune response to protein antigens is a well-known limitation in BLT mice.66
Transgenic DR4 murine immunogenicity studies
Twelve female 6–8-week-old DR4 transgenic mice (Abb Knockout/Transgenic HLA-DR4; B6.129S2-H2-Ab1tm1GruTg(HLA-DRA/H2-Ea,HLA-DRB1*0401/H2-Eb)1Kito; Taconic Farms, Germantown, NY) were divided into four groups of three each and immunized with 50 μl subcutaneous injections of wild-type LST using one of the following four schemes: (i) initial immunization with 100 μg enzyme in CFA, followed by 100 μg boosts in incomplete Freund’s adjuvant (IFA) on days 14 and 28; (ii) initial immunization with 20 μg enzyme in CFA, followed by 20 μg boosts in IFA on days 14 and 28; (iii) initial immunization with 100 μg enzyme in PBS buffer, followed by 100 μg boosts in PBS buffer on days 7, 14, 21, and 28; (iv) initial immunization with 20 μg enzyme in PBS buffer, followed by 20 μg boosts in PBS buffer on days 7, 14, 21, and 28. Serum IgG antibody titers against wild-type LST were measured on days 13, 20, 27, 34, and 62. Five weeks after the final boost, all 12 mice exhibited equivalent maximum ELISA signals at a 1:40 serum dilution and all signals were within 20% at a 1:160 dilution (Supplementary Figure S5). Mice were housed without further manipulation until week 23 of the study, at which time serum IgG antibody titers were again measured and mice were divided into two experimental arms having equivalent average antibody titers (Supplementary Figure S4). Note that during the week 9 to week 23 recovery period, two mice (the lowest titer 100 μg no adjuvant and one of the high titer 100 μg adjuvant) began suffering hair loss, weight loss, and reduced mobility and were sacrificed as per the IACUC approved protocol. At week 24, one arm was rechallenged with 100 μg wild-type LST in IFA and the other arm with 100 μg variant F4a in IFC. At week 26, mice were sacrificed and splenocytes were harvested and pooled for each group. Proliferation assays were conducted as described above. Anti-lysostaphin antibody titers remained high at the end of the 19-week washout period just prior to the final restimulation (average EC50 at week 24 was between 1:2,000 and 1:4,000 serum dilution; see Supplementary Figure S4a). Thus, the memory immune response could not be readily quantified using antibody titers, and instead the ex vivo proliferative response of mouse splenocytes was used to assess the memory immune response against wild type LST and the F4a variant.
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases 1R21AI098122, the National Institute of General Medical Sciences R01-GM-098977, and a pilot grant from the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P30GM106394. Generation of humanized immune system mice was supported by the NIH IDeA program 5P30GM103415-03 and Norris Cotton Cancer Center P30 CA023108-27. HZ was supported in part by funds from the Dartmouth Cystic Fibrosis Research Development Program. RSS was supported in part by a Thayer Innovation Program Fellowship from the Thayer School of Engineering.
KEG, CB-K, YC, HZ, LM, KB, and SNF designed the studies. YC and CB-K designed the enzymes. KB, HZ, WL, RSS, DCO, BB, JF, and KEG conducted the experiments. KEG, KB, HZ, RSS, WL, BB, JF, and SNF analyzed the data. LM and SNF contributed key materials and tools. KEG, CB-K, and YC wrote the manuscript, and all other authors contributed to the writing.
Karl E. Griswold and Chris Bailey-Kellogg are Dartmouth faculty and co-members of Stealth Biologics, LLC, a Delaware biotechnology company. Leonard Moise is an employee of and holds stock options in EpiVax, a privately owned biotechnology company located in Providence, RI. These authors acknowledge that there is a potential financial conflict of interest related to their associations with these companies, and they hereby affirm that the data presented in this paper is free of any bias. This work has been reviewed and approved as specified in Karl E. Griswold’s and Chris Bailey-Kellogg’s Dartmouth conflict of interest management plans. The remaining authors declare no conflict of interest.
H.Z. and Y.C. contributed equally to this work.
References
- Pantosti A, Venditti M. What is MRSA? Eur Respir J. 2009;34:1190–1196. doi: 10.1183/09031936.00007709. [DOI] [PubMed] [Google Scholar]
- Defres S, Marwick C, Nathwani D. MRSA as a cause of lung infection including airway infection, community-acquired pneumonia and hospital-acquired pneumonia. Eur Respir J. 2009;34:1470–1476. doi: 10.1183/09031936.00122309. [DOI] [PubMed] [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- Fowler VG, Jr, Miro JM, Hoen B, Cabell CH, Abrutyn E, Rubinstein E, ICE Investigators Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA. 2005;293:3012–3021. doi: 10.1001/jama.293.24.3012. [DOI] [PubMed] [Google Scholar]
- Klein EY, Sun L, Smith DL, Laxminarayan R. The changing epidemiology of methicillin-resistant Staphylococcus aureus in the United States: a national observational study. Am J Epidemiol. 2013;177:666–674. doi: 10.1093/aje/kws273. [DOI] [PubMed] [Google Scholar]
- CDC 2013Antibiotic Resistance Threats in the United States, 2013 Centers for Disease Control and Prevention; Atlanta, GA. [Google Scholar]
- Lee BY, Singh A, David MZ, Bartsch SM, Slayton RB, Huang SS. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) Clin Microbiol Infect. 2013;19:528–536. doi: 10.1111/j.1469-0691.2012.03914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet. 2006;368:874–885. doi: 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO 2014Antimicrobial Resistance: Global Report on Surveillance, 2014 WHO; Geneva. [Google Scholar]
- Taubes G. The bacteria fight back. Science. 2008;321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- Thallinger B, Prasetyo EN, Nyanhongo GS, Guebitz GM. Antimicrobial enzymes: an emerging strategy to fight microbes and microbial biofilms. Biotechnol J. 2013;8:97–109. doi: 10.1002/biot.201200313. [DOI] [PubMed] [Google Scholar]
- Pastagia M, Schuch R, Fischetti VA, Huang DB. Lysins: the arrival of pathogen-directed anti-infectives. J Med Microbiol. 2013;62 Pt 10:1506–1516. doi: 10.1099/jmm.0.061028-0. [DOI] [PubMed] [Google Scholar]
- Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong SL.2012. Endolysins as antimicrobials. In: Lobocka, M and Szybalski, WT (eds). Advances in Virus Research, vol 83: Bacteriophages, Pt B 299–365. [DOI] [PubMed]
- Kokai-Kun JF.2012. Lysostaphin: a silver bullet for staph. In: Tegos, G (ed). Antimicrobial Drug Discovery, 1st edn CABI; Wallingford Oxfordshire, UK, pp. 147–165. [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28:482–490. doi: 10.1016/j.it.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Baker MP, Jones TD. Identification and removal of immunogenicity in therapeutic proteins. Curr Opin Drug Discov Devel. 2007;10:219–227. [PubMed] [Google Scholar]
- Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol. 2013;149:534–555. doi: 10.1016/j.clim.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Szweda P, Schielmann M, Kotlowski R, Gorczyca G, Zalewska M, Milewski S. Peptidoglycan hydrolases-potential weapons against Staphylococcus aureus. Appl Microbiol Biotechnol. 2012;96:1157–1174. doi: 10.1007/s00253-012-4484-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:1603–1612. doi: 10.1128/AAC.01625-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Schmelcher M, Harty WJ, Foster-Frey J, Donovan DM. Chimeric Ply187 endolysin kills Staphylococcus aureus more effectively than the parental enzyme. FEMS Microbiol Lett. 2013;342:30–36. doi: 10.1111/1574-6968.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovitch DC, Griswold KE. Fusion with a cell wall binding domain renders autolysin LytM a potent anti-Staphylococcus aureus agent. FEMS Microbiol Lett. 2015;362:1–7. doi: 10.1093/femsle/fnu035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob Agents Chemother. 2011;55:738–744. doi: 10.1128/AAC.00890-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rubio L, Martinez B, Donovan DM, Garcia P, Rodriguez A. Potential of the virion-associated peptidoglycan hydrolase HydH5 and its derivative fusion proteins in milk biopreservation. PLoS One. 2013;8:e54828. doi: 10.1371/journal.pone.0054828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vipra AA, Desai SN, Roy P, Patil R, Raj JM, Narasimhaswamy N. Antistaphylococcal activity of bacteriophage derived chimeric protein P128. BMC Microbiol. 2012;12:41. doi: 10.1186/1471-2180-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker AS, Choi Y, Griswold KE, Bailey-Kellogg C. Structure-guided deimmunization of therapeutic proteins. J Comput Biol. 2013;20:152–165. doi: 10.1089/cmb.2012.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Friedman AM, Bailey-Kellogg C. A divide-and-conquer approach to determine the Pareto frontier for optimization of protein engineering experiments. Proteins. 2012;80:790–806. doi: 10.1002/prot.23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firczuk M, Mucha A, Bochtler M. Crystal structures of active LytM. J Mol Biol. 2005;354:578–590. doi: 10.1016/j.jmb.2005.09.082. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer JR, Jesdale BM, George JA, Kouttab NM, De Groot AS. Prediction of well-conserved HIV-1 ligands using a matrix-based algorithm, EpiMatrix. Vaccine. 1998;16:1880–1884. doi: 10.1016/s0264-410x(98)00173-x. [DOI] [PubMed] [Google Scholar]
- Zhao H, Blazanovic K, Choi Y, Bailey-Kellogg C, Griswold KE. Gene and protein sequence optimization for high-level production of fully active and aglycosylated lysostaphin in Pichia pastoris. Appl Environ Microbiol. 2014;80:2746–2753. doi: 10.1128/AEM.03914-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvat RS, Moise L, Bailey-Kellogg C, Griswold KE. A high throughput MHC II binding assay for quantitative analysis of peptide epitopes. J Vis Exp. 2014;85:e51308. doi: 10.3791/51308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J, del Guercio MF, Southwood S, Sette A. The HLA molecules DQA1*0501/B1*0201 and DQA1*0301/B1*0302 share an extensive overlap in peptide binding specificity. J Immunol. 2002;169:5098–5108. doi: 10.4049/jimmunol.169.9.5098. [DOI] [PubMed] [Google Scholar]
- Hill JA, Wang D, Jevnikar AM, Cairns E, Bell DA. The relationship between predicted peptide-MHC class II affinity and T-cell activation in a HLA-DRbeta1*0401 transgenic mouse model. Arthritis Res Ther. 2003;5:R40–R48. doi: 10.1186/ar605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160:3363–3373. [PubMed] [Google Scholar]
- Salvat RS, Parker AS, Choi Y, Bailey-Kellogg C, Griswold KE. Mapping the Pareto optimal design space for a functionally deimmunized biotherapeutic candidate. PLoS Comput Biol. 2015;11:e1003988. doi: 10.1371/journal.pcbi.1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvat RS, Parker AS, Guilliams A, Choi Y, Bailey-Kellogg C, Griswold KE. Computationally driven deletion of broadly distributed T cell epitopes in a biotherapeutic candidate. Cell Mol Life Sci. 2014;71:4869–4880. doi: 10.1007/s00018-014-1652-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovitch DC, Parker AS, Makokha CD, Desrosiers J, Kett WC, Moise L. Design and analysis of immune-evading enzymes for ADEPT therapy. Protein Eng Des Sel. 2012;25:613–623. doi: 10.1093/protein/gzs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I. The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice) Int Immunol. 2009;21:843–858. doi: 10.1093/intimm/dxp050. [DOI] [PubMed] [Google Scholar]
- Tonomura N, Habiro K, Shimizu A, Sykes M, Yang YG. Antigen-specific human T-cell responses and T cell-dependent production of human antibodies in a humanized mouse model. Blood. 2008;111:4293–4296. doi: 10.1182/blood-2007-11-121319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Bian HJ, Molina M, Han J, Magram J, Saar E. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J Exp Med. 1996;183:2635–2644. doi: 10.1084/jem.183.6.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depil S, Angyalosi G, Moralès O, Delacre M, Delhem N, François V. Peptide-binding assays and HLA II transgenic Abeta degrees mice are consistent and complementary tools for identifying HLA II-restricted peptides. Vaccine. 2006;24:2225–2229. doi: 10.1016/j.vaccine.2005.11.048. [DOI] [PubMed] [Google Scholar]
- Walsh S, Shah A, Mond J. Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob Agents Chemother. 2003;47:554–558. doi: 10.1128/AAC.47.2.554-558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Yoo TH, Dixit A, Iverson BL, Forsthuber TG, Georgiou G. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108:1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding FA, Liu AD, Stickler M, Razo OJ, Chin R, Faravashi N. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther. 2005;4:1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005;3:991–1000. doi: 10.1111/j.1538-7836.2005.01309.x. [DOI] [PubMed] [Google Scholar]
- Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci USA. 2014;111:8571–8576. doi: 10.1073/pnas.1405153111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmerdam PA, Plaisance S, Vanderlick K, Vandervoort P, Brepoels K, Collen D. Elimination of a human T-cell region in staphylokinase by T-cell screening and computer modeling. Thromb Haemost. 2002;87:666–673. [PubMed] [Google Scholar]
- Cizeau J, Grenkow DM, Brown JG, Entwistle J, MacDonald GC. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J Immunother. 2009;32:574–584. doi: 10.1097/CJI.0b013e3181a6981c. [DOI] [PubMed] [Google Scholar]
- King C, Garza EN, Mazor R, Linehan JL, Pastan I, Pepper M. Removing T-cell epitopes with computational protein design. Proc Natl Acad Sci USA. 2014;111:8577–8582. doi: 10.1073/pnas.1321126111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvat RS, Choi Y, Bishop A, Bailey-Kellogg C, Griswold KE. Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads. Biotechnol Bioeng. 2015;112 7:1306–18. doi: 10.1002/bit.25554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2:Unit 2.9. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Deane CM. FREAD revisited: accurate loop structure prediction using a database search algorithm. Proteins. 2010;78:1431–1440. doi: 10.1002/prot.22658. [DOI] [PubMed] [Google Scholar]
- Ponder JW.2004TINKER - Software Tools for Molecular Design Washington University School of Medicine; Saint Louis, MO. . http://dasher.wustl.edu/tinker/ . [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaldon P, Argos P. Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences. Proteins. 1988;4:99–122. doi: 10.1002/prot.340040204. [DOI] [PubMed] [Google Scholar]
- Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S. Amber, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- Chen CY, Georgiev I, Anderson AC, Donald BR. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci USA. 2009;106:3764–3769. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. 2009;83:7305–7321. doi: 10.1128/JVI.02207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaudy J, Schotte R, Legrand N, Spits H. Critical assessment of human antibody generation in humanized mouse models. J Immunol Methods. 2014;410:18–27. doi: 10.1016/j.jim.2014.06.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.