

Abstract
The synthesis of a C(1)-C(24) advanced southern hemisphere fragment towards the total synthesis of spirastrellolide E has been achieved. Highlights of the route include a highly convergent Type I Anion Relay Chemistry (ARC) tactic for fragment assembly, in conjunction with a directed, regioselective gold-catalyzed alkyne functionalization to generate the central unsaturated [6,6]-spiroketal.
Keywords: Spirastrellolide, Anion Relay Chemistry, Brook Rearrangement, Gold Catalysis, Total Synthesis
In 2003 Anderson and colleagues reported the isolation, partial structural determination, and disclosure that spirastrellolide A (1; Figure 1) was a selective (1 nM) and extremely potent inhibitor of phosphotase 2A.1 The complete connectivity of 1, however, was not established until 2004,2 with the stereochemical relationship and absolute configuration of the core remaining unknown until the 2007 isolation and X-ray characterization of the closely related spirastrellolide B (2; Figure 1).3 Later that year, the complete stereostructure of 1–7, including absolute configuration, was established via chemical degradation of spirastrellolide D (4).4
Figure 1.

The spirastrellolides A-G
The combination of structural complexity, biological activity, and, at the outset, unknown relative stereochemistry led to considerable interest in the synthetic community,5–27 culminating in the first total synthesis of spirastrellolide A (2008) by the Paterson28–31 group, and the total syntheses of spirastrellolide F (2009, 2011)32–34 and later A (2013),35 both by Furstner and colleagues.
Our interest in the spirastrellolides began in 2007, resulting early on in completed approaches to advanced fragments for a southern hemisphere relevant to spirastrellolide A and B22,23 and a northern hemisphere relevant to B and E.26 With these routes established, we considered a total synthesis venture; however it quickly became apparent that our first generation southern hemisphere synthesis, totaling 33 steps for the longest linear sequence, was not amenable to advancing ample material for a successful synthetic campaign. We therefore set out to design a second-generation route. Herein, we report the result of that effort, which has now led to a significantly improved approach to a related southern hemisphere congener for spirastrellolide E, featuring both a decrease in the longest linear step count and an increase in the overall yield.
Our initial 2007 spirastrellolide venture highlighted the use of Type II Anion Relay Chemistry (ARC)36 to construct advanced spirastrellolide A southern hemisphere intermediate 11 from fragments 8–10 (Scheme 1). Unfortunately, this approach precluded installation of the C(14) methyl group until after spiroketalization, a tactic requiring three steps. Moreover, when attempting to install the C(23)-C(25) fragment 15, the undesired stereoisomer predominated, which required an oxidation/reduction/reprotection sequence.
Scheme 1.
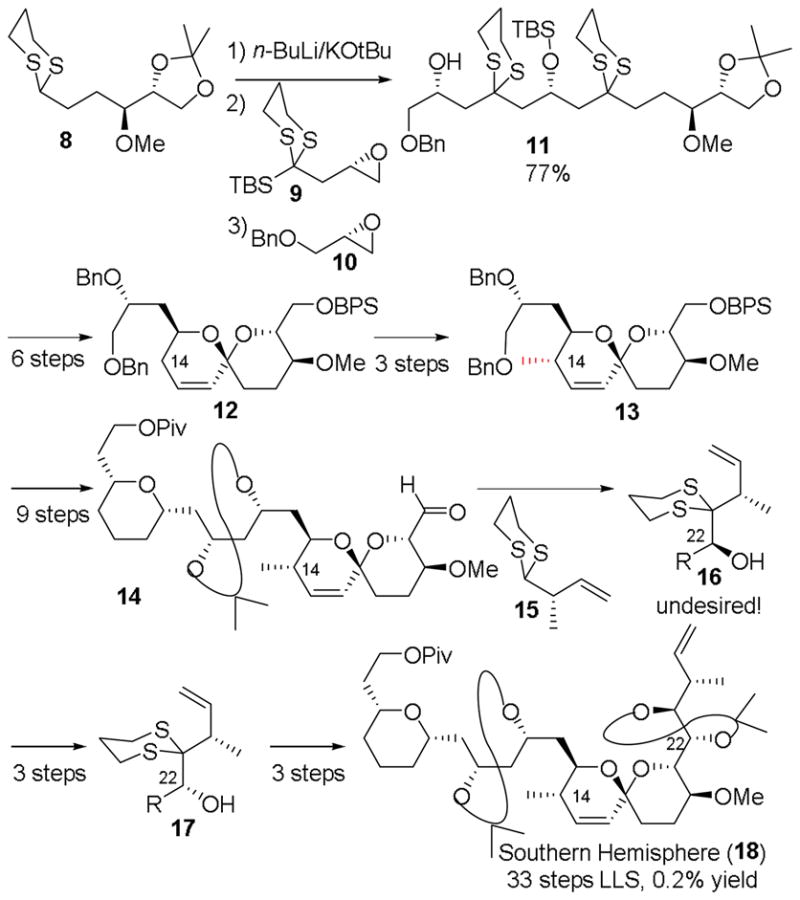
First-generation analysis of the C(1)-C(26) fragment of the spirastrellolides.
To address these shortcomings, we present here a second-generation synthetic analysis, now aimed at spirastrellolide E (Scheme 2). As a new subtarget, we selected C(1)-C(24) fragment 19 (Scheme 2A), guided by the observations of both the Patterson and Furstner groups that union with northern hemispheres might best be accomplished at C(24).8,20,21 A key strategic consideration in our second generation analysis was to alter the bonds forged by the ARC protocol, thus permitting incorporation of the C14 methyl group as part of an appropriately designed epoxide (24). Thus, the revised strategy clearly showcases the flexibility inherent in the ARC tactic.
Scheme 2.

Retrosynthetic Analysis.
Moreover, inspired by our recent northern hemisphere synthesis,26 we envisioned access to the southern hemisphere [6,6]-spiroketal via a gold-catalysed cyclization. However, given the difficulties often encountered with control of regioselectivity in gold-catalyzed spiroketalizations, we chose to exploit the method of Aponick and co-workers,37 which employs a substrate propargylic carbinol to direct the site of ring closure, via an intermediate allene (Scheme 2B).
With this scenario in mind, the southern hemisphere 19 would arise from an appropriately functionalized propargylic triol 20. Inclusion of a carbinol directing group conveniently revealed an aldehyde alkynylation retron, simplifying construction of 20 to two fragments: alkyne 21 and aldehyde 22. Alkyne 21, in turn, would be constructed in 8 steps using a strategy adapted from the Patterson spirastrellolide A synthesis,30 while aldehyde 22 appeared as an ideal substrate for Type I Anion Relay Chemistry,36 employing TES-dithiane 23 with epoxides 2438 and 25.39
Our initial approach to aldehyde 22 is outlined in Scheme 3. Treatment of TES-dithiane 23 with n-butyllithium, followed by addition of epoxide (−)-24 in a mixture of tetrahydrofuran (THF) and ether (3:1), and in turn a solvent-mediated Brook rearrangement of the intermediate lithium alkoxide, triggered by the addition of hexamethylphosphramide (HMPA) in the presence of epoxide (−)-25, smoothly furnished the three component adduct (+)-26 in 60–70% yield.
Scheme 3.

Synthesis of aldehyde 22.
With the ARC product (+)-26 in hand, we turned to dithiane removal, which proved to be non-trivial. A wide range of conditions (cf. the Stork reagent,40 mercury salts, NCS/AgNO3, 41 iodine/sodium bicarbonate,42 methyl iodide43) led either to poor or inconsistent results. Eventually we discovered that a combination of NBS, silver perchlorate, and 2,6-lutidine41,44 furnished a reliable and reproducible yield of 70–80% of the desired ketone (+)-27.
Having established conditions for dithiane removal, we next performed a stereocontrolled anti-reduction of the derived hydroxyketone to form the desired anti diol in 70% yield as the only observed diastereomer (Scheme 3). At this stage, all that remained was a series of functional group manipulations to arrive at the aldehyde fragment; this however also proved non-trivial. Attempted protection of the diol as a bis-TBS ether resulted in partial migration of the C(13) TES group in 27. Unfortunately, the resulting products proved inseparable by column chromatography. Switching to bis-MOM protection (i.e. 29) alleviated this problem, albeit the MOM protecting group was deemed not ideal from the point of view of a total synthesis, given the harsh conditions typically required for removal. Notwithstanding these issues, we continued with the synthesis to validate several critical late stage transformations. Removal of the benzyl group and oxidation to aldehyde (−)-22 again proved troublesome due to TES migration and partial deprotection. Nonetheless, the stage was set to attempt the union via alkynylation.
Two observations proved important. First, at this stage of our spirastrellolide synthetic venture, the required configuration of the C(15) propargylic hydroxyl was unknown. Second, a number of groups had reported a strong stereochemical dependence of the propargylic stereogenic center on spiroketalizations.37,45 We therefore decided to initially pursue a non-selective alkynylation, separate the diastereomers, and then subject each to gold catalysis (Scheme 4). To this end, treatment of alkyne (−)-2146 with lithium diisopropylamide (LDA) in THF followed by addition of aldehyde (−)-22 in THF, furnished a diastereomeric mixture of propargylic alcohols [(+)-30a and (+)-30b (1.7:1)] in a combined yield of 75%. Pleasingly, the diastereomers proved readily separable via routine flash column chromatography. Each isomer in turn was subjected to PMB removal to furnish the spiroketalization precursors (+)-31a and (+)-31b.
Scheme 4.

First attempts at southern hemisphere end game.
It quickly became evident that our originally designed spirocyclization substrates were not optimal. We had anticipated that spiroketalization conditions could be found that would permit simultaneous removal of both the C(13) TES group, a prerequisite for spiroketalization, as well as removal of the C(22) TES group, a requirement for a planned Suzuki union of the Northern and Southern hemispheres. In practice, however, the required use of methanol as either a solvent or cosolvent for the spiroketalization, along with a mild acid, lead to slow decomposition of the substrate. Only traces of spiroketals were observed under a variety of conditions.
Undaunted, we examined a differentially protected construct, in which the two alcohols required for spiroketalization could be revealed selectively without affecting the C(22) TES group. Such a scenario would greatly facilitate optimization of the key cyclization tactic. This approach would also permit replacement of the C(13) TES group, which had earlier led to significant difficulties due to its lability.
The revised strategy is illustrated in Scheme 5. Here we target differentially protected spiroketalization precursor 32. Importantly, the requisite advanced ARC adduct (35, Scheme 6) could be accessed employing the same components as the previous route, by merely changing the order of addition of the two epoxides, again demonstrating the flexibility of Anion Relay Chemistry.
Scheme 5.

Revised retrosynthesis
Scheme 6.

Implementation of the revised retrosynthetic analysis.
Implementation of the revised strategy is outlined is Scheme 6. Pleasingly, the ARC union proceeded smoothly with the inverted order of addition to furnish three component adduct (+)-35 in 68% yield. Dithiane hydrolysis as before then led to (+)-36.
With (+)-36 in hand, we now required a tactic to reduce the C(11) ketone with concomitant differential protection of the resulting diol. This requirement was conveniently achieved by exploiting the Evans-Tishchenko reaction47 with benzaldehyde, followed in turn by TBS protection of the remaining hydroxyl, removal of the benzyl group, and oxidation of the resultant hydroxyl48 to furnish aldehyde (+)-33. Notably, yields for the TES-free synthetic route were uniformly excellent. Alkynylation exploiting the previously established protocol then furnished a mixture of diastereomeric alcohols (+)-32a and (+)-32b (1.4:1) in 85% combined yield, which again could be readily separated and subjected to a two stage deprotection to remove the benzoyl and PMB groups to furnish spiroketalization precursors (+)-38a and (+)-38b. At this stage, we were also able to confirm the configurations of the C(15) hydroxyl group in 32a and 32b by conversion of 38a to the bisacetonide, with concomitant removal of the C(22) TES ether. The relative stereochemistry of the resulting bisacetal was determined by NMR, utilizing the method of Rychnovsky.49
With the differentially protected adducts 38a and 38b in hand, we turned to the critical spiroketalization (Scheme 7). Treatment of the cis isomer (+)-38a with cationic gold catalyst 39, first prepared by Echavarren,50 employing methylene chloride as the solvent proceeded smoothly to furnished the desired spiroketal (19). The stereostructure of the spiroketal was assigned by extensive 2D NMR analysis.51 Particularly important was observation of an nOe between the hydrogens on C(13) and C(21), which provided evidence both for the expected double carbinol addition to the alkyne, as well as the stereochemistry at the ketal center. A similar nOe was observed in a related system by Paterson.19 We were thus confident that the spiroketalization had proceeded as predicted, thereby signaling completion of the C(1)-C(24) southern hemisphere fragment for a prospective total synthesis of spirastrellolide E (5).
Scheme 7.
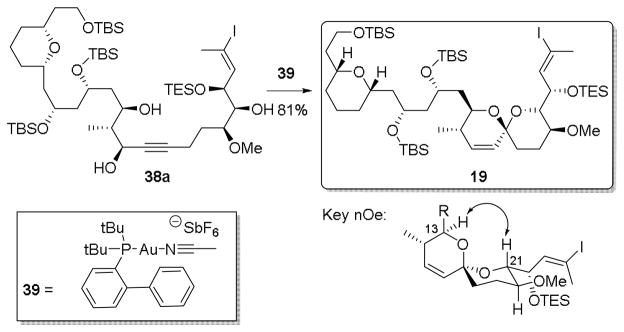
Completion of the southern hemisphere.
The anti isomer (+)-38b however did not lead to the anticipated spiroketalization product under otherwise identical conditions. Structural determination of the observed product, along with the development of a rationale for the stereochemical dependence on reaction efficiency, is currently under investigation.
In summary, we have achieved a significantly improved second generation synthesis of a spirastrellolide E C(1)-C(24) southern fragment, now involving a longest linear sequence of 19 steps and an overall yield of 2%. With streamlined routes to both hemispheres now available, efforts turned to the total synthesis of spirastrellolide E, which will be reported in due course.
Supplementary Material
Acknowledgments
This Tetrahedron Letters is presented to honor and remember Professor Harry Wasserman (Yale University), great friend, true gentleman, and scientist par excellence: Harry, thank you for everything.
Financial support for this work was provided by the National Institutes of Health, Grant G.M.-29028. The authors would like to acknowledge Dr. George Furst for help in obtaining NMR spectra, as well as Dr. Rakesh Kohli for providing high resolution mass spectrometry spectra.
Footnotes
Experimental procedures and characterization of new compounds, as well as detailed NMR analysis of compound 19 can be found in the online version of this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Williams DE, Roberge M, Van Soest R, Andersen RJ. J Am Chem Soc. 2003;125:5296–7. doi: 10.1021/ja0348602. [DOI] [PubMed] [Google Scholar]
- 2.Williams DE, Lapawa M, Feng X, Tarling T, Roberge M, Andersen RJ. Org Lett. 2004;6:2607–10. doi: 10.1021/ol0490983. [DOI] [PubMed] [Google Scholar]
- 3.Warabi K, Williams DE, Patrick BO, Roberge M, Andersen RJ. J Am Chem Soc. 2007;129:508–9. doi: 10.1021/ja068271i. [DOI] [PubMed] [Google Scholar]
- 4.Williams DE, Keyzers Ra, Warabi K, Desjardine K, Riffell JL, Roberge M, Andersen RJ. J Org Chem. 2007;72:9842–5. doi: 10.1021/jo7018174. [DOI] [PubMed] [Google Scholar]
- 5.Chandrasekhar S, Rambabu C, Reddy AS. Org Lett. 2008;10:4355–7. doi: 10.1021/ol801771s. [DOI] [PubMed] [Google Scholar]
- 6.Chen JLY, Brimble MA. Chem Commun (Camb) 2010;46:3967–9. doi: 10.1039/c0cc00056f. [DOI] [PubMed] [Google Scholar]
- 7.Chen JLY, Brimble MA. J Org Chem. 2011;76:9417–28. doi: 10.1021/jo201729t. [DOI] [PubMed] [Google Scholar]
- 8.Fürstner A, Fasching B, O’Neil GW, Fenster MDB, Godbout C, Ceccon J. Chem Commun (Camb) 2007;3:3045–7. doi: 10.1039/b707835h. [DOI] [PubMed] [Google Scholar]
- 9.Fürstner A, Fenster MDB, Fasching B, Godbout C, Radkowski K. Angew Chem Int Ed Engl. 2006;45:5510–5. doi: 10.1002/anie.200601655. [DOI] [PubMed] [Google Scholar]
- 10.Fürstner A, Fenster MDB, Fasching B, Godbout C, Radkowski K. Angew Chem Int Ed Engl. 2006;45:5506–10. doi: 10.1002/anie.200601654. [DOI] [PubMed] [Google Scholar]
- 11.Keaton KA, Phillips AJ. Org Lett. 2008;10:1083–6. doi: 10.1021/ol702955m. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Hsung RP. Org Lett. 2005;7:2273–6. doi: 10.1021/ol050653q. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Yang JH, Ko C, Hsung RP. Tetrahedron Lett. 2006;47:6121–3. [Google Scholar]
- 14.Pan Y, De Brabander J. Synlett. 2006:853–6. [Google Scholar]
- 15.Paterson I, Anderson EA, Dalby SM, Lim JH, Loiseleur O, Maltas P, Moessner C. Pure Appl Chem. 2007;79:667–76. [Google Scholar]
- 16.Paterson I, Anderson EA, Dalby SM, Genovino J, Lim JH, Moessner C. Chem Commun (Camb) 2007:1852–4. doi: 10.1039/b700827a. [DOI] [PubMed] [Google Scholar]
- 17.Paterson I, Anderson EA, Dalby SM, Lim JH, Maltas P, Moessner C. Chem Commun (Camb) 2006:4186–8. doi: 10.1039/b612697a. [DOI] [PubMed] [Google Scholar]
- 18.Paterson I, Anderson EA, Dalby SM, Loiseleur O. Org Lett. 2005;7:4121–4. doi: 10.1021/ol051403c. [DOI] [PubMed] [Google Scholar]
- 19.Paterson I, Anderson EA, Dalby SM, Loiseleur O. Org Lett. 2005;7:4125–8. doi: 10.1021/ol051405x. [DOI] [PubMed] [Google Scholar]
- 20.Paterson I, Dalby SM, Maltas P. Isr J Chem. 2011;51:406–19. [Google Scholar]
- 21.Paterson I, Maltas P, Anderson EA. Pure Appl Chem. 2013;85:1133–47. [Google Scholar]
- 22.Smith AB, III, Kim DS. Org Lett. 2007;9:3311–4. doi: 10.1021/ol071282b. [DOI] [PubMed] [Google Scholar]
- 23.Smith AB, III, Smits H, Kim DS. Tetrahedron. 2010;66:6597–605. doi: 10.1016/j.tet.2010.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Forsyth CJ. Org Lett. 2006;8:2997– 3000. doi: 10.1021/ol0609457. [DOI] [PubMed] [Google Scholar]
- 25.Wang C-C, Tang Y, Yang K, Li X-Y, Wu Y-B, Hsung RP. Tetrahedron. 2013;69:8284–90. [Google Scholar]
- 26.Wang X, Paxton TJ, Li N, Smith AB., III Org Lett. 2012;14:3998–4001. doi: 10.1021/ol301795a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang JH, Liu J, Hsung RP. Org Lett. 2008;10:2525–8. doi: 10.1021/ol8008057. [DOI] [PubMed] [Google Scholar]
- 28.Paterson I, Anderson EA, Dalby SM, Lim JH, Genovino J, Maltas P, Moessner C. Angew Chem Int Ed Engl. 2008;47:3016–20. doi: 10.1002/anie.200705565. [DOI] [PubMed] [Google Scholar]
- 29.Paterson I, Anderson EA, Dalby SM, Lim JH, Genovino J, Maltas P, Moessner C. Angew Chem Int Ed Engl. 2008;47:3021–5. doi: 10.1002/anie.200705566. [DOI] [PubMed] [Google Scholar]
- 30.Paterson I, Anderson EA, Dalby SM, Lim JH, Maltas P, Loiseleur O, Genovino J, Moessner C. Org Biomol Chem. 2012;10:5861–72. doi: 10.1039/c2ob25100k. [DOI] [PubMed] [Google Scholar]
- 31.Paterson I, Anderson EA, Dalby SM, Lim JH, Maltas P. Org Biomol Chem. 2012;10:5873–86. doi: 10.1039/c2ob25101a. [DOI] [PubMed] [Google Scholar]
- 32.O’Neil GW, Ceccon J, Benson S, Collin MP, Fasching B, Fürstner A. Angew Chem Int Ed Engl. 2009;48:9940–5. doi: 10.1002/anie.200906121. [DOI] [PubMed] [Google Scholar]
- 33.Benson S, Collin MP, O’Neil GW, Ceccon J, Fasching B, Fenster MDB, Godbout C, Radkowski K, Goddard R, Fürstner A. Angew Chem Int Ed Engl. 2009;48:9946–50. doi: 10.1002/anie.200906122. [DOI] [PubMed] [Google Scholar]
- 34.Benson S, Collin MP, Arlt A, Gabor B, Goddard R, Fürstner A. Angew Chem Int Ed Engl. 2011;50:8739–44. doi: 10.1002/anie.201103270. [DOI] [PubMed] [Google Scholar]
- 35.Arlt A, Benson S, Schulthoff S, Gabor B, Fürstner A. Chem - a Eur J. 2013;19:3596–608. doi: 10.1002/chem.201203965. [DOI] [PubMed] [Google Scholar]
- 36.Smith AB, III, Wuest WM. Chem Commun (Camb) 2008:5883–95. doi: 10.1039/b810394a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aponick A, Li CY, Palmes JA. Org Lett. 2009;11:121–4. doi: 10.1021/ol802491m. [DOI] [PubMed] [Google Scholar]
- 38.Mori K, Iwasawa H. Tetrahedron. 1980;36:87–90. [Google Scholar]
- 39.Available in two steps from known alkene, see reference 30.
- 40.Stork G, Zhao K. Tetrahedron Lett. 1989;30:287– 90. [Google Scholar]
- 41.Corey EJ, Erickson BW. J Org Chem. 1971;36:3553–60. [Google Scholar]
- 42.Russell GA, Ochrymowycz LEOA. J Org Chem. 1969;34:3618–24. [Google Scholar]
- 43.Fetizon M, Jurion M. J Chem Soc, Chem Commun. 1972:382–3. [Google Scholar]
- 44.Nicolaou KC, Patron AP, Ajito K, Richter PK, Khatuya H, Bertinato P, Miller RA, Tomaszewski MJ. Chem - a Eur J. 1996;2:847– 68. [Google Scholar]
- 45.Fang C, Pang Y, Forsyth CJ. Org Lett. 2010;12:4528–31. doi: 10.1021/ol101833h. [DOI] [PubMed] [Google Scholar]
- 46.Prepared by a modification of a route developed by Paterson. See the supporting information.
- 47.Evans DA, Hoveyda AH. J Am Chem Soc. 1990;112:6447–9. [Google Scholar]
- 48.Careful oxidation with DMP was necessary to avoid facile benzoate elimination to give the corresponding α,β-unsaturated aldehyde, which was inseperable.
- 49.Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–8. [Google Scholar]
- 50.Nieto-Oberhuber C, Muñoz MP, López S, Jiménez-Núñez E, Nevado C, Herrero-Gómez E, Raducan M, Echavarren AM. Chemistry. 2006;12:1677–93. doi: 10.1002/chem.200501088. [DOI] [PubMed] [Google Scholar]
- 51.See suporting information for NMR analysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.