

Abstract
A nitrosopurine-ene reaction easily assembles the asmarine pharmacophore and transmits remote stereochemistry to the diazepine-purine hetereocycle. This reaction generates potent cytotoxins that exceed the potency of asmarine A (1.2 μM IC50) and supersede the metabolites as useful leads for biological discovery.
Keywords: alkaloids, terpenoids, nitroso, purine, ene reaction
Asmarines A and B (1&2, Figure 1) were identified in 1998 by Kashman and co-workers as the bioactive constituents of a Red Sea sponge (Raspailia sp.) extract, exhibiting cytotoxicity against several cancer cell lines with a minimum EC50 of 1.2 μM and 120 nM, respectively.[1] The asmarines are unique among alkaloids by virtue of the embedded N-hydroxypurine diazepine (primary pharmacophore)[2] connected by an ethyl bridge to a clerodane decalin (putative secondary pharmacophore). Biosynthetically, the asmarines derive from agelasines (e.g. 3), which are thought to exert cytotoxicity by Na+/K+ ATPase inhibition[3] or by membrane disruption.[4] However, the uncharged purines 1 and 2 are likely to possess different mechanisms than 3 (vide infra). Thus, the driving force for investigation of the asmarines is to determine their mechanism of action and chemical reactivity within the cell. However, no material remains from the original isolation work[5] and the exact identity of the sponge is unknown.[1b] Chemical synthesis is therefore the most feasible way to access material for biological study.\
Figure 1.
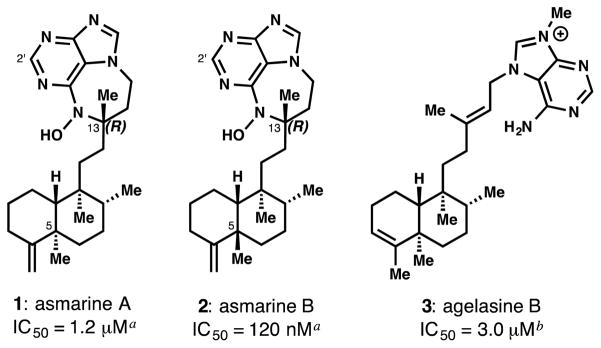
Asmarines may be derived from agelasines. aagainst HT-29 cells; bagainst MCF-7 cells [3b].
Procurement of these cytotoxic molecules represents a challenge for synthesis despite efforts by Kashman, Schauss, Tashiro and Gundersen.[6] Their difficulty arises partly from the remoteness of the stereogenic tert-alkyl N-hydroxyamine moiety (see Figure 1), which frustrates stereocontrol relative to the clerodane subunit. Furthermore, synthesis of the tert-alkyl N-hydroxy-diazepinepurine pharmacophore is a challenge in itself[6a,b,d] and only one route exists. This successful strategy by Kashman[2] utilizes a carbocationic mode of ring closure (30% HBr/AcOH at 100 °C), which limits functional group compatibility and stereocontrol, and delivers analogs (e.g. 4–7) that, while bioactive, reach a maximum potency of 4 μM[7] (GI50, HT-29, see Figure 2a). We aimed to secure efficient access to the asmarines in order to understand their SAR and to provide simplified, high potency analogs for further biological study.
Figure 2.

a. Prior analog work. b. Stereochemical relay and high potency analogs via nitrosopurine-ene reaction. acytotoxicity against HT-29 cells (EC50 more appropriate).
A biomimetic strategy to close the seven-membered ring from an agelasine-type intermediate 8 (Figure 2b) appeared to offer high efficiency. However, control of the diazepine stereochemistry during C-N bond formation presented a serious obstacle to this strategy. We wondered if a nitrosoene reaction[8] of linear precursor 8 might relay stereochemistry[9] of the decalin core to the tert-alkyl amine stereocenter in 9, and achieve the necessary Markovnikov selectivity.[10] Here we report 1) the successful implementation of this strategy to relay clerodane stereochemistry to the remote stereocenter, 2) the ability of this strategy to probe the role of stereochemistry in SAR, and 3) the use of this nitrosopurine-ene reaction to synthesize simplified high potency asmarine analogs that exhibit cytotoxicity at nanomolar levels against multiple cancer cell lines.
A short route to the targeted nitrosopurine (8) began with 6-chloro-4,5-pyrimidinediamine (11) and 4-iodo-1-butyne (12). Diamine 11 was monoformylated to amide 13, and 12 was subjected to the Wipf-modified[11] Negishi carboalumination,[12] followed by alkylation of the intermediate organoaluminum with gaseous formaldehyde[13] to yield iodoalcohol 14. Formamide 13 was alkylated with iodide 14 and heating effected ring closure to chloropurine 15.[14] This alcohol was converted to allylbromide 16 (see Supporting Information), which was then trapped with the enolate generated from dissolving metal reduction of (methyl)-Wieland-Miescher ketone[15] ketal 17 (99% ee) in presence of bis(2-methoxyethyl)amine[16] to yield ketone 18 as a single stereoisomer. Selective displacement of the arylchloride of 18 with hydroxylamine proved challenging since condensation with the decalin ketone to form an oxime occurred competitively. We reasoned that the oxime might be cleaved later, and thus decided to push the reaction towards the bis-hydroxyaminated product 19 – an unforeseen but providential choice (see Table 1). Having obtained the agelasine scaffold 19, oxidation of the 6-N-hydroxyaminopurine (HAP) moiety to a nitrosopurine was explored in a model system (see Figure 3).
Table 1.
Solvent and substituent control stereoinduction.
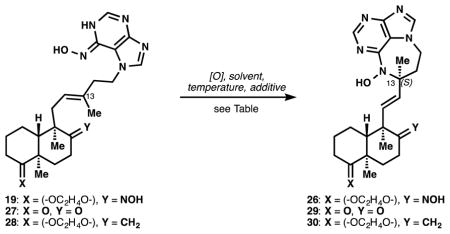
| |||||
|---|---|---|---|---|---|
| Entry | Y | Solvent | Temperature | Additive | 13-(S) : 13-(R) |
| 1 | NOH | DMF | 22 °C | – | 1.5 : 1.0 |
| 2 | NOH | CH2Cl2 | 22 °C | – | 1.5 : 1.0 |
| 3 | NOH | EtOH | 22 °C | – | 3.8 : 1.0 |
| 4 | NOH | EtOH | −20 °C | – | 3.0 : 1.0 |
| 5 | NOH | 1:9 MeOH/CH2Cl2 | 22 °C | NH4OH (1%) | 2.1 : 1.0 |
| 6 | NOH | EtOH | 22 °C | AcOH (1 equiv.) | 2.7 : 1.0 |
| 7 | NOH | TFE | 22 °C | – | 2.3 : 1.0 |
| 8 | NOH | 1:1 EtOH/H2O | 22 °C | – | 4.7 : 1.0 |
| 9 | NOH | 1:3 EtOH/H2O | 22 °C | – | 7.9 : 1.0 |
| 10 | O | 1:4 EtOH/H2O | 22 °C | – | 1.2 : 1.0 |
| 11 | CH2 | 1:4 EtOH/H2O | 22 °C | – | 1.1 : 1.0 |
Figure 3.

Proof-of-principle for nitrosopurine-ene reactions.
The parent compound 6-nitrosopurine 20[17] proved unstable and in our hands could not be isolated. However, we found that treatment of 6-N-hydroxyaminopurine 21 with MnO2 in DMSO and in the presence of excess tetramethylethylene gave the expected tert-alkyl methallyl hydroxylamine 22. A variety of oxidants (I2, Mn(OAc)3, PhI(OAc)2) gave similar results and on large scale PhI(OAc)2 proved more amenable to purification. When these oxidation conditions were applied to N-hydroxyaminopurine 23 in a mixture of methanol and dichloromethane, the targeted 7-membered diazepine purine 24 was obtained as the sole product with no trace of the 8-membered diazocine. Notably, this reaction could be preformed on gram-scale, demonstrating that the chemistry of nitrosopurines (e.g. 25) is a practical means of procuring material.
When applied to agelasine-type compound 19, we also obtained the diazepine 26 exclusively, but initial experiments generated this as a 1:1 mixture of diastereomers (entry 1, Table 1). Fortunately, we discovered two influences on diastereoselectivity in the ene reaction. There was a marked solvent effect associated with diastereoselectivity, whereby more polar protic solvents increased selectivity, and water proved the most selective (ethanol was added for solubility). This effect was not due to protonation or hydrogen bonding, since acetic acid had no pronounced effect (entry 6), nor did the strongly hydrogen bond-donating solvent trifluoroethanol[18] (entry 7) compared to ethanol (entry 3). There was also a stark and serendipitous substituent effect: although the incidental oxime caused high stereoselectivity, the ketone 27 and exocyclic methylene 28 showed almost no selectivity in formation of 29 and 30, respectively (entries 10 and 11). Application of the polarized diradical (PD, 31) model for the nitroso-ene reaction[19] of 19 suggests that developing positive charge at carbon in the transition state might be stabilized by the proximal oxime (Figure 4). Conformer 32 should be lower in energy than 33, which suffers a steric clash between the methyl and the aromatic ring. These polarized transition states would be more populated in solvents of high polarity (e,g, water), as predicted by Houk.[19]
Figure 4.
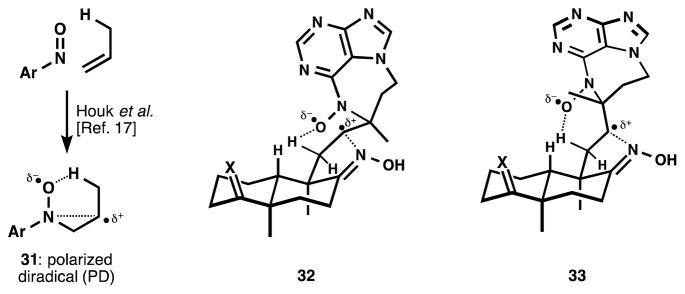
Water might increase the polar character in a nitrosoene polarized diradical (PD) and the transition state leading to it. Conformers of different energy are stabilized by the proximal oxime; X = (-OC2H4O-).
This strategy allowed us to directly probe the contribution of diazepine stereochemistry to cytotoxicity. The major diastereomer 26 (Figure 5) exhibited higher potency against HT-29 cells (EC50 = 8 μM) than the minor isomer 34 (17 μM), even though 34 corresponds to the stereochemistry of the natural asmarines (determined by x-ray analysis,[20] see Supporting Information). Since the difference in potency is not profound, the stereochemistry in the diazepine seems to play only a minor role in the mechanism of action.
Figure 5.

Activity (EC50, 48 h) against HT-29 cells. Although there is clear SAR, stereochemistry does not play a major role. X = (-OC2H4O-)
Synthesis also allowed us to probe the role of absolute stereochemistry. We were surprised to find that ent-26 and ent-34 possessed similar biological activity as their enantiomers (EC50 = 10 μM for both). Similarly, diketones 35 and ent-35 possess activity that differs only two-fold. Whereas this similarity might suggest a non-specific mechanism of action, it seems more likely that the differences in potencies reflect cell-permeability, affecting accessibility to the target. As such, replacement of the oxime with a more lipophilic methylene delivers compounds with greater potency (36 and 37). In this case, the ‘natural’ diazepine 37 is five-fold more potent than the ‘unnatural’ diastereomer 36, and in fact matches the potency against HT-29 cells reported for asmarine A (EC50 = 1 μM). As control compounds, we also tested the N-H purine 38 and the uncyclized N-hydroxyaminopurine 19. Both compounds were unable to effectively induce cell death after 72 h. The inactivity of 19 and 37 suggests a different mechanism than the inhibition of ATPases ascribed to the agelasines and their simplified analogs,[21] although further study will be necessary to rule out this target.
Since the ultimate goal of our work was procurement of material for biological study and it was clear that stereochemistry had little influence on the activity of the asmarines, we adapted the route shown in Scheme 1 to the late-stage, divergent appendage of unnatural hydrophobic cores. As shown in Figure 6, a short convergent sequence from 13 and 39 was devised to arrive at iodide 40 on gram scale. From 39, a variety of substituents were added by a very effective Negishi coupling[22] - a noteworthy step given the acidic proton on the hydroxyamino group and our observation that the N-hydroxyamino purine will chelate metals. Each adduct (41–48) was then cyclized via the nitrosopurine-ene reaction to its N-hydroxydiazepine purine 50–57 to generate a small library of cytotoxic compounds.
Scheme 1.

Union of clerodane and purine cores, and a nitrosopurine-ene reaction with remote stereocontrol.
Figure 6.

A late-stage divergent route to unnatural asmarines and their activities (EC50, 48 h) against HT-29 and HeLa cells. Potencies are on a logarithmic scale.
Each molecule in Figure 6 was screened for cytotoxicity against HT-29, Jurkat, and HeLa cells. While compounds with truncated sidechains (24, 49–53) show cytotoxicity, the potency is weak, with or without an unsaturated linker (24 vs. 49). Small rings like a methylenecyclobutane can be generated in the ene reaction, highlighting the mildness of the reaction conditions, and in stark contrast to the previously reported high temperature/strong acid method of ring closure. Therefore, esters (52) and amino acid motifs can be incorporated (53); the β,γ-unsaturated amino ester carbamate does not migrate into conjugation under the reaction conditions. This latter example shows some small stereoselectivity (2:1) associated with the presence of the carbamate (compare to methyl, 52), supporting the idea that a Lewis basic group is necessary for stereoinduction. However, none of these short, polar side chains show significant potency. In contrast, large hydrophobic groups similar to the clerodane cores of asmarines A and B induce high potency. For example, compounds with 1- and 2-naphthyl substituents (54 and 55) possess single digit μM activity; 54 is equipotent to asmarine A (1.1 μM against HT-29). 1-Adamantyl (56) and 4-biphenyl (57) asmarines are more potent still with sub-micromolar activity against all three cell lines (471 nM–714 nM; 56 also inhibits HL60 cells at 199 nM, see Figure 8), approaching the 120 nM IC50 value reported for asmarine B (2) against HT29 cells. In fact, this study represents a rare example of the deprioritization of isolated metabolites in light of near-equipotent but simpler analogs. Readily-accessible asmarines 56 and 57 supersede the scarce metabolites 1 and 2 as useful tools for biochemical inquiry.
Figure 8.

1-adamantyl-asmarine (55) and 4-biphenyl-asmarine (56) possess nM activity against seven cell lines.
Whereas there is some latitude in the choice of hydrophobic lobe, the N-hydroxy diazepine purine is more conservative. Acyclic tert-alkyl N-hydroxyaminopurine 19 was completely inactive (Figure 7), indicating that a ring is necessary. However, the ring-expanded N-hydroxy diazocine purine 51 was similarly inactive. This 8-membered ring was the unexpected anti-Markovnikov product of nitrosopurine-ene reaction of cyclopropane 42, which reacts with ‘twix’ selectivity and avoids the alternative, highly strained methylenecyclopropane (Figure 7).
Figure 7.
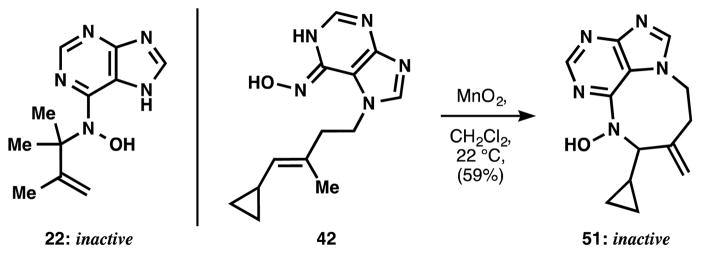
Structural specificity for activity.
The potency of 56 and 57 is general, showing sub-micromolar cytotoxicity against HL60 (leukemia), HEK 293, MCF7 (breast cancer), and MDA-MB 231 (breast cancer) cell lines, in addition to the HT29, Jurkat, and Hela cell lines (Figure 8). The activity of 56 is surprising, since the saturated analog was reported by Kashman to posses very weak activity against two cancer cell lines (NSCL A549 and PANC1, EC50 > 27 μM). The activity of 57 is noteworthy since installation of functional groups on the aromatic rings should allow a variety of pull down experiments.
This work builds a platform for the discovery of the mechanism of action of the asmarines. We are entertaining three hypotheses: non-covalent, covalent, and radical. The latter two possibilities are supported by some experimental data. Kashman[1b] and Tashiro[6c] have reported the instability of the N-hydroxypurine upon acylation, wherein methanol adds to the purine ring and cleaves the N-O bond (2→58, Figure 9). This mode of reactivity may also be effected in vivo by acetylation or phosphorylation. Alternatively, we observed that the tert-alkyl N-hydroxypurine is readily oxidized with mild reagents. The acyclic purine 22 generates stable nitroxide radical 59, a vibrant red-orange crystalline solid whose structure was confirmed by x-ray analysis.[23] Interestingly, the nitroxides of the diazepines (e.g. 24) are very unstable, cannot be isolated, and instead appear to disproportionate to the corresponding N-H diazepines, suggesting that, if generated, they may react with a target in vivo.
Figure 9.
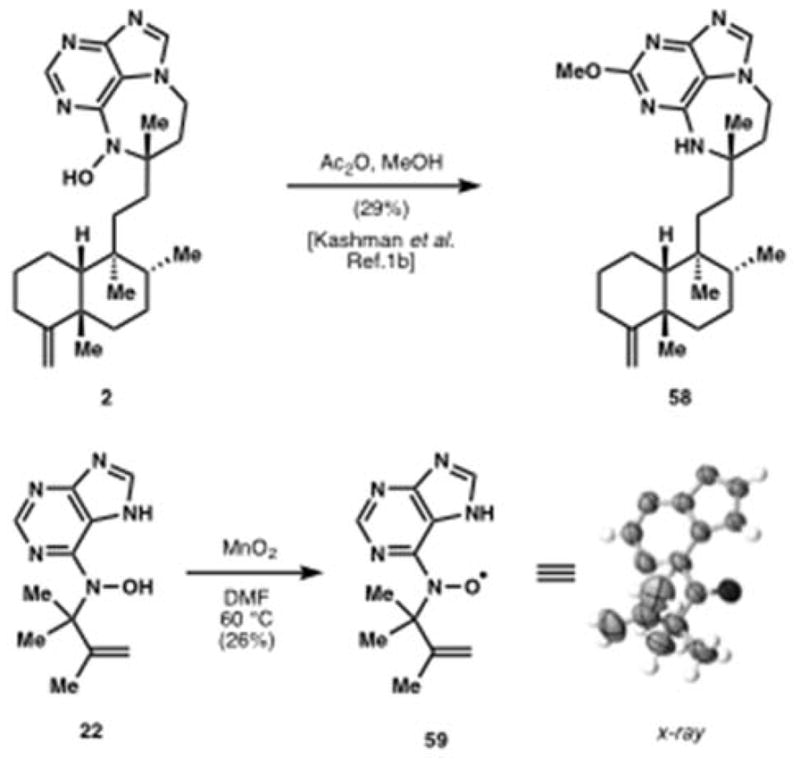
Reactivity of potential relevance to bioactivity.
To summarize, we have established a chemical platform for the study of asmarine cytotoxins, enabled by an unusual but highly practical nitrosopurine-ene reaction. This reaction exhibits both high regioselectivity for the Markovnikov product and exhibits high chemoselectivity. Identification of a Lewis base (oxime) as a stereoelectronic control element in the nitroso-ene reaction may be generally useful for linear stereocontrol. Use of the nitrosopurine-ene reaction as a simplifying element in the synthesis of cytotoxic asmarine analogs enables very short syntheses with few redox manipulations and no protecting groups. Notably, this route is diversifiable at a late stage, and has generated potent analogs with cytotoxicity in the nanomolar range. We have also found that 1) increased potency may result from cell permeability, but the N-hydroxypurine diazepine is required; 2) the similar potency of 54 and 55 suggests the hydrophobic moiety does not play a major role in target binding; 3) stereochemistry at the diazepine plays a minor, but not insignificant role in potency; 4) the N-O bond is required for activity, since an N-H analog is inactive; and 5) the seven-membered ring is required for activity, since acyclic variants and a diazocine analog show no cytotoxicity. These findings, coupled with the development of a short, scalable and divergent route to asmarine analogs, and especially the identification of potent biphenyl asmarine 57 lay the groundwork for identification of the mechanism of action of these enigmatic metabolites.
Supplementary Material
Footnotes
This work was supported by the NIH (GM104180) and the NSF GRFP (K.K.W.; DGE-1346837). We thank Dr. C. E. Moore and Prof. A. L. Rheingold (UCSD) for x-ray crystallographic analysis. We thank TSRI, Eli Lilly, Boehringer Ingelheim, Amgen, the Baxter Foundation and the Sloan Foundation for additional financial support.
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Kanny K. Wan, Department of Chemistry, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla CA 92037 (USA)
Kotaro Iwasaki, Department of Chemistry, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla CA 92037 (USA).
Jeffrey C. Umotoy, Department of Molecular and Experimental Medicine and Chemical Physiology, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla CA 92037 (ISA)
Dennis W. Wolan, Email: wolani@scripps.edu, Department of Molecular and Experimental Medicine and Chemical Physiology, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla CA 92037 (ISA)
Ryan A. Shenvi, Email: rshenvi@scripps.edu, Department of Chemistry, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla CA 92037 (USA)
References
- 1.a) Yosief T, Rudi A, Stein Z, Goldberg I, Gravalos GMD, Schleyer M, Kashman Y. Tetrahedron Lett. 1998;39:3323–3326. [Google Scholar]; b) Yosief T, Rudi A, Kashman Y. J Nat Prod. 2000;63:299–304. doi: 10.1021/np9902690. [DOI] [PubMed] [Google Scholar]
- 2.Pappo D, Shimony S, Kashman Y. J Org Chem. 2005;70:199–206. doi: 10.1021/jo048622g. [DOI] [PubMed] [Google Scholar]
- 3.a) Nakamura H, Wu H, Ohizumi Y, Hirata Y. Tetrahedron Lett. 1984;25:2989. [Google Scholar]; b) Pimentel AA, Felibertt P, Sojo F, Colman L, Mayora A, Silva ML, Rojas H, Dipolo R, Suarez AI, Compagnone RS, Arvelo F, Galindo-Castro I, De Sanctis JB, Chirino P, Benaim G. Cancer Chemother Pharmacol. 2012;69:71. doi: 10.1007/s00280-011-1677-x. [DOI] [PubMed] [Google Scholar]; c) Fathi-Afshar R, Allen TM, Krueger CA, Cook DA, Clanachan AS, Vriend R, Baer HP, Cass CE. Can J Physiol Pharmacol. 1989;67:276. doi: 10.1139/y89-045. [DOI] [PubMed] [Google Scholar]
- 4.Vik A, Hedner E, Charnock C, Tangen LW, Samuelsen Ø, Larsson R, Bohlin L, Gundersen LL. Bioorg Med Chem. 2007;15:4016. doi: 10.1016/j.bmc.2007.03.086. [DOI] [PubMed] [Google Scholar]
- 5.Kashman Yoel. personal communication.
- 6.a) Pappo D, Rudi A, Kashman Y. Tetrahedron Lett. 2001;42:5941. [Google Scholar]; b) Ohba M, Tashiro T. Heterocycles. 2002;57:1235. [Google Scholar]; c) Rodgen SA, Schaus SE. Angew Chem Int Ed. 2006;45:4929. doi: 10.1002/anie.200601076. [DOI] [PubMed] [Google Scholar]; d) Vik A, Gundersen LL. Tetrahedron Lett. 2007;48:1931. [Google Scholar]
- 7.Although labeled as cytotoxicity in Ref.[2], the use of GI50 indicates cytostatic effect. Based on the activity of our analogs, we assume the authors observed cytotoxicity and meant to use EC50.
- 8.Adam W, Krebs O. Chem Rev. 2003;103:4131. doi: 10.1021/cr030004x. [DOI] [PubMed] [Google Scholar]
- 9.a) Keck GE, Webb RR. J Org Chem. 1982;47:1302. [Google Scholar]; b) Adam W, Degen HG, Krebs O, Saha-Möller CR. J Am Chem Soc. 2002;124:12938. doi: 10.1021/ja026640e. [DOI] [PubMed] [Google Scholar]; c) Oppolzer W, Poli G, Starkemann C, Bernardinelli G. Tetrahedron Lett. 1988;29:3559. [Google Scholar]; d) Kreβe G, Vasella A, Felber H, Ritter A, Ascherl B. Recl Trav Chim Pays-Bas. 1986;105:295. [Google Scholar]; e) Adam W, Bottke N. J Am Chem Soc. 2000;122:9846. [Google Scholar]; f) Adam W, Bottke N, Krebs O, Lykakis I, Orfanopoulos M, Stratakis M. J Am Chem Soc. 2002;124:14403. doi: 10.1021/ja027800p. [DOI] [PubMed] [Google Scholar]
- 10.Keck GE, Webb RR, Yates JB. Tetrahedron Lett. 1981;37:4007. [Google Scholar]
- 11.Wipf P, Lim S. Angew Chem Int Ed. 1993;32:1068. [Google Scholar]
- 12.Chen J, Ma S. J Org Chem. 2009;74:5595. doi: 10.1021/jo900389m. [DOI] [PubMed] [Google Scholar]
- 13.Okukado N, Negishi E. Tet Lett. 1978;27:2357. [Google Scholar]
- 14.Montgomery JA, Hewson K. J Org Chem. 1961;26:4469. [Google Scholar]
- 15.Tokoroyama T. Synthesis. 2000;5:611. [Google Scholar]
- 16.Donohoe TJ, Guyo PM, Helliwell M. Tetrahedron Lett. 1999;40:435. [Google Scholar]
- 17.Duthaler RO, Roberts JD. J Am Chem Soc. 1978;100:4969. [Google Scholar]
- 18.Giner-Sorolla A. J Hetero Chem. 1970;7:75. [Google Scholar]
- 19.Leach AG, Houk KNK. J Am Chem Soc. 2002;124:14820. doi: 10.1021/ja012757b. [DOI] [PubMed] [Google Scholar]
- 20.Cambridge Crystallographic Data Centre (CCDC) #1036462
- 21.Gundersen LL. Phytochem Rev. 2013;12:467. [Google Scholar]
- 22.Negishi EI, Hu Q, Huang Z, Qian M, Wang G. Aldrichimica Acta. 2005;38:2005. [Google Scholar]
- 23.Cambridge Crystallographic Data Centre (CCDC) #1036478
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.