

Abstract
In vivo data suggest that monocytes participate critically in cross-presentation, but other data suggest that lymph node resident dendritic cells (DCs) mainly cross-present. Here, we utilized a three-dimensional model of a blood vessel wall that endogenously supports DC development from human monocytes, and we incorporated dying autologous cells in the subendothelial matrix of the model. Flu-infected dying cells promoted monocytes to become mature DCs and cross-present cell-associated Ags for the activation of CTLs. Similar responses were induced by loading the dying cells with the TLR7/8 ligand ssRNA, whereas dying cells loaded with TLR3 ligand were less efficient. Monocyte-derived DCs that developed in this model cross-presented Ag to T cells efficiently regardless of whether they engulfed detectable amounts of labeled dying cells. Unexpectedly, the monocyte-derived cells that directly engulfed dying cells in vitro were not the major APCs stimulating CD8+ lymphocytes. Instead, bystander DCs acquired more robust capacity to cross-prime through receipt of MHC class I/peptide from the phagocytic, monocyte-derived cells. In mice, lymph node-homing monocyte-derived DCs processed Ags from engulfed cells and then transferred MHC class I/peptide complexes to confer cross-priming capacity to MHC class I-deficient lymph node resident CD8α+ DCs. Thus, natural or synthetic TLR7/8 agonists contained within dying cells promote the conversion of monocytes to DCs with capacity for cross-presentation and for “cross-dressing” other DCs. These data reveal a way in which migratory monocyte-derived DCs and other DCs, like lymph node resident DCs, both mediate cross-presentation.
Many viral Ags are best detected and combated within the immune system through cross-priming, a mechanism by which professional APCs, most notably dendritic cells (DCs),4 acquire exogenous Ags, likely often from dying parenchymal cells, for presentation through the MHC class I (MHC I) pathway to CD8+ T cells (1, 2). Monocytes readily mobilize into peripheral tissues and give rise to lymph node-homing DCs under inflammatory conditions (3), and monocytes newly recruited to tissues conditioned by various adjuvants participate critically in cross-priming (4). It has been suggested from studies in mice that monocyte-derived DCs directly primed CD8+ T cells because monocytes that selectively lacked MHC I are unable to support cross-priming (4). This observation seems inconsistent with other findings that a population of DCs in mouse lymph nodes that expresses CD8α, and hence called CD8α+ DCs, has been shown to manifest superior capacity for cross-presentation (5), and that these DCs selectively recognize and engulf dying cells (6). Because CD8α+ DCs are lymph node resident cells that do not survey peripheral tissues, they may in many instances collaborate with other DCs that mobilize from the periphery to gain access to Ag through transfer between DCs (7–10). However, whether and how monocyte-derived DCs may cooperate with these DCs, or truly cross-present Ag on their own as suggested (4), have not been fully determined. Monocyte-derived DCs cultured in GM-CSF and IL-4 for several days to generate immature DCs can cross-present influenza A Ags derived from dying cells (11). However, immature DCs derived from GM-CSF/IL-4-treated monocytes are more akin to tissue resident DCs, and they may not model the response of monocytes newly arriving at a site of viral infection. Thus, much remains to be explored regarding how newly recruited monocytes may participate in cross-presentation of viral Ags or whether newly recruited human monocytes do so at all.
To investigate the potential role of newly recruited monocytes in acquiring, processing, and presenting Ag in the periphery to support cross-priming, we took advantage of an in vitro model of a simple connective tissue that consists of an endothelium grown on a matrix of type I collagen. In this model, freshly isolated human monocytes readily transmigrate across the endothelium to differentiate into macrophages that remain within the subendothelial matrix and DCs that retraverse, or reverse transmigrate, across the endothelial layer (12). Within the collagen, we grew cell lines that were autologous to the transmigrating monocytes; these cell lines were induced to undergo apoptosis and in some cases were infected with influenza A virus (flu) as a model virus or were loaded with synthetic TLR ligands that mimic viral stimuli to evaluate how these signals delivered to monocytes through dying cells might influence cross-presentation. We show that, in particular, TLR7/8 ligands (ssRNA) present within dying cells strongly promote monocytes to become cross-priming APCs. However, monocyte-derived DCs that engulfed Ag-bearing dying cells were surprisingly poor stimulators of CTLs themselves, but they supported cross-priming by acting as efficient suppliers of Ag to other DCs. Subsequent in vivo studies in mice then indicated that migratory monocyte-derived DCs supplied intact MHC I/peptide to lymph node DCs, allowing MHC I-deficient CD8α+ DCs to cross-prime CD8+ T cells.
Materials and Methods
Lymphoblastoid cell lines (LCLs)
EBV-transformed B cells, or LCLs, were generated as described (13). A suspension of LCLs in RPMI 1640 containing 20% FBS was mixed with bovine type I collagen (Inamed), a 10× stock of M199 (Lonza), 0.05 N NaOH, and HEPES buffer at relative volumes of 2.5:8:1:2.5, respectively. To monitor phagocytosis, LCLs were labeled with 5 µmol/L CFSE (Molecular Probes) for 15 min at 37°C. The mixture of collagen and cells at a volume of 50 µl was dispensed into microtiter wells and allowed to polymerize. The following day, the LCL/collagen constructs were irradiated at 600 rad. Then HUVECs were applied atop the collagen (30 × 104 HUVECs per well), a density that allowed them to rapidly achieve confluence.
Plasmids pCAGGS-M (encoding influenza MP) and pCAGGS-GFP were kindly given by Dr. Garcia-Sastre (Mount Sinai School of Medicine). To produce LCL lines that stably expressed MP, plasmids were linearized with Drd I and mixed at a 2:1 ratio of pCAGGS-M to pCAGGS-GFP and used to transfect LCLs. One LCL line was derived from an HLA-A201+ donor, and another was from an HLA-A201− donor. HLA-A201 was typed as previously described (14). The cultured cells were sorted to select those that were GFP+. MP expression was confirmed by immunoblot using mouse MP Ab (Serotec).
Virus infection and poly(I:C) or ssRNA loading of LCLs
In some experiments, PBMCs were directly infected with the influenza A PR/8 virus before addition to endothelium, using a multiplicity of infection (MOI) of 0.5, as described (15). LCL were infected (MOI = 25) with live PR/8 in serum-free medium for 1 h. Free virus was removed by centrifugation and diluted in 10% human serum. The infected cells were cultured in this medium for 1 day, then mixed with collagen and plated in microtiter wells. The amount of poly(I:C) or ssRNA loaded into cells was estimated as described previously (16), using fluorescence detection rather than radiolabeling.
Flow cytometry and immunofluorescence
Abs used for flow cytometric staining included mAbs to CD3, CD4, CD8, CD14, CD16, CD86, and HLA-DR (BD Pharmingen); CCR7 (R&D Systems); anti-hemagglutinin (HA) (gift from Dr. Thomas Moran, Mount Sinai School of Medicine); anti-granzyme B (eBioscience); and mAb UPC10 as isotype-matched control (Sigma-Aldrich). Detection reagents included FITC-conjugated or PE-conjugated rabbit anti-mouse Ig (Dako) and streptavidin-allophycocyanin (Caltag Laboratories). Nucleoprotein was stained with FITC-labeled anti-nucleoprotein mouse mAb (United States Biological) (15). To confirm and quantify apoptosis, the LCLs incorporated in collagen were retrieved from the collagen with collagenase D and stained with biotin-conjugated anti-annexin V (Sigma-Aldrich), followed by detection with streptavidin-allophycocyanin (Caltag Laboratories). Propidium iodide was added before data acquisition.
Transendothelial migration and reverse transmigration assays
Adult peripheral blood was collected according to guidelines approved by the Institutional Review Board of Mount Sinai School of Medicine and subjected to density gradient centrifugation on Ficoll. Transmigration assays were conducted based on previously described methods (12). In brief, collagen with or without LCLs was polymerized as described above and HUVECs were applied and cultured in M199 containing 20% FBS until confluent (2–3 days) before addition of PBMCs. For experiments, whole PBMCs or monocytes purified by use of the “untouched monocytes” isolation kit from Miltenyi Biotec were applied to the endothelium, incubated for 1.5–2 h, then washed thoroughly in medium to remove nontransmigrated cells from above the endothelium, and continued in culture until specified. After this wash, some wells received 5 µg/ml ssRNA-DR/LyoVec (InvivoGen), 10 µg/ml poly(I:C) (Amersham), or both in combination. Reverse-transmigrated cells were collected from the above endothelium by gently pipetting in the presence of 1 mM EGTA (12).
Transfer of Ag and coculture of DCs
Monocytes were purified using CD14 microbeads (Miltenyi Biotec) and cultured in GM-CSF (50 ng/ml) and IL-4 (34 ng/ml) (PeproTech) for 6 days. They were labeled with 2 µM CFSE and matured in the presence of 10 ng/ml TNF-α, 10 ng/ml IL-1β, 1000 IU IL-6 (PeproTech), and 0.5 µg/ml PGE2 (Sigma-Aldrich) for 2 days as reported (17).
HLA-A201+ monocyte-derived reverse-transmigrated and subendothelial cells were generated as described above. Subendothelial cells were positively selected by using CD14 microbeads after digestion of the collagen. Both reverse-transmigrated and subendothelial cells were labeled with HLA-DR-allophycocyanin and mixed with the CFSE-labeled mature DCs from the same or another donor (HLA-A201−) at the ratio of 1:1 overnight (18). Then the CFSE+ mature DCs and allophycocyanin-positive, CFSE-negative reverse-transmigrated or subendothelial cells were sorted into separate tubes, to be tested as APCs for induction of CTLs. These sorted cells were cocultured with autologous T cells at the ratio of 1 APC to 50 T cells for 6 days, and ELISPOT for IFN-γ-producing CTLs was conducted as described below. To assess transfer of MHC I/peptide by FACS, matured DCs and reverse-transmigrated cells were cocultured overnight and stained with mouse anti-HLA-A201 (clone BB7.2 from Abcam). Mouse IgG2b (R&D Systems) was used as an isotype control, and PE-conjugated goat anti-mouse IgG (Dako) was used as the secondary Ab.
IFN-γ ELISPOT assay
Whole PBMCs or “untouched monocytes” from HLA-A201+ donors were cocultured with LCLs in the endothelial/collagen model. Bulk T cells were isolated by negative selection with anti-HLA II magnetic beads (Dynal Biotech). CD8+ naive T cells were further sorted to purify cells that were HLA-DR−CD8+CD45RA+CD27+ (19, 20). The reverse-transmigrated cells were used as candidate APCs to coculture with autologous T cells, the MP-specific T cell line, or CD8+CD45RA+CD27+ naive T cells in the presence of 20 U/ml recombinant human IL-2. MP-restricted CD8+ T cells were prepared by culturing HLA-A201+ PBMCs in the presence of influenza matrix peptide, GILGFVFTL. The proliferated cells were restimulated with irradiated autologous LCLs pulsed with the above peptide for two cycles and cloned by coculture with T2 cells pulsed with the same peptide.
After 7 days of T cell/Ag-presenting cell coculture (20:1), naive T cells were restimulated using the same donor’s candidate APCs from the same condition for another 5 days. The proliferated cells were collected for assessment of CTL activity by ELISPOT.
ELISPOT assays for IFN-γ (reagents from Mabtech) release from single Ag-specific CD8+ T cells were performed as described (15, 21, 22). Serum-free T2 cells (American Type Culture Collection CRL-1992, a TAP−/−HLA-A201+class II− cell line) were pulsed for 1 h with 1 µM influenza matrix peptide, GILGFVFTL. Primed CD8+CD45RA+CD27+ naive T cells were mixed with peptide-pulsed T2 cells at the ratio of 1:1. IFN-γ spots were developed using the HRP-3-amino-9-ethylcarbazole system after being cultured for 20 h. Where specified, background reactivity was determined using T2 cells pulsed with the HLA-A201-restricted epitope SLYNTVATL from HIV Gag protein.
Studies on mouse monocytes and DCs
Female Ly5.2 (CD45.1+) C57BL/6 mice and β2-microglobulin (β2m) knockout mice were purchased from the National Cancer Institute and The Jackson Laboratory, respectively, and used at ~8 wk of age in accordance with protocols approved by the Institutional Animal Care and Use Committee at the Mount Sinai School of Medicine. Recruited monocytes were obtained from CD45.1+ donors 22 h after i.p. administration of 4% thioglycolate, and lymphocytes were depleted with anti-CD3/anti-CD19 Mini-MACS (Miltenyi Biotec). These cells were subsequently cultured in the presence of intact chicken OVA (grade V, Sigma-Aldrich) and the SINFEKL OVA257–264 peptide (AnaSpec) in the presence of a GM-CSF-containing supernatant to preserve DC potential in culture (23). Alternatively, OVA-expressing melanoma cells B16-F10 (kindly given by Dr. Shu-Hsia Chen, Mount Sinai School of Medicine) were loaded with ssRNA by eletropheresis as described above. These cells were i.p injected into 4% thioglycolate-treated mice for the final 5 h of the 22-h thioglycolate treatment. Retrieved monocytes were cultured as described above but without addition of OVA or OVA peptide. Subsequently, 1 × 106 of these monocyte-derived cells were injected into the scapular skin (23) of β2m-deficient mice. The draining brachial and axillary lymph nodes or distal mesenteric lymph nodes were harvested 36 h later, and then digested in collagenase D (Roche). Lymph node cells were pooled from five recipients per treatment, respectively, and stained for CD11c, CD45.1, CD45.2, and CD8α using Fluor-conjugated mAbs from BD Biosciences. CD8α+CD11c+CD45.2+, CD8α−CD11c+CD45.2+, or CD8α−CD11c+CD45.2− DCs were sorted by flow cytometry and then cultured in the presence of CFSE-labeled OT-I T cells that recognize SIINFEKL in the context of presentation by H-2Kb at a DC-to-T cell ratio of 2:1 (with or without OVA peptide), a modified version of previous protocols (24). Dilution of CFSE by live OT-I T cells was monitored by flow cytometry.
Statistics
Student’s t test was used to analyze the differences between specified groups.
Results
Establishment of the model
We established a three-dimensional culture model to investigate a scenario in which cells that die in peripheral tissues are cleared by phagocytes. In this model, LCLs derived from HLA-A201− and HLA-A201+ donors were established and seeded within type I collagen matrix in microtiter wells. Then HUVECs were applied on the top of the collagen (Fig. 1A). Some of the LCLs growing within the collagen underwent spontaneous apoptosis, with 20– 25% of LCLs annexin V+ 72–96 h after they were seeded (Fig. 1, B and C). Apoptosis in most of the LCLs could be induced by low-dose gamma-irradiation (Fig. 1C, right panel) that led to death ~2 days later, and this method was employed in subsequent experiments. When LCLs were infected with influenza A virus before their incorporation in the collagen, viral HA was detected (Fig. 1D). This model, therefore, possessed features suitable to investigate how newly recruited monocytes respond to autologous dying cells that are actively infected or not with virus.
FIGURE 1.

A model containing autologous cells in subendothelial connective tissue. A, Low-power micrograph showing a cross-section of HUVECs grown on type I collagen with LCLs growing within the matrix. B, Higher power image shows the fragmenting nucleus of one LCL undergoing spontaneous death (arrow), surrounded by many living LCLs. C, Staining for propidium iodide and annexin V illustrates the degree of spontaneous apoptosis within LCLs and the extent to which it can be augmented by low dose gamma-irradiation. D, HA staining to monitor LCLs 16 h after infection with influenza A (P/R8) relative to staining with isotype-matched control mAb. Staining was done on nonpermeabilized (left) and permeabilized cells (right).
Monocytes efficiently engulf dying cells, but few monocyte-derived migratory DCs carry apoptotic cell fragments
We confirmed our previous observations that monocytes were the vast majority of cells that traversed endothelium in this model (12), even in the presence of dying cells. Less than 1% of the transmigrated cells were comprised of myeloid human blood DCs, plasmacytoid DCs, or γδ T cells that might serve as alternative sources of DCs (data not shown). Additionally, the presence of LCLs in the collagen, whether infected or not with influenza A virus, had no effect on the number of cells that reverse transmigrated (data not shown); approximately half of initially entering monocytes later reverse transmigrate (12). To assess how efficiently monocytes that crossed the endothelium engulfed apoptotic cells in subendothelial collagen, irradiated LCLs infected or not with flu were labeled with CFSE and incorporated in the HUVEC/collagen cultures. Only a small fraction (6–20%) of monocyte-derived cells carrying CFSE+ LCL fragments retraversed the endothelium (12) (Fig. 2A). In comparison, monocyte-derived cells that remained in the collagen to become macrophages (12) much more efficiently engulfed LCLs (Fig. 2A). Since the fraction of reverse-transmigrated cells that carried CFSE+ material was low, we conducted additional experiments to ensure that proteolytic degradation did not cause us to underestimate the fraction of monocyte-derived cells that acquired CFSE+ cellular debris (data not shown). The low level at which reverse-transmigrated cells carried material acquired from LCLs contrasts markedly with the nearly uniform labeling of reverse-transmigrated cells by uptake of latex beads implanted in subendothelial collagen (12). Thus, only a minority of reversetransmigrated monocyte-derived DCs phagocytized material from dying cells, and most of the apoptotic debris was consumed by macrophages.
FIGURE 2.

Characterization of apoptotic cell uptake and its effects on phenotype and Ag presentation of monocyte-derived cells. A, LCLs were CFSE labeled, irradiated, and incorporated into collagen beneath HUVEC monolayers. The capture and presence of CFSE+ LCL-derived material (bold lines) by reverse-transmigrated (R/T) and subendothelial (S/E) monocyte-derived cells, respectively, were analyzed. Shaded profiles indicate the fluorescence associated with reverse-transmigrated (top) or subendothelial cells (bottom) from cultures wherein LCLs were incorporated in the collagen without CFSE labeling. In the subendothelial population, monocyte-derived cells were distinguished from LCLs by CD14 counterstaining. B, Cell surface expression of HLA-DR and CD86 on reverse-transmigrated monocyte-derived cells after some of these cells phagocytized apoptotic, autologous LCLs (LCLs, no Flu) or influenza A virus-infected LCLs (LCL/Flu) incorporated within collagen, or after they were directly infected with 0.5 MOI live influenza A virus PR/8 (Direct Flu) or were only mock infected (No LCLs, no Flu) before addition to endothelium. C, ELISPOT to quantify the induction of IFN-γ during a CTL assay by the HLA-A201-restricted MP58–66 peptide (GILGFVFTL)-specific CD8+ T cell line primed by HLA-A201+ reverse-transmigrated cells derived from HUVEC/collagen cultures containing LCLs without influenza MP Ag (background reactivity control, open bar) or cultures in which HLA-A201− LCLs expressed MP with or without flu infection (black bars), or from reverse-transmigrated cells derived from monocytes directly infected with flu (gray bars). Spontaneous activity in the absence of APC activation is shown by the stippled bar. Data shown are from one experiment, representative of four conducted. D, HLA-A201+ monocyte-derived reverse-transmigrated cells derived from the conditions described in C were cocultured with autologous CD45RA− memory T cells for 7 days. IFN-γ-producing CD8+ CTLs were assayed by ELISPOT using T2 cells (TAP−/−, HLA-A201+) pulsed with influenza A virus MP58–66 peptide as target cells. The ratio of T cells to target cells was 1:1. Data shown are representative of five independent experiments. E, Flow cytometric cell sorting of reversetransmigrated and subendothelial monocyte-derived cells derived from HUVEC/collagen cultures containing flu-infected LCLs was done to compare their relative efficacy in stimulating CTL responses by cross-presentation. The open bar shows background reactivity observed with reverse-transmigrated cells when T2 cells were pulsed with control HIV Gag peptide rather than with MP peptide. Measurement by ELISA of TNF-α (F) and IFN-α (G) concentrations in conditioned medium collected from cultures 48 h after monocytes were added to the endothelium. Mo + flu indicates monocytes directly infected with influenza A; LCL + flu, LCLs infected with flu and loaded in collagen. Where specified, media was collected without addition of PBMCs as a source of monocytes. Otherwise, monocytes were present in the cultures for 48 h at the time of supernatant collection. In some such cultures, LPS was added after monocyte transmigration across the endothelium, as described (12). Bar sets to the right of each graph show cultures wherein TLR agonists ssRNA, poly(I:C), or both were added to the culture medium (open bars) or LCLs were loaded with the ligands by electroporation (filled bars). Concentrations of these cytokines were measured in three independent experiments; one representative experiment is shown.
ssRNA and natural viral signals contained within dying cells support cross-presentation by monocyte-derived DCs
Reverse-transmigrated monocyte-derived cells from cultures containing virally infected, apoptotic LCLs robustly up-regulated surface HLA-DR and CD86 (Fig. 2B). In many donors, up-regulation of such molecules on these reverse-transmigrated cells exceeded the magnitude at which they were increased when the input monocytes were directly infected with flu before their addition to endothelium and collection as reverse-transmigrated cells (Fig. 2B). In contrast, reverse-transmigrated monocyte-derived cells from cultures containing irradiated LCLs in the collagen that were not infected with flu possessed low surface HLA-DR and CD86 (Fig. 2B).
Whether these changes have functional relevance was next probed by examining the induction of specific CTL reactivity. We established an HLA-A201− LCL line that expressed influenza A virus matrix protein (MP) to eliminate the possibility that LCLs may present Ag to T cells directly. These cells were named MPLCL. MP-LCL cells that were not infected with flu virus or the same parental cells infected with flu were incorporated into the collagen matrices beneath HUVEC monolayers. HLA-A201+ monocyte-derived reverse-transmigrated cells or subendothelial cells were then prepared and collected as test APCs. Reverse-transmigrated cells from cultures containing MP-LCL rather weakly presented MP to MP58–66 peptide-specific T cells (Fig. 2C) or to freshly isolated autologous memory T cells from the same donor (Fig. 2D). The capacity of reverse-transmigrated cells to promote CTL reactivity increased to at least 3-fold above background reactivity after they phagocytized flu-infected LCLs (Fig. 2, C and D, LCL/Flu) and approached the magnitude of the T cell response seen when monocytes were directly infected with flu before the generation of reverse-transmigrated cells (Fig. 2, C and D, Direct Flu). Reverse-transmigrated cells were an order of magnitude more potent at provoking CTL reactivity than subendothelial monocytederived cells (Fig. 2E).
The effect of flu-infected LCLs on monocytes was not due to shedding of free virus by the LCLs, since viral genomes could not be detected in supernatants of the LCL cultures, and expression of nucleoprotein, detectable in newly infected cells, was also not observed (data not shown). Furthermore, the profiles of TNF (Fig. 2F) and IFN-α (Fig. 2G) production were strikingly distinct between cultures wherein monocytes were directly infected with flu compared with those obtaining flu Ags through interaction with LCLs. Finally, the high level of serum in our cultures would serve as a major impediment to the propogation of live influenza infection from cell to cell. Instead, uptake of dying cells by recruited monocytes permitted them to access pathogen-associated stimulants as well as Ag present within the apoptotic cell.
These data indicate that flu-infected apoptotic cells generate signals that can be passed on and perceived by monocyte-derived cells in a manner that provokes changes in their phenotype. TLR ligands may particularly be critical elements acquired through the uptake of flu-infected LCLs that led to these changes and cross-presentation. Monocytes express TLR8 constitutively, express TLR7 weakly or not at all, and lack TLR3 and TLR9 (25). TLR3 is up-regulated by monocytes cultured in GM-CSF and IL-4 while they remain highly positive for TLR8 (25). Likewise, in our model, expression of TLR3 was detectable by 16 h after incoming monocytes interacted with flu-infected LCLs (data not shown).
To determine whether viral TLR ligands could account for the effects of flu infection on stimulating cross-presentation, we loaded LCLs by electroporation (16) with the TLR3 ligand poly(I:C), the TLR7/8 ligand ssRNA, or the two combined. These LCLs were then incorporated in the collagen underlying HUVEC monolayers. PBMCs were added, and the fate of the transmigrating monocytes was traced for 48 h. HLA-DR expression was increased in monocyte-derived reverse-transmigrated cells when they took up LCLs loaded with poly(I:C) cell-bound but loading LCLs with ssRNA was even more potent (Fig. 3A). Poly(I:C) added in soluble form to the culture medium at ~50× more than the estimated dose contained within the LCLs conversely down-regulated HLA-DR, whereas addition of high-dose ssRNA to the culture up-regulated HLA-DR, although less so than the LCL-associated ssRNA (Fig. 3A). LCL-associated Ags were efficiently cross-presented by HLA-A201+ monocyte-derived reverse-transmigrated cells to autologous naive or memory T cells, respectively (Fig. 3. B and C). LCLs loaded with poly(I:C) very poorly induced primary, naive CD8+ T cells to become CTLs, but ssRNA was as effective at doing so as the flu-infected LCLs (Fig. 3, B and C). No additive effects were seen when poly(I:C) and ssRNA were dually loaded, and, indeed, for memory T cells, there was a partial inhibitory effect of poly(I:C) loading (Fig. 3C). Similar results were obtained when we tested purified monocytes or started with the bulk PBMC fraction that enriches mainly for monocytes after transmigration (12). Overall, these data indicate that TLR7/8 signals present within dying cells are a superior stimulus for the conversion of some monocytes to cross-presenting cells capable of priming naive CD8+ T cells. For this reason, along with the fact that reverse-transmigrated cells were previously shown to become DCs (12), we use the term “reverse-transmigrated DCs” to refer to these cells.
FIGURE 3.

Effect of loading LCL with model TLR ligands. A, Expression of HLA-DR on monocyte-derived reverse-transmigrated cells from cultures wherein subendothelial collagen contained electroporated LCLs without TLR ligand loading (gray profiles); LCLs electroporetically loaded with poly(I:C), ssRNA, or both ligands (bolded line, open profiles, labeled “cell-bound”) amounting to ~125 ng of each TLR ligand per well; or cultures in which soluble (“free”) poly(I:C), ssRNA, or both were added to the medium (5 µg/ml ssRNA or 10 µg/ml poly(I:C). B and C, IFN-γ ELISPOT to detect CTL induction in autologous HLA-A201+CD45RA+ naive T cells after two rounds of stimulation (B) and IFN-γ ELISPOT to detect CTL induction in HLA-A201+CD45RA− memory T cells (C) stimulated for 7 days by reverse-transmigrated DCs from cultures lacking LCLs (plain) or cultures containing LCLs without MP (LCL) or MP-positive LCLs (MP-LCL) that were not infected with flu or loaded with TLR ligands (LCL); or were infected with flu (LCL/Flu); or were loaded with poly(I:C) (PIC), ssRNA, or both (cell-bound) at the concentrations described in A. All of the conditions that used LCLs transduced to express MP are bracketed. Reactivity in cultures containing LCL without MP (LCL) or flu infection is indicative of nonspecific background. Alternatively, soluble TLR ligands (Free) were added to the culture medium. IFN-γ-producing CD8+ T cells were determined by ELISPOT using T2 cells (TAP−/−, HLA-A201+) pulsed with influenza A virus MP58–66 peptide as target cells. Data shown are from one experiment representative of the outcome of three independent experiments conducted.
Transfer of Ag between monocyte-derived DCs plays an important role in cross-presentation
A puzzling aspect of the data is that very few reverse-transmigrated monocyte-derived DCs can be demonstrated to have engulfed apoptotic cells, even though most reverse-transmigrated cells moved to a more mature APC phenotype (Fig. 2). To determine whether the small number of reverse-transmigrated DCs that engulfed dying cells were the only DCs robustly presenting Ag, we sorted reverse-transmigrated DCs into groups that were CFSE+ or CFSE− after inclusion of CFSE+ LCLs in the collagen (as in Fig. 2). Surprisingly, CFSE− monocyte-derived DCs cross-presented the cell-associated MP Ag (Fig. 4A) from LCLs markedly better than did their CFSE+ counterparts, opposite to what we anticipated. The fraction of CD8+ CTLs that expressed granzyme at the end of the stimulation period was also elevated in groups wherein the APCs were CFSE− rather than CFSE+ reverse-transmigrated cells (35% vs 16% granzyme+ T cells, respectively) (Fig. 4B). This finding suggests that uptake of a detectable quantity of dying (CFSE+) cells fails to support, and may even reduce, robust crosspresentation. The data also raise the question as to how CFSE− reverse-transmigrated cells gained access to Ag.
FIGURE 4.
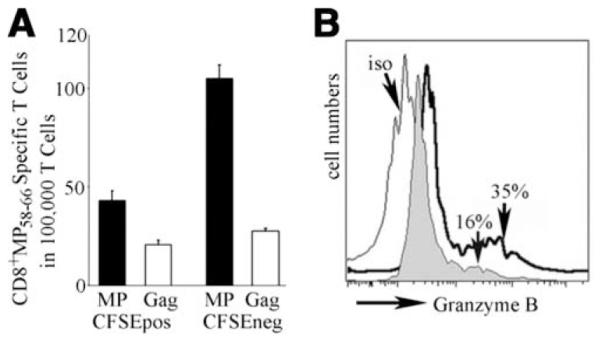
Analysis of the relationship between LCL engulfment and CTL induction by monocyte-derived reverse-transmigrated DCs. A, Purified monocytes were added to HUVEC/collagen cultures containing CFSElabeled flu-infected LCLs, and reverse-transmigrated cells were collected and sorted using flow cytometry into CFSE+ (indicating engulfment of LCLs) and CFSE− reverse-transmigrated cells. Then these populations were cocultured with HLA-A201+ autologous T cells for 7 days. IFN-γ-producing CD8+ T cells were determined by ELISPOT using T2 target cells (TAP−/−, HLA-A201+) pulsed with influenza A virus MP58–66 peptide or HIV Gag peptide (to establish background reactivity). Data shown are from one experiment that is representative of two conducted. B, Intracellular granzyme B was stained in the proliferated T cells (gated on CD8+ T cells) stimulated by CFSE+ (gray profile) or CFSE− reverse-transmigrated cells (bold line). Open profile indicates PE-conjugated isotype control staining. The bracketed area demarcates the portion of the histogram that positively detected granzyme+CD8+ T cells.
Thus, we tested the possibility that Ag might be donated by some cells from the culture system to other DCs. To examine this in a way where we could be certain that the recipient DCs did not have access to flu-infected LCLs directly, we collected reversetransmigrated monocyte-derived DCs from cultures wherein MP-LCL were grown in the collagen under various conditions and then cocultured them with the same donor’s DCs derived from GM-CSF/IL-4-treated monocytes that had matured so their endocytic uptake pathways were down-regulated. Even without the capacity for endocytosis, mature DCs that never interacted with MP-expressing LCLs could induce CTL reactivity. This ability was acquired from reverse-transmigrated cells derived from cultures wherein LCLs were infected with flu or loaded with ssRNA (Fig. 5A). This “Ag donation” was more efficient when reverse-transmigrated cells rather than subendothelial monocyte-derived macrophages were used as “Ag donors” (Fig. 5B), even though the latter phagocytosed most of the Ag (Fig. 2A).
FIGURE 5.

Transfer of the capacity for CTL induction between DCs. A, IFN-γ ELISPOT assessing the activation of CD8+ MP-specific peripheral blood T cells by a population of mature CFSE+ GM-CSF/IL-4 monocyte-derived DCs after they were cocultured overnight with the same donor’s reverse-transmigrated cells derived from endothelial/LCL/collagen cultures and then reisolated by cell sorting before mixing with T cells. The x-axis shows the type of LCLs that were originally present in the tissue for engulfment by monocyte-derived reverse-transmigrated cells used as the source of “donor DCs” for the overnight cultures. LCL + Flu indicates reverse-transmigrated cells made from HUVEC/collagen cultures containing flu-infected LCLs in the subendothelial matrix. TW indicates that the reverse-transmigrated DCs were separated from the other population of mature DCs by a transwell membrane. IFN-γ-producing CTLs were assayed by ELISPOT using T2 target cells (TAP−/−, HLA-A201+) pulsed with influenza A virus MP58–66 peptide (filled bars) or HIV Gag peptide (open bars, to establish background reactivity). B, Same experiments were designed as A, except here reverse-transmigrated (filled bars) and subendothelial (open bars) cells were compared as the source of “donor DCs” for the overnight cultures. C, Design of two different sources of DC coculture. D, FACS staining of HLA-A201 molecules on two different sources of DCs before coculture and after coculture overnight (bold line). Data are representative of the outcomes in two independent experiments. E, Similar experiment as designed in A but here the sorted, assayed DCs were derived from HLA-A201− donors. The x-axis shows the type of LCLs that were originally present in the collagen for engulfment by monocyte-derived HLA-A201+ reverse-transmigrated (RT) cells used as the source of “donor DCs” during overnight culture with HLA-A201− recipient DCs. “No coculture” specifies conditions when RT cells were not used as Ag donors, but instead HLA-A201− DCs were used alone (with or without MP peptide) to establish background reactivity. Data are representative of two experiments conducted.
To assess whether recipient DCs might obtain intact MHC I/peptide complexes from “donor” reverse-transmigrated DCs, we cocultured Ag-experienced HLA-A201+ reverse-transmigrated cells with HLA-A201− mature DCs derived from monocytes treated with GM-CSF and IL-4 (Fig. 5C). Use of anti-HLA-A201-specific mAb revealed that transfer of MHC I/peptide complex indeed occurred (Fig. 5D). Moreover, HLA-A201− DCs that became HLA-A201+ through coculture with reverse-transmigrated HLA-A201+ cells acquired the capacity to stimulate HLA-A2-restricted MP58–66-specific T cells to release IFN-γ (Fig. 5E), and this was evident even with the relatively higher background in this experimental design likely due to alloreactivity between HLA-A2− DCs and the T cell line. DCs that were cocultured with reverse-transmigrated cells from cultures containing LCLs without intracellular TLRs were far less effective at inducing CTL reactivity (Fig. 5, A, B, and E). Moreover, DCs that were separated from reverse-transmigrated cells by a transwell were unable to acquire robust MP-specific CTLs (Fig. 5A), suggesting a need for close proximity between the two APC populations in the transfer of Ag-presentation capacity. Taken together, we conclude that appropriately stimulated monocyte-derived DCs, much more than monocyte-derived macrophages, can transfer intact MHC I/peptide complexes to other DCs in close proximity. Appropriate stimulation for monocyte-derived DCs includes uptake of dying cells bearing the TLR ligand ssRNA.
Finally, we wondered if monocyte-derived DCs might have the capacity to transfer MHC/peptide to other DCs in vivo. In brief, newly recruited monocytes were obtained from the peritoneal cavity of CD45.1+ C57BL/6 mice and exposed to OVA/OVA peptide in vitro and then transferred to CD45.2+ MHC I-deficient mice (β2m knockout mice) by injection intracutaneously. Alternatively, OVA-expressing B16-F10 cells were loaded with ssRNA in vitro and then injected i.p into mice 17 h after thioglycolate injection and 5 h before mice were terminated for collection of recruited monocyte-derived cells. As described previously (23), after transfer of these cells i.c. into recipient mice, some of the transferred monocyte-derived cells developed into DCs with restricted homing to the draining lymph node, detected as CD45.1+ cells in the lymph node of CD45.2+ recipients (Fig. 6A). We harvested the draining and nondraining lymph nodes from the β2m knockout recipients 36 h after injection of monocyte-derived cells into the skin, prepared single cell suspensions, and sorted the recipient CD8α− and CD8α+ CD45.2+ lymph node DCs (Fig. 6, B and C) or CD45.2−CD11c+ donor monocyte-derived DCs (Fig. 6C). These sorted cells were cultured in vitro with OVA-restricted CD8+ OT-I T cells, essentially as described (24), to determine whether recipient lymph node DCs acquired the ability to present MHC I-restricted Ag to CD8+ T cells from MHC I+ monocytederived DCs. Indeed, CD8α+ DCs, but not CD8α− DCs, of recipient MHC I-deficient origin presented OVA to OVA-restricted OT-I T cells, but only when this population was derived from a skin-draining lymph node downstream of the injection of OVAbearing monocyte-derived cells (Fig. 6B). The extent of CD8+ T cell proliferation (a single cell division) was reduced when we used monocyte-derived DCs that took up OVA-expressing tumor cells (Fig. 6C) compared with the OVA peptide-loaded monocytederived DCs (Fig. 6B). Monocyte-derived DCs were able to crosspresent OVA from the B16 cell line on their own when recovered from the lymph node, and they induced a somewhat stronger proliferative response than did the recipient CD8α+ β2m-deficient DCs. If monocyte-derived cells were transferred but not OVAloaded, if OVA-expressing tumor cells were transferred without engulfment by monocytes, or if nondraining lymph node were examined, sorted CD8α− or CD8α+ DCs did not promote OT-I T cell proliferation (data not shown). These data suggest that migratory monocyte-derived DCs can promote cross-presentation in two ways: by directly cross-priming CD8+ lymph node T cells after acquiring dead cells in tissues or by cross-dressing resident CD8α+ DCs.
FIGURE 6.

In vivo transfer of Ag-presenting capacity to MHC I-deficient CD8α+ DCs by migratory MHC I+ monocyte-derived DCs. A, CD45.1+ monocyte-derived cells collected from the peritoneal lavage of thioglycolate-treated wild-type C57BL/6 mice were loaded with OVA Ag and injected i.c. into the scapular skin of β2m-deficient CD45.2+ mice. The appearance of the CD45.1+ monocyte-derived cells into the recipient brachial lymph nodes was monitored by flow cytometry, where a population of CD45.1+ cells was detected (right panel, gated box) in contrast to the same lymph nodes from β2m-deficient mice that did not receive CD45.1+ monocyte-derived cells (left panel). B, CD8α+ and CD8α− CD45.2+ β2m-deficient lymph node DCs were sorted from lymph nodes that either did not receive monocyte-derived cells (not shown) or received WT CD45.1+ monocyte-derived cells that were loaded or not loaded (not shown) ex vivo with OVA peptide. Dot plots show the gates that were used to sort after initially gating on CD45.2+ cells. DCs from these sorts were cocultured with OT-I CFSE-labeled T cells in vitro and then proliferation was monitored 68 h later. a and b are used to match gates for DCs that were sorted (left) and later cocultured with T cells for assessment of proliferation (right). C, A similar experimental design as in A was conducted except OVA-B16 was loaded with ssRNA and injected into the peritoneum as a source of Ag, instead of peptide loading as in A. CD8α+ β2m-deficient DCs (gate c) and DCs from the injected monocyte-derived cells recovered in the draining lymph nodes (gate d) were purified by flow cytometric cell sorting and then cocultured with OT-I CFSE-labeled T cells in vitro for 4 days (right panel). These samples (black profiles, gates c or d) were compared with wild-type spleen DCs loaded with soluble OVA, as a positive control (open profiles, +OVA), or to CD8α+ DCs from β2m-deficient mice sorted from nondraining mesenteric lymph nodes and then cultured with OT-1 T cells in the absence (gray line) or presence of OVA257–264 peptide (thin lines, +Pep).
Discussion
Lymph node resident DCs are pivotal for cross-presentation in many instances (5, 6), and it has been argued that DCs migrating to lymph nodes from the periphery mainly serve to donate Ag to lymph node resident DCs (CD8α+ DCs in mice) (7, 8). Newly recruited monocytes may in many instances contribute substantially to the population of DCs that acquires Ag in the context of inflammation (3, 4, 26–29) and therefore may serve as a source of migratory DCs that deliver Ag to CD8α+ DCs. Indeed, monocytes have recently been implicated as crucial participants in the induction of CTL responses through cross-presentation in vivo (4), but it was proposed that they directly presented Ag to CD8+ T cells, rather than transfer Ag to CD8α+ DCs, because the monocytederived DCs had to express MHC I to promote cross-priming (4). The findings reported herein offer an additional mechanism to the previous interpretation, as we show that monocyte-derived DCs also donate intact, functional MHC I/peptide complexes to other DCs, including CD8α+ DCs in vivo, thereby using a mechanism previously termed “cross-dressing” (30–32) to support cross-priming.
We draw these conclusions after taking in vitro and in vivo approaches to delineate in detail the steps involved in how newly recruited monocytes, in both humans and mice, contribute to crosspresentation. Our work in vitro utilized a previously developed three-dimensional model (12) to examine cross-presentation by human monocytes. As much as possible, the model is self-driven by endogenous signals from intercellular interactions to allow the environment of the culture system to serve as the primary influence over the differentiation and behavior of DCs that develop within it. Our culture system incorporated dying cells that expressed influenza MP, which has well-defined CTL epitopes, even when none of the cells were infected with flu. In this model, most of the monocyte-derived cells that engulfed dying cells after traversing the endothelium remained in the subendothelial matrix as macrophages (12). A small, but detectable fraction of monocyte-derived cells took up cellular material acquired from dying subendothelial cells and joined the reverse transmigratory population of DCs. CTL reactivity to Ags expressed by the dying cells was augmented if they had been naturally infected with flu, fitting with evidence in the mouse that the presence of TLR ligands within dying cells can promote DC maturation and cross-presentation (16).
In mouse CD8α+ DCs, TLR3 engagement by viral components within dying cells strongly supported cross-priming (16). In our model, human monocyte-derived cells also up-regulated TLR3, but loading dying cells with the TLR3 agonist poly(I:C) was not as effective at facilitating monocyte maturation to cross-priming DCs than were flu-infected dying cells. In contrast, dying cells that were previously loaded with ssRNA before uptake by monocyte-derived cells induced a CTL response to influenza MP that was as strong as the CTL response mounted against dying cells infected naturally with flu. The failure of TLR3 agonists to augment the CTL-inducing activity in our model as well as previously is likely explained by the difference in sources of DCs and species studied, as well as by the fact that we focused on how monocytes responded to TLR signals within dying cells immediately after recruitment into a model tissue, rather than using already differentiated immature DCs.
The strong response to ssRNA-loaded dying cells is most likely mediated by TLR8, rather than by TLR7. Both TLRs mediate recognition of ssRNA (25), but only TLR8 is expressed by monocytederived cells. Use of purified monocytes in our model system was effective for mediating cross-presentation, indicating that TLR-7+ plasmacytoid DCs were not required. Stimulation of human monocytes with TLR8 agonists induces IL-12 and blocks IL-10 production (33), and stimulation through TLR8 is especially efficacious in supporting the overall Ag-presenting capacity of monocytes from human newborns (34). Furthermore, vaccinations in nonhuman primate models have identified TLR7/8 agonists as superior adjuvants when the agonists are complexed with the Ag of interest (35, 36), in agreement with our findings that a TLR7/8-targeting stimulus is a superior agonist for monocyte conversion to cross-presenting DCs and that soluble TLR agonists were markedly less effective than those present within the dying cells at supporting DC maturation and CTL reactivity. Our data underscore the possibility that the efficacy of these vaccines may relate to how stimulatory they are for differentiating monocyte-derived cells. Thus, vaccines that deliver Ags in the form of cells, such as altered cell lines that serve as antitumor vaccines (37), may particularly benefit from a step in which the cells used during vaccination are loaded with a TLR8 stimulus like ssRNA, as we did here.
Previous studies demonstrated that DC uptake of cells bearing viral-derived signals promotes cross-presentation, but these studies did not exclude the possibility that the presentation itself was being conducted by DCs in the culture other than the DCs that really engulfed the dying cells. In one of these studies, only 20–25% of the DCs that cross-presented dying cells had engulfed them (11), whereas 70% of mouse CD8α+ DCs had engulfed dying cells in another study (16); each body of work leaves open the possibility that “bystander” DCs in the culture that had not engulfed dying cells played a key role in presentation. In our system, only a minority of monocyte-derived cells that reverse transmigrated (<20%) could be demonstrated to have engulfed detectable fragments of labeled dying cells. When we determined whether this minority of cells accounted for the bulk of cross-priming, we were surprised to find that the DCs that had engulfed detectable amounts of dying cells cross-presented notably more poorly than did those that did not bear evidence of having acquired cellular material from LCLs. Whereas it is possible that the most phagocytic APCs are inherently the least capable of robust cross-presentation, it is also possible that the uptake of certain phagocytic material itself induces suppressed capacity for cross-presentation.
One way to get around this problem is for such DCs to share or transfer Ag. Coculture experiments between two populations of DCs illustrated that presentation capacity, and intact HLA-201 itself, could be transferred between and from reverse-transmigrated DCs to other, already matured DCs and vice versa. Experiments in vivo indicated that monocyte-derived DCs that emigrate to lymph nodes can share intact MHC I/peptide (or at least intact β2m and peptide) with other DCs, especially the CD8α+ lymph node DCs, although in our experimental design their ability to transfer peptide/MHC I complexes was lower when the monocyte-derived cells processed sources of Ag from dying cells as compared with peptide loading. The common element between our in vitro model and the in vivo study are the migratory monocyte-derived cells bearing Ag, and we do not suggest that our in vitro model contains human equivalents of CD8α+ DCs. Instead, in vitro, it appears that Ag sharing takes place between monocyte-derived DCs in close proximity that had and had not engulfed dying cells, respectively. However, in vivo, the highly organized environment of the lymph node wherein a number of DC subsets reside may alter the types of DCs that interact or come into close proximity. Alternatively, through competition, certain interactions may be favored that cannot be modeled in vitro. Furthermore, many DC types may have received MHC/peptide by transfer, but CD8α+ DCs may be restricted in presenting it, as they also have an advantage in crosspresentation in general.
The overall mode of Ag exchange observed herein has been previously described between DCs or other cell types and is sometimes called “cross-dressing” (30–32, 38– 41). Much remains to be determined mechanistically about the process: phagocytosis of membrane has been implicated (39), or alternatively exosomes may mediate transfer in an αL integrin-dependent manner (41). Our findings differ from the conclusions of Smyth et al., in that we (but not they) found that CD8α+ DCs were efficient in presenting Ag obtained by cross-dressing (32). Since there may be multiple mechanisms that give rise to cross-dressing, it is possible that the relative efficiency of presentation through cross-dressing may differ depending on context. Here, we argue that cross-dressing of lymph node DCs (such as CD8α+ DCs) can serve as a way in which populations of DCs that survey inflamed tissues and subsequently emigrate to lymph nodes (namely monocyte-derived DCs) functionally support cross-priming. Our in vitro data, wherein only a small fraction of flu-presenting DCs were bearing evidence of having engulfed dying cells that served as a source of Ag, suggest that one purpose of cross-dressing may be to overcome the relatively poorer cross-presentation by monocyte-derived cells that directly engulf dying cells and to spread Ag presentation to a much larger number of DCs that would otherwise not directly come into contact with a given source of Ag. However, in our experiments in vivo in which tumor cells expressing OVA were used as a source of Ag, we did not observe that the CD8α+ DCs presented transferred Ag better than the initial monocyte-derived DCs themselves, if we assume that the monocyte-derived DCs recovered from the lymph node are the same monocyte-derived cells that engulfed the OVA-bearing tumor cells. Thus, the purpose of Ag transfer may not always be to overcome poor cross-presentation by the initially engulfing cells. Instead, the ability of migratory monocyte-derived DCs to promote cross-presentation in two ways may protect the host against the possibility that a microorganism would evolve a means to shut down the direct pathway of cross-presentation but leave the pathway involving MHC/peptide transfer intact, or vice versa.
In summary, this study allows us to put together a sequence of events related to cross-presentation that occurs soon after monocytes leave the bloodstream. A few of these steps have been separately proposed before in various mouse or human models, but here we monitored the process from start to finish to place these steps in order and to evaluate their experimental validity and relevance to humans in reference to how newly recruited monocytes as a source of DCs during infection and inflammation contribute to cross-priming. As a first step in the sequence, some monocytederived cells recently recruited to tissues engulf dying cells in the environment. Most of these phagocytic cells differentiate to macrophages, but some develop stronger Ag presentation capacity and the migratory abilities attributed to DCs and leave the tissue. The up-regulation of Ag presentation capacity and costimulatory molecules by these cells is a function of the contents of the ancillary cells (here, LCLs) that they acquire in the subendothelial tissue. In particular, ssRNA, likely acting through TLR8, is a potent cellassociated cue to produce mature DCs from newly recruited monocytes that induce CTL responses. For human monocyte-derived cells in vitro, uptake of dying cells bearing Ag had a negative impact on the ability to cross-prime, and the best inducers of CTLs were the DCs that cannot be linked directly to having engulfed the dying cells but instead acquired the Ag, including intact MHC/peptide, from the phagocytic DCs through cross-dressing. Thus, the DC that actually presents the Ag to T cells most efficiently is distinct from the DC that engulfed the cell that supplied it. Transfer of intact MHC-peptide between DCs in vivo can explain why monocytes need to express MHC I to support cross-presentation (4) beyond a role for direct cross-presentation themselves.
Acknowledgments
We thank Drs. Adolfo Garcia-Sastre, Tom Moran, and Shu-Hsia Chen (Mount Sinai School of Medicine) for providing plasmids, Abs, and tumor cell lines, and we are grateful to Dr. Davor Frleta (Baylor Institute of Immunology, Dallas, TX) for helpful discussions.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by the Cancer Research Institute, National Institutes of Health Grant AI49653, and Defense Advanced Research Planning Agency contract W81XWH-04-C-0139. The prime contractor of the latter contract is the company VaxDesign. G.J.R. and Mount Sinai School of Medicine are subcontractors of this award. C.Q. later was supported by Grant JK2006A01 from the Chinese Academy of Medical Sciences.
- DC
- dendritic cells
- MHC I
- MHC class I
- LCL
- lymphoblastoid cell line
- MP
- influenza A virus matrix protein
- MP-LCL
- lymphoblastoid cell line expressing influenza A virus matrix protein
- MOI
- multiplicity of infection
- HA
- hemagglutinin
- β2m
- f32-microglobulin
Disclosures
G.J.R. works collaboratively with VaxDesign, the sponsor of this research. She has received stock options from VaxDesign. G.J.R. and Mount Sinai School of Medicine have applied for a patent with VaxDesign for technology in which vascular and connective tissue is reconstructed from human cells for the purposes of vaccine testing and selection. If this technology were licensed to a commercial entity, then G.J.R. and M.M. would benefit financially.
References
- 1.Bevan MJ. Cross-priming. Nat. Immunol. 2006;7:363–365. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 2.Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001;1:126–134. doi: 10.1038/35100512. [DOI] [PubMed] [Google Scholar]
- 3.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 4.Le Borgne M, Etchart N, Goubier A, Lira SA, Sirard JC, van Rooijen N, Caux C, Ait-Yahia S, Vicari A, Kaiserlian D, Dubois B. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity. 2006;24:191–201. doi: 10.1016/j.immuni.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 5.den Haan JM, Lehar SM, Bevan MJ. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, Takahara K, Steinman RM, Inaba K. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 2002;195:1289–1302. doi: 10.1084/jem.20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 8.Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR. Cutting edge: conventional CD8α+ dendritic cells are generally involved in priming CTL immunity to viruses. J. Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- 9.He Y, Zhang J, Donahue C, Falo LD., Jr. Skin-derived dendritic cells induce potent CD8+ T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. 2006;24:643–656. doi: 10.1016/j.immuni.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randolph GJ. Migratory dendritic cells: sometimes simply ferries? Immunity. 2006;25:15–18. doi: 10.1016/j.immuni.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 12.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- 13.Bartido SM, Zier K. T-cell responses to multiple antigens presented by RNA-transfected APCs: a possible immunomonitoring tool. Cancer Immunol. Immunother. 2004;53:100–109. doi: 10.1007/s00262-003-0434-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krausa P, Brywka M, 3rd, Savage D, Hui KM, Bunce M, Ngai JL, Teo DL, Ong YW, Barouch D, Allsop CE, et al. Genetic polymorphism within HLA-A*02: significant allelic variation revealed in different populations. Tissue Antigens. 1995;45:223–231. doi: 10.1111/j.1399-0039.1995.tb02444.x. [DOI] [PubMed] [Google Scholar]
- 15.Qu C, Moran TM, Randolph GJ. Autocrine type I IFN and contact with endothelium promote the presentation of influenza A virus by monocyte-derived APC. J. Immunol. 2003;170:1010–1018. doi: 10.4049/jimmunol.170.2.1010. [DOI] [PubMed] [Google Scholar]
- 16.Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Sousa C. Reis e. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 17.Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 18.Mendoza-Naranjo A, Saez PJ, Johansson CC, Ramirez M, Mandakovic D, Pereda C, Lopez MN, Kiessling R, Saez JC, Salazar-Onfray F. Functional gap junctions facilitate melanoma antigen transfer and cross-presentation between human dendritic cells. J. Immunol. 2007;178:6949–6957. doi: 10.4049/jimmunol.178.11.6949. [DOI] [PubMed] [Google Scholar]
- 19.De Rosa SC, Roederer M. Eleven-color flow cytometry: a powerful tool for elucidation of the complex immune system. Clin. Lab. Med. 2001;21:697–712. vii. [PubMed] [Google Scholar]
- 20.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuber B, Levitsky V, Jonsson G, Paulie S, Samarina A, Grundstrom S, Metkar S, Norell H, Callender GG, Froelich C, Ahlborg N. Detection of human perforin by ELISpot and ELISA: ex vivo identification of virus-specific cells. J. Immunol. Methods. 2005;302:13–25. doi: 10.1016/j.jim.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Rotta G, Edwards EW, Sangaletti S, Bennett C, Ronzoni S, Colombo MP, Steinman RM, Randolph GJ, Rescigno M. Lipopolysaccharide or whole bacteria block the conversion of inflammatory monocytes into dendritic cells in vivo. J. Exp. Med. 2003;198:1253–1263. doi: 10.1084/jem.20030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, Carbone FR. Epidermal viral immunity induced by CD8α+ dendritic cells but not by Langerhans cells. Science. 2003;301:1925–1928. doi: 10.1126/science.1087576. [DOI] [PubMed] [Google Scholar]
- 25.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 26.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 27.Ginhoux F, Tacke F, Angeli V, Bogunovic M, Loubeau M, Dai XM, Stanley ER, Randolph GJ, Merad M. Langerhans cells arise from monocytes in vivo. Nat. Immunol. 2006;7:265–273. doi: 10.1038/ni1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villadangos JA. Hold on, the monocytes are coming! Immunity. 2007;26:390–392. doi: 10.1016/j.immuni.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity. 2007;26:519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 30.Dolan BP, Gibbs KD, Jr., Ostrand-Rosenberg S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J. Im- munol. 2006;177:6018–6024. doi: 10.4049/jimmunol.177.9.6018. [DOI] [PubMed] [Google Scholar]
- 31.Dolan BP, Gibbs KD, Jr., Ostrand-Rosenberg S. Tumor-specific CD4+ T cells are activated by “cross-dressed” dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J. Immunol. 2006;176:1447–1455. doi: 10.4049/jimmunol.176.3.1447. [DOI] [PubMed] [Google Scholar]
- 32.Smyth LA, Harker N, Turnbull W, El-Doueik H, Klavinskis L, Kioussis D, Lombardi G, Lechler R. The relative efficiency of acquisition of MHC:peptide complexes and cross-presentation depends on dendritic cell type. J. Immunol. 2008;181:3212–3220. doi: 10.4049/jimmunol.181.5.3212. [DOI] [PubMed] [Google Scholar]
- 33.Bekeredjian-Ding I, Roth SI, Gilles S, Giese T, Ablasser A, Hornung V, Endres S, Hartmann G. T cell-independent, TLR-induced IL-12p70 production in primary human monocytes. J. Immunol. 2006;176:7438–7446. doi: 10.4049/jimmunol.176.12.7438. [DOI] [PubMed] [Google Scholar]
- 34.Levy O, Suter EE, Miller RL, Wessels MR. Unique efficacy of Toll-like receptor 8 agonists in activating human neonatal antigen-presenting cells. Blood. 2006;108:1284–1290. doi: 10.1182/blood-2005-12-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wille-Reece U, Flynn BJ, Lore K, Koup RA, Kedl RM, Mattapallil JJ, Weiss WR, Roederer M, Seder RA. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA. 2005;102:15190–15194. doi: 10.1073/pnas.0507484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wille-Reece U, Flynn BJ, Lore K, Koup RA, Miles AP, Saul A, Kedl RM, Mattapallil JJ, Weiss WR, Roederer M, Seder RA. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after prime-boost immunization in nonhuman primates. J. Exp. Med. 2006;203:1249–1258. doi: 10.1084/jem.20052433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dranoff G. GM-CSF-secreting melanoma vaccines. Oncogene. 2003;22:3188–3192. doi: 10.1038/sj.onc.1206459. [DOI] [PubMed] [Google Scholar]
- 38.Herrera OB, Golshayan D, Tibbott R, Salcido Ochoa F, James MJ, Marelli-Berg FM, Lechler RI. A novel pathway of alloantigen presentation by dendritic cells. J. Immunol. 2004;173:4828–4837. doi: 10.4049/jimmunol.173.8.4828. [DOI] [PubMed] [Google Scholar]
- 39.Xu H, Dhanireddy KK, Kirk AD. Human monocytes as intermediaries between allogeneic endothelial cells and allospecific T cells: a role for direct scavenger receptor-mediated endothelial membrane uptake in the initiation of alloimmunity. J. Immunol. 2006;176:750–761. doi: 10.4049/jimmunol.176.2.750. [DOI] [PubMed] [Google Scholar]
- 40.de Heusch M, Blocket D, Egrise D, Hauquier B, Vermeersch M, Goldman S, Moser M. Bidirectional MHC molecule exchange between migratory and resident dendritic cells. J. Leukocyte Biol. 2007;82:861–868. doi: 10.1189/jlb.0307167. [DOI] [PubMed] [Google Scholar]
- 41.Segura E, Guerin C, Hogg N, Amigorena S, Thery C. CD8+ dendritic cells use LFA-1 to capture MHC-peptide complexes from exosomes in vivo. J. Immunol. 2007;179:1489–1496. doi: 10.4049/jimmunol.179.3.1489. [DOI] [PubMed] [Google Scholar]