

Abstract
Histone deacetylases catalyze the hydrolysis of an acetyl group from post-translationally modified acetyl-lysine residues in a wide variety of essential cellular proteins, including histones. As these lysine modifications can alter the activity and properties of affected proteins, aberrant acetylation/deacetylation may contribute to disease states. Many fundamental questions regarding the substrate specificity and regulation of these enzymes have yet to be answered. Here, we optimize an enzyme-coupled assay to measure low micromolar concentrations of acetate, coupling acetate production to the formation of NADH which is measured by changes in either absorbance or fluorescence. Using this assay, we measured the steady-state kinetics of peptides representing the H4 histone tail, and demonstrate that a C-terminally conjugated methylcoumarin enhances the catalytic efficiency of deacetylation catalyzed by Co(II)-HDAC8 compared to peptide substrates containing a C-terminal carboxylate, amide, and tryptophan by 50-fold, 2.8-fold, and 2.3-fold, respectively. This assay can be adapted for a high-throughput screening format to identify HDAC substrates and inhibitors.
Keywords: histone deacetylase 8, HDAC8, acetate detection assay, fluorescence, catalysis
Introduction
Histone (or acetyl-lysine) deacetylases (HDACs) are a family of 18 enzymes that catalyze the deacetylation of acetylated lysine side chains.[1-2] Acetylation is a post-translational modification identified on over 3,100 lysines within the mammalian proteome [3] that alter the activities and properties of modified proteins.[4] As many of these proteins are essential to cellular processes,[5-6] aberrant acetylation and deacetylation may contribute to disease states.[7] Attesting to the role of HDACs in diseases are two HDAC inhibitors (Vorinostat and Romidepsin) that have been approved by the FDA for the treatment of T-cell Lymphoma,[8] though the mechanism of action for these drugs is not well understood. One complicating factor to understanding the biological function and regulation of protein deacetylation is the lack of identified HDAC isozyme-substrate pairs. Determining the substrate specificity of HDACs would provide insight into cellular homeostasis and development of isozyme-specific inhibitors.
There are four classes of HDAC enzymes. Classes I, II, and IV use an active site divalent metal ion cofactor to catalyze deacetylation, yielding lysine and acetate as products.[9] Class III HDACs use NAD+ as a cofactor and produce 2’-O’acetylribose, nicotinamide, and the deacetylated protein.[10] Current in vitro assays for the measurement of HDAC activity use environmentally sensitive fluorophores,[9, 11-17] HPLC methods,[18] free amine reactive reagents,[11] radiolabeled acetate,[19-20] and mass spectrometry.[21-23] While these assays are useful, each has associated limitations. In particular, many of these techniques can only be used to measure deacetylation of short peptide substrates rather than the biologically-relevant acetylated proteins. Furthermore, in the most frequently used assay, the Fluor de Lys assay, the methylcoumarin substituent alters substrate recognition.[23] For assays that can be adapted to measure deacetylation of proteins, many cannot be adapted to high throughput formats, are hard to quantify, or require specialized equipment.
Here we optimize an enzyme-coupled assay for the measurement of low micromolar acetate concentrations that can be used to evaluate the activity of class I, II, and IV HDACs. This assay couples the formation of acetate to the production of NADH that is monitored via an absorbance or fluorescence signal. This assay quantitatively measures deacetylation independent of the substrate size or structure, does not require specialized equipment, and can be adapted to real-time and high throughput formats. Using this assay, we demonstrate that cobalt(II)-bound histone deacetylase 8 (Co(II)-HDAC8) catalyzes deacetylation of a peptide sequence from the H4 histone tail containing a C-terminal methylcoumarin fluorophore with a kcat/KM value that is 50-fold, 2.8-fold, and 2.3-fold greater than the kcat/KM value for deacetylation of the same peptide containing a C-terminal carboxylate, amide, or tryptophan, respectively. The loss of catalytic efficiency for deacetylation of the C-terminal carboxylate peptide is a result of a 2.3-fold increase in the value of KM and a 22-fold decrease in the kcat value. These data demonstrate that interactions between HDAC8 and the C-terminal moiety are important for substrate recognition and efficient chemistry.
Materials and Methods
Reagents
ATP, Coenzyme A, NAD+, L-malic acid, citrate synthase (CS), and malate dehydrogenase (MDH) were purchased from Sigma. The acetic acid detection kit was purchased from R-biopharm. Fluor de Lys peptide and the developing reagent were purchased from Enzo Life Sciences. The unlabeled peptides (Ac-KGGAKac-COO−, Ac-KGGAKac-NH2, and Ac-KGGAKacW-NH2) were purchased from Peptide2.0 (>85 % purity). Cobalt and magnesium were purchased as ICP standards from GFS Chemicals and the acetic acid standard was purchased from the Ricca Chemical Company. Chelex 100 resin was purchased from Bio-Rad. HDAC3/NCOR1 was purchased from Enzo Life Sciences. Ethylenediaminetetraacetic acid (EDTA) was purchased from Sigma-Aldrich at >99 % purity. All other materials were purchased from Fisher and were of a purity >95 % unless otherwise noted.
Acetyl-CoA synthetase preparation
The chitin tagged acetyl-CoA synthetase (ACS) plasmid (Acs/pTYB1)[24] was a generous gift from Professor Andrew Gulick (Hauptman-Woodward Institute). To increase the yield of protein, the gene for ACS was subcloned from the chitin-tagged ACS plasmid into a pET vector containing a His6x affinity tag. The ACS gene from Acs/pTYB1was amplified using the polymerase chain reaction to add XhoI and XbaI restriction sites. The amplified DNA segment was digested using XhoI and XbaI and ligated into a pHD4 vector[25] containing a T7 RNA polymerase promoter and a C-terminal TEV (Tobacco Etch Virus) protease sequence followed by a His6x motif to form the pHD4-ACS-TEV-His6x expression vector. The plasmid sequence was confirmed by sequencing at the University of Michigan DNA Sequencing Core.
The pHD4-ACS-TEV-His6x vector was transformed into BL21(DE3) cells. An overnight culture (12.5 mL/L) was used to inoculate autoinduction TB medium (12 g/L tryptone, 24 g/L yeast extract, 4.6 g/L KH2PO4, 20.6 g/L K2HPO4, 4 g/L lactose, 1 g/L glucose, 10 mL/L glycerol) that was supplemented with 100 μg/mL ampicillin and grown at 30°C for 20 hours prior to harvest. The cells were pelleted by centrifugation (9,000 × g, 10 min) and then resuspended and lysed in low imidazole buffer (30 mM HEPES, 150 mM NaCl, 20 mM imidazole, 1 mM TCEP, pH 8) using an M110L microfluidizer (Microfluidics). The lysate was cleared by centrifugation (39,000 × g, 45 min) and the supernatant was loaded onto a 12 mL GE Chelating Sepharose column charged with NiCl2. The ACS was eluted with a gradient of low (20 mM) to high (200 mM) imidazole buffer. Fractions containing ACS were identified using SDS-PAGE chromatography and were concentrated to <1 mL using 30,000 MWCO Amicon Ultra-15 centrifugal units and loaded onto a GE HiPrep 16/60 Sephacryl S200 HR size exclusion column equilibrated with size exclusion buffer (30 mM HEPES, 150 mM NaCl, 1 mM TCEP, pH 8). The fractions containing ACS were collected, combined with his-tagged TEV(S219V) protease (0.5 mg per liter of culture) purified in our lab using the method of Tropea et al. [26] and dialyzed against >500 fold excess of low imidazole buffer at 4°C overnight. The dialyzed ACS was run over a second Ni2+-charged Sepharose column, and the flow-through containing ACS was collected and concentrated to ~2 mM. The protein was then flash frozen in liquid nitrogen and stored at −80°C. Frozen ACS can be used for at least 12 months with little to no effect on the assay.
HDAC8 expression and purification
HDAC8 was expressed and purified as previously described[9] with the exception that a 20 mL DEAE Sepharose column was used after the second Chelating Sepharose column to remove excess metal from HDAC8. This column utilized a gradient from low to high salt buffer (50 mM HEPES, 10 μM ZnSO4, 1 mM TCEP, 50 mM NaCl, 5 mM KCl, pH 7.8 and 50 mM HEPES, 10 μM ZnSO4, 1 mM TCEP, 1 M NaCl, 5 mM KCl, pH 7.8, respectively).
Fluor de Lys assay
All assays were performed in metal free tubes using metal free tips before being quenched into 96 well black plates (Corning plate# 3638). The Fluor de Lys assay was performed as previously described.[9] Briefly, HDAC8 was reconstituted with stoichiometric cobalt(II) at a final concentration of 10 μM and incubated on ice for 1 hour. Fluor de Lys HDAC8 deacetylase substrate (0 to 100 μM) was resuspended in HDAC8 assay buffer (50 mM HEPES, 137 mM NaCl, 2.7 mM KCl, pH 8) and incubated at 30°C for 5 minutes. Reactions were initiated by adding 0.5 μM Co(II)-HDAC8 and the reaction was quenched by a10-fold dilution into 0.05x Enzo developer II and 1.2 μM Trichostatin A (TSA) in HDAC8 assay buffer at 0, 30, and 60 seconds. Samples were incubated at room temperature for 15 minutes and the fluorescence was measured using a Polarstar Galaxy fluorometer (ex. = 340 nm; em. = 450 nm and 380 nm). The initial rate of deacetylation was determined from the time-dependent increase in the fluorescence ratio (450 nm/380 nm) and the concentration of product was calculated using a standard curve.
Acetate assay kit
Acetate standard curves were made by diluting the Ricca acetic acid standard with HDAC8 assay buffer (above). The acetic acid detection kit (R-biopharm) was used according to the instructions except that the reaction volume and coupled solution volume were decreased 10-fold and no additional water was added to dilute the reaction. Solutions 1, 2, 3 and 4 in the kit were preincubated at room temperature for 20 minutes before being mixed with acetate. The reaction was incubated at room temperature for 40 minutes and then NADH fluorescence (ex. = 340 nm, em. = 460 nm) was measured using a Polarstar Galaxy fluorometer in a 96 well plate.
Optimized stopped coupled acetate assay
To remove contaminating metals from peptide substrates, ~6 % (v/v) hydrated Chelex 100 was added to the Ac-KGGAKac-NH2 and Ac-KGGAKacW-NH2 peptides and incubated at room temperature for three hours. The Ac-KGGAKacW-NH2 peptide concentration was determined from the absorbance measurement using an ND-1000 Spectrophotometer (Nanodrop) with a calculated extinction coefficient of 5500 M−1 cM−1.[27] Additionally, the concentration of peptides containing a free amine (lysine) was measured using the fluorescamine assay described below. Peptide substrates without a fluorophore (0 – 1600 μM) were preincubated in HDAC8 assay buffer (above) at 30°C for 10 min. The reactions were initiated by adding 0.5 μM (final concentration) Co(II)-HDAC8 or HDAC3/NCOR1, and quenched by addition of 0.37 % (v/v, final concentration) HCl after 0, 30, 60, and 90 minutes of incubation. The reactions were flash frozen within 20 minutes of quenching and stored at −80°C. Upon thawing, the reactions were neutralized by addition of 0.6% (w/v, final concentration) NaHCO3. The coupler mixture (50 mM HEPES, 400 μM ATP, 10 μM NAD+, 30 μM CoA, 0.07 U/μL CS, 0.04 U/μL MDH, 50 μM ACS, 100 mM NaCl, 3 mM KCl, 50 mM MgCl, 2.5 mM L-malic acid, pH 8) were incubated for 20 minutes at room temperature and added to each quenched reaction (at a ratio of 10 μL coupler mix/65 μL reaction) in a 96 well black plate. The reactions were incubated at room temperature for 40 minutes and the NADH fluorescence (ex. = 340 nm, em. = 460 nm) was measured. The steady state kinetic parameters for the Ac-KGGAKac-COO− peptide were determined from fitting the Michaelis-Menten equation to the concentration dependence of HDAC-catalyzed deacetylation. Substrate inhibition is observed for the peptides Ac-KGGAKac-NH2 and Ac-KGGAKacW-NH2, therefore the kinetic parameters for these substrates were determined by fitting Equation 1 to the dependence of the initial velocities on peptide concentration. Equation 1 was derived from rearrangement of the Briggs-Haldane to report the value of kcat/KM and the standard error directly from the output.
| Equation 1 |
Optimized continuous coupled acetate assay
The 96 well plates were soaked (>3 hours) in 100 mM divalent metal-free EDTA to strip the plate of contaminating metal. The continuous assay buffer (50 mM HEPES, 400 μM ATP, 10 μM NAD+, 30 μM CoA, 0.07 U/μL CS, 0.04 U/μL MDH, 50 μM ACS, 127 mM NaCl, 2.7 mM KCl, 2.5 mM L-malic acid, pH 8) was incubated with Chelex resin for 1 hour at room temperature. The mixture was clarified by centrifugation at 16,800 × g for 2 minutes and the supernatant was collected. Then 6 mM magnesium was added to the buffer and the mixture was incubated for 20 minutes to allow NAD+/malate and NADH/OAA to equilibrate. The peptide (100 μM final concentration Ac-KGGAKac-NH2) in HDAC8 assay buffer was added to this mixture at a ratio of 2:1, respectively. The reaction was initiated with the addition of Co(II)-HDAC8 (0.5 – 1 μM final concentration) and deacetylation was measured from the time-dependent increase in NADH fluorescence (ex. = 340 nm, em. = 460 nm).
Fluorescamine assay
The peptide substrate Ac-KGGAKac-COO− (0 - 1600 μM) was preincubated in HDAC8 assay buffer (above) at 30°C for 10 minutes. The reactions were initiated by adding 0.5 μM HDAC8, and quenched by addition of 1 μM TSA after 0, 30, 60, and 90 minutes of incubation. The reactions were used immediately or flash frozen and stored at −20°C. Upon thawing, solutions were filtered through Pall 10K mwco NanosepMF Centrifugal devices to remove HDAC8. The flow-through (80 μL) was mixed with 50 μL of 1 M boric acid (pH 9) and the mixture was added to a Corning 96 well black plate. 33 μL of 4.3 mM fluorescamine (dissolved in acetone) was then added to the sample, incubated at room temperature for 10 minutes, and fluorescence (ex. = 340 nm, em. = 460 nm) was measured.[28] A standard curve was created using N-α-acetyl lysine methyl ester (0 – 5 μM). To measure the peptide concentration, peptides were diluted into 1 M borate, pH 9, and 0.56 mM fluorescamine was added. The mixture was incubated at room temperature for 10 min and the fluorescence (ex. = 340 nm, em. = 460 nm) was measured. The peptide concentration was determined from the standard curve.
Results
Assay
An acetate detection assay was initially described in the Official Collection of Assays according to § 35 of German food law [29] and a kit containing the assay components is distributed by the R-biopharm company (Figure 1). This assay system couples enzymatic reactions that produce one molecule of citrate, CoA, and NADH per molecule of acetate. The NADH concentration is monitored using fluorescence and/or absorbance, allowing determination of the acetate concentration from a spectroscopic signal. This assay is optimized to measure millimolar concentrations of acetate with a detection limit of ~100 μM (Figure 2 and S1), which is not sufficiently sensitive to measure the steady state kinetic parameters of many enzymes, including HDACs.
Figure 1.

Assay scheme. Acetate is a product of deacetylation catalyzed by HDAC8. Acetate, ATP, and CoA are converted into acetyl-CoA, AMP, and inorganic pyrophosphate by acetyl-CoA synthetase. Acetyl-CoA and oxaloacetate are converted into citrate and CoA catalyzed by citrate synthase. Simultaneously, malate dehydrogenase catalyzes equilibration of NAD+ and malate with NADH and oxaloacetate. When a molecule of oxaloacetate is removed from solution by the citrate synthase reaction, a molecule of NADH is formed. NADH concentrations are quantified using absorbance or fluorescence.
Figure 2.

Comparison of R-biopharm kit and optimized assay for detection of acetate. The fluorescence change as a function of acetate concentration is measured using the R-biopharm kit (△) and the optimized acetate assay (□). The fluorescent signal is normalized to 0 μM acetate using the optimized assay. The signal from the optimized assay is much larger in the μM range, which is required to measure steady state turnover catalyzed by HDAC8. This signal is accurate to ~1 μM with a coefficient of variance equal to 2.2. The R-biopharm kit standard curve is linear at higher concentrations of acetate (250 – 1250 μM acetate) (Figure S1).
Optimization of Assay for Measuring HDAC Activity
The KMvalues for HDAC-catalyzed hydrolysis of acetyl-lysine residues in peptides are typically in the low to mid micromolar range. Therefore, to measure the initial rate (≤10 %) of the reaction, the detection limit should be in the low micromolar range. To optimize the detection limit for the acetate-coupled assay, the signal to noise ratio was improved by: (1) using highly purified recombinant ACS, which decreased the background signal; (2) lowering the concentration of L-malic acid and NAD+ to decrease the background signal due to the equilibrium formation of OAA and NADH; and (3) increasing the ratio of the sample to the coupling solution volume to improve the signal intensity. With these alterations, a linear standard curve from 0 to 50 μM acetate with a limit of detection of ~1 μM (coefficient of variance 2.2; Figure 2) was produced using this assay. This assay can be altered to measure larger concentrations of acetate (>50 μM) by adding higher concentrations of the limiting reagents CoA and NAD+. This standard curve indicates that the optimized coupled assay is sensitive enough to measure the steady state kinetic parameters for HDACs and other enzymes.
Based on the assay design, each molecule of acetate should yield one molecule of NADH. To test this, the fluorescence change from the addition of acetate to the coupled assay was compared with the fluorescence of a comparable concentration of NADH (Figure 3). The slopes of the standard curves for NADH and acetate were 770 ± 61 and 790 ± 50 fluorescence units per μM, respectively. The equivalence of these slopes indicates that there is a one to one relationship between the concentration of acetate and the signal created by the production of NADH, allowing calculation of the acetate concentration from the fluorescence change.
Figure 3.

Standard curve for the acetate assay compared to NADH fluorescence. The fluorescence change observed upon addition of acetate to the coupled assay (■) is compared to the NADH fluorescence under comparable conditions (50 mM HEPES, 100 mM NaCl, 3 mM KCl, 50 mM MgCl, pH 8) (▲). The two slopes are equal, indicating that in the coupled assay one mole of NADH is formed per mole of acetate. The higher background observed in the acetate standard curve is a result of NADH formed before addition of acetate due to the equilibrium of the malate dehydrogenase-catalyzed reaction.
Stopped Assay
We first optimized the acetate assay in a stopped format to measure HDAC8 activity. After reacting HDAC8 with the substrate of interest, the reaction was quenched by the addition of HCl and flash frozen in liquid nitrogen. Upon thawing, the pH was neutralized by addition of NaHCO3. Using the Fluor de Lys assay to measure activity, the HCl solution quenches HDAC8 activity immediately (<10 sec), and HDAC8 activity is not restored upon neutralization (data not shown). The acetate concentration in this sample is then measured using the coupled assay. Under these optimized conditions, formation of NADH from the addition of acetate occurs within minutes. Upon mixing the coupling enzymes with the assay substrates, NAD+ and L-malic acid equilibrate to form NADH and oxaloacetate. This equilibration is complete in 20 minutes (Figure 4), forming ~4 μM NADH, consistent with the equilibrium constant for the reaction catalyzed by malate dehydrogenase.[30] The reaction of up to 20 μM acetate is complete within 30 minutes and the signal remains stable for over an hour. The limiting step in this assay is the formation of citrate and CoA catalyzed by citrate synthase. Therefore, the rate of acetate production can be increased by the addition of higher concentrations of CS, if needed.
Figure 4.

Time course for the formation of acetate. At −25 minutes the assay reagents are mixed and incubated at room temperature (~25°C). Equilibration is complete in <20 minutes, forming ~4 μM NADH. The reaction is initiated (t = 0 min) by addition of 20 μM acetate. The reaction reaches equilibrium at ~30 min and the signal is stable for at least one hour.
To demonstrate the effectiveness of the acetate assay in measuring deacetylation, we compared the rate of HDAC8-catalyzed deacetylation determined using the coupled assay with the fluorescamine assay. Fluorescamine is a reagent that increases in fluorescence intensity upon reaction with primary amines[31] and therefore a fluorescent signal is coupled to the formation of lysine generated by HDAC-catalyzed deacetylation. We measured the reactivity of Co(II)-HDAC8 with an unlabeled peptide (Ac-KGGAKac-COO−) mimicking the H4 histone K16 acetylation site (H4 K16ac). HDAC8-catalyzed deacetylation of this peptide (200 μM peptide, 0.5 μM HDAC8) measured by the coupled acetate assay and the fluorescamine assay yielded comparable rates within experimental error of 0.0021 ± 0.0003 μM s−1 and 0.0027 ± 0.0008 μM s−1, respectively. Therefore, both assays measure the deacetylation rate and are viable for measuring HDAC8 activity, though the fluorescamine assay is less accurate for substrates containing multiple lysine side chains due to a higher signal to noise ratio.
To demonstrate that this optimized stopped assay can serve as a general acetate assay, we measured deacetylation of 100 μM Ac-KGGAKac-NH2 catalyzed by another HDAC isozyme, 0.5 μM HDAC3/NCOR1 [Figure S3]. The initial rate for this reaction was 0.015 ± 0.0016 μM s−1, comparable to the value measured for HDAC8 (see below).
Continuous Assay
We next evaluated whether the coupled acetate assay could be carried out as a continuous, real-time assay to measure HDAC8 activity. Since HDAC8 is sensitive to inhibition by metals,[9] and monovalent cations [15], the acetate coupling solutions were reformulated with concentrations of NaCl and KCl typically used to assay HDAC8 activity (Biomol unpublished)[15] and treated with Chelex resin prior to addition of magnesium. The concentration of magnesium was decreased to 2 mM to minimize inhibition of HDAC8 activity (~2-fold inhibition under these conditions) [Figure S2]. To counteract the loss in activity of the coupling enzymes due to the lower concentration of magnesium, the concentration of these enzymes was increased by 2.3-fold to yield a final rate for the coupling reactions of 0.046 μM s−1. These assay conditions were used to measure HDAC8-catalyzed deacetylation of 100 μM Ac-KGGAKac-NH2 peptide yielding rates of 0.018 ± 0.00013 μM s−1 and 0.028 ± 0.00024 μM s−1 at 0.5 μM and 1 μM HDAC8, respectively [Figure 5]. The linear dependence on the HDAC8 concentration demonstrates that the assay rate is not limited by the coupling reactions. Furthermore, the HDAC8 activity measured using the stopped assay (0.5 μM HDAC8 and 100 μM Ac-KGGAKac-NH2) is 0.033 ± 0.0034 μM s−1 which is within the two-fold of the stopped assay measured rate, and represents the difference expected due to magnesium inhibition of Co(II)-HDAC8.
Figure 5.

Continuous assay to measure HDAC8 activity. The initial rates for deacetylation of the Ac-KGGAKAc-NH2 peptide (100 μM) measured using the acetate assay in a continuous formate are 0.011 ± 0.00041 μM s−1 and 0.022 ± 0.00043 μM s−1 for 0.5 μM (□) and 1 μM (○) Co(II)-HDAC8, respectively. A side-by-side acetate assay using the stopped formate (0.5 μM Co(II)-HDAC8, 100 μM Ac-KGGAKac-NH2) has an initial rate of 0.018 ± 0.0021 μM s−1(■).
Additionally, we measured the HDAC8-catalyzed deacetylation of H3/H4 tetramer acetylated using acetic anhydride, where ~100% of the lysines were acetylated as indicated by mass spectrometry [data not shown]. The initial rate for deacetylation of ~0.076 μM acetylated H3/H4 tetramer catalyzed by 0.5 μM Co-(II)-HDAC8 is 0.0021 ± 0.0001 μM s−1 [Figure S4], demonstrating that this assay can measure deacetylation of both peptide and protein substrates.
Reactivity of HDAC8 with peptides
Using a mass spectrometric assay, Gurard-Levin et al. [23] previously demonstrated that HDAC8 catalyzes deacetylation of a p53 peptide mimic containing a C-terminal methylcoumarin fluorophore significantly faster than a comparable peptide with a C-terminal cysteine followed by an amide terminus. To further analyze the recognition of the peptide C-terminus by HDAC8, we measured Co(II)-HDAC8-catalyzed deacetylation using the stopped coupled assay of an H4 peptide mimic containing varied C-termini, including: a carboxylate (Ac-KGGAKac-COO−); an amide (Ac-KGGAKac-NH2); and a tryptophan capped by an amide (Ac-KGGAKacW-NH2) (Figure 6 and Table 1). The steady state kinetic parameters for Ac-KGGAKac-COO− were determined from fitting the Michaelis Menten equation to the concentration dependence of activity, yielding values of kcat/KM, kcat and KM of 56 M−1 s−1, 0.041 s−1 and 730 μM, respectively (Figure 6). For the Ac-KGGAKac-NH2 and Ac-KGGAKacW-NH2 peptides, modest to substantial inhibition was observed at higher peptide concentrations. This inhibition is not due to metal contamination of the peptide[9] as pre-incubation of the peptide with Chelex 100 resin had no effect on the observed activity (Figure 6). Therefore, these data indicate that the activity is inhibited by high substrate concentrations. As many HDAC isozymes form multi-protein complexes,[32-33] the peptides may inhibit HDAC8 activity by binding to non-active site protein-protein interaction sites. For these peptides the kinetic parameters were determined by fitting an equation including terms for substrate inhibition (Equation 1) to the data; the values of kcat/KM for deacetylation of Ac-KGGAKacW-NH2 and Ac-KGGAKac-NH2 are 1200 ± 250 M−1s−1 and 980 ± 47 M−1s−1 parameters are coupled, independent values for these parameters could not be accurately determined. Substrate inhibition by the Ac-KGGAKac-NH2 peptide appears to be cooperative with a Hill coefficient (n) that is larger than 1. In general, substrate inhibition significantly complicates the analysis of the reactivity of HDAC8 with peptide libraries measured at a single peptide concentration.
Figure 6.

Dependence of HDAC8-catalyzed deacetylation on the concentration of H4 K16ac peptide mimics. The steady state kinetics of deacetylation catalyzed by HDAC8 were measured at 30°C in 50 mM HEPES, 137 mM NaCl, 2.7 mM KCl, pH 8, 0.85 μM HDAC8. A) The solid line is a fit of the Michaelis-Menten to the initial rates for deacetylation of a peptide ending in a carboxylic acid (Ac-KGGAKac-COO−) using the values listed in Table 1. B) The peptide ending in an amide (Ac-KGGAKac-NH2). C) The peptide ending in a tryptophan and capped with an amide (Ac-KGGAKacW-NH2). Equation 1 was fir to the data for the Ac-KGGAKacW-NH2 and Ac-KGGAKacW-NH2. The solid lines shown are the best fits with the following values: Ac-KGGAKac-NH2, kcat/KM, KM, KI, and n values of 980 ± 47 M−1 s−1, 90 ± 21 μM, 190 ± 15 μM, and 3.9 ± 0.6, respectively and Ac-KGGAKacW-NH2, kcat/KM, KM, and KI values of 1250 ± 250 M−1 s−1, 890 ± 2100 μM, and 440 ± 1600 μM respectively. As the KM and KI values are highly correlated, these values are not accurate.
Table 1.
Structure and kinetic constants for HDAC8-catalyzed deacetylation.a
| Peptide |
kcat/KM
(M−1 S−1) |
kcat
(S−1) |
KM
(μM−1) |
|---|---|---|---|
|
2800a
± 200 |
0.90a
± 0.03 |
320a
± 20 |
|
56
± 14 |
0.041b
± 0.0042 |
730b
± 160 |
|
980c
± 47 |
N.D.d | N.D.d |
|
1200c
± 250 |
N.D.d | N.D.d |
The steady-state kinetic parameters and standard errors for HDAC8-catalyzed are measured as described in the legend of Figure 6.
Values reported in ref. [25].
Kinetic parameters determined using Equation 1, including substrate inhibition.
N.D. indicates values that were not determined as a result of substrate inhibition.
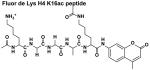
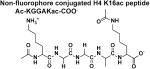
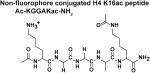
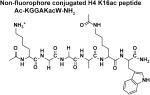
The four peptides differ in their C-terminus; the Fluor de Lys peptide contains a methylcoumarin fluorophore while the non-fluorophore conjugated H4 K16ac peptides contain a C-terminal carboxylate, carbamide, or tryptophan followed by a carbamide. The kinetic constants of these peptides suggest that the differences in the peptides affect both substrate binding and chemistry. Standard error is reported.
These data demonstrate that the structure of the C-terminal moiety significantly affects the reactivity of Co(II)-HDAC8 with peptide substrates. Furthermore, a comparison of these data to the kcat/KM value of 2800 M−1 s−1 [25] for HDAC8-catalyzed deacetylation of the Ac-KGGAKac-methylcoumarin peptide demonstrates that the methylcoumarin fluorophore enhances (up to 50-fold) the catalytic efficiency of HDAC8-catalyzed deacetylation (Table 1).
Discussion
The current HDAC assays have limitations that do not allow in-depth and/or high throughput analysis of HDAC substrate specificity. The first kinetic measurements of HDAC activity employed radioactively labeled peptides and proteins and detected the formation of radiolabeled acetate.[19-20] While this assay is effective and sensitive, radioactive peptide substrates are expensive to produce, and many radiolabeled protein substrates are acetylated non-specifically, preventing the determination of detailed kinetics at specific sites. Recently, the Fluor de Lys assay (Biomol) has become popular for measuring HDAC kinetics.[9, 11-17] This assay uses a methylcoumarin fluorophore-conjugated peptide, which upon deacetylation becomes a substrate for the serine protease trypsin, cleaving the fluorophore and altering the fluorescence spectrum of the methylcoumarin. The rate of deacetylation is measured by the change in the fluorescence signal.[12] However, the methylcoumarin fluorophore can interact with HDACs and alter the kinetics of deacetylation [23](as discussed later). Thus, the results obtained using this method may not accurately report the selectivity of HDACs for native peptides and proteins. Additionally, because the fluorophore is located immediately on the C-terminal side of the acetyl-lysine moiety, this method cannot be used to determine the preference for sequences downstream of the acetylated lysine. Many of these limitations were solved by a mass spectrometric assay developed by the Mrksich group; this assay uses MALDI mass spectrometry of peptides attached to a gold surface to observe the mass difference caused by deacetylation.[23] This assay is effective for peptide substrates and can be carried out in a high throughput manner, however a MALDI mass spectrometer is required. Additionally, this method requires the inclusion of a cysteine in the peptide sequence to conjugate the peptide to the plate via a maleimide linkage. HPLC separation of acetylated and deacetylated peptides on a C18 column is another method employed to determine deacetylation kinetics.[18] This assay quantifies product formation by absorbance (230 or 280 nm) and can measure the deacetylation of any peptide, yet it suffers from labor intensive techniques such as the determination of peak elution times making it not ideal for a high throughput format. Furthermore, it is not easily transitioned to assaying the deacetylation of proteins due to the difficulty of separating full length proteins differing by a single acetylated lysine. The assay presented in this paper provides an alternative that overcomes many of the limitations of the previous assays. This acetate assay is versatile; either fluorescence or absorbance can be measured using a cuvette or 96-well plate and in a stopped or continuous format. The continuous method is well-suited for high-throughput screening. Peptide substrates for the assay are inexpensive to purchase as they do not require conjugated fluorophores, and results can be quantified using an acetic acid standard curve. Furthermore, the assay can measure deacetylation of proteins (Figure S4). However, the acetate assay cannot be used with fluorescent peptides (i.e. methylcoumarin-conjugated peptides) that absorb and emit at wavelengths similar to NADH, nor can it be used on cell lysate extracts containing NADH, other metabolites, or various other enzymes. Overall, this assay provides a stable and sensitive platform for the measurement of deacetylation with few limitations. Finally, as this assay has been optimized to measure micromolar acetate concentrations, it can be used to assay other enzymes that produce acetate as a product and have KM values in the micromolar range, including other HDAC isozymes (Figure S3).
While most kinetic measurements of HDAC8 have been performed using the Fluor de Lys assay,[9, 11, 13-15] the Mrksich group used a mass spectrometric assay to demonstrate that a peptide mimicking the p53 transcription factor was deacetylated significantly slower than the same peptide containing a methyl-coumarin fluorophore.[23] However, the steady state kinetic parameters were not determined for these substrates. Using the optimized coupled assay that measures the formation of acetate, we demonstrated that substitution of the coumarin fluorophore with a carboxylate lowered the value of kcat/KM for Co(II)-HDAC8 by 50-fold, resulting from a 22-fold reduction in the value of kcat and a 2.3-fold increase in the KM value (Table 1). As the value of KM is relatively high (730 μM) and the value of kcat is relatively low (0.041 s−1) compared to other enzymes acting under diffusion control,[34] it is likely that substrate dissociation is faster than deacetylation, indicative of a rapid equilibrium substrate binding model. This assumption is further validated by the increase in kcat measured for trifluoro-acetyl lysine substrates, [18, 35] suggesting that deacetylation is the rate-limiting step for kcat. Based on this assumption, KM reflects KD for the peptide. Therefore, ΔΔGbinding, calculated from the alteration in the KM values5, indicative of the additional binding affinity conferred by the methylcoumarin fluorophore relative to a carboxylate at the C-terminus, is equal to ~0.45 kcal/mol. This alteration in binding energy is modest but within the range of energy due to the addition of a single pi-pi interaction[36-37] or hydrogen bond.[34] Crystal structures of a methylcoumarin-conjugated peptide bound to HDAC8 visualize interactions between the methylcoumarin and the side chain of Tyr100.[13, 38] These structures suggest that the enhanced binding energy for the methylcoumarin peptide results from a combination of interactions between the C-terminus of the peptide and the hydrophobic cavity formed by the L1, L7, and L8 loops, and interactions between the aromatic C-terminal residue of the peptide and Tyr100 on the L2 loop of HDAC8.
The steady state kinetic parameters for deacetylation catalyzed by HDAC8 demonstrate that the significant (50-fold) enhancement in Co(II)-HDAC8 kcat/KM for the Ac-KGGAKac-methylcoumarin peptide compared to the Ac-KGGAK-COO− peptides is largely due to an increase in the kcat value. The change in the stabilization of the transition state relative to the unbound ground state (ΔΔG‡)6 of 2.3 kcal/mol likely results from a combination of altered electrostatic and pi-pi interactions between the peptide and HDAC8 that enhance optimal positioning of the peptide and side chains in the active site to efficiently catalyze deacetylation. The effects of the C-terminal interactions of the peptide with Co(II)-HDAC8 on kcat and KM are consistent with data demonstrating that L2 loop residues are important for both binding and catalysis; mutations at Asp101 in HDAC8 lead to both higher KM and lower kcat values,[13] compared to the wildtype enzyme. However, these mutations do not lead to observable alterations in the crystal structure of inhibitor-bound HDAC8,[13, 38] suggesting that the activity decrease may be due to an alteration in the HDAC8 dynamics. The catalytic efficiency (kcat/KM) of Co(II)-HDAC8 with the Ac-KGGAKac-NH2 peptide is enhanced 9-fold (ΔΔG‡ = 1.6 kal/mol)6 compared to the Ac-KGGAKac-COO− peptide indicating that electrostatic interactions between the C-terminus and HDAC8 impair peptide binding and/or reactivity. Addition of a tryptophan (Ac-KGGAKacW-NH2) or methylcoumarin moiety to the peptide increases net transition state stabilization by 0.1 – 0.5 kcal/mol, providing an estimate of the enhancement of the catalytic efficiency by base stacking with Tyr100. Previous studies performed by the Mrksich lab show that HDAC8 catalyzes deacetylation of peptides containing a phenylalanine on the C-terminal side of the acetyl lysine faster than substrates containing any other amino acid, including tryptophan, at that position.[21, 23] These data suggest that both amino acid hydrophobicity and volume play a role in substrate preference. However, a direct comparison of the reactivity of peptides in this paper compared to those in Gurard-Levin et al. [21, 23] is complicated by differences in peptide length, the sequence at other positions of the peptide, and method of kinetic measurement.
Interestingly, Co(II)-HDAC8-catalyzed deacetylation of acetyl-lysine peptides have low values for kcat/KM(56 – 2800 M−1 s−1) compared to enzymes that are limited by diffusion,[34] suggesting either that these peptides are poor substrates for this enzyme or that the low activity is biologically relevant for control of enzyme activity.[39] The in vivo catalytic efficiency could be enhanced either by additional interactions with the protein substrates or by additional cofactors or binding partners. Furthermore, the measured rate constants for Co(II)-HDAC8-catalyzed deacetylation of Ac-KGGAKac-COO− and high concentrations of Ac-KGGAKac-NH2 peptides are similar to the rates measured for deacetylation catalyzed by various class II HDAC isozymes (HDAC7, 10)[17, 40] suggesting that the low activity of these isozymes in the Fluor de Lys assay may reflect decreased enhancement of reactivity by aromatic C-terminal moieties.
Supplementary Material
Acknowledgements
We thank Dr. Eric Drake and Dr. Andrew Gulick for their generous contribution of the ACS plasmid and subsequent help in ACS purification. We thank Byungchul Kim for his help in using 96 well plates for this assay. We also thank members of the Fierke lab for their insight, discussion, and critiques of the material within the paper. This material was supported, in whole or in part, by the National Institutes of Health Grants, NIGMS GM40602 (CAF), T32-GM-008353 (NAW), and T32-GM-008597 (CAP), and the National Science Foundation Graduate Fellowship DGE0718128 (NAW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ΔΔGbinding = RT ln(KM Fluor de Lys peptide) − RT ln(KM non-fluorophore conjugated peptide)
ΔΔG‡ (kcat/KM) = RT ln(kcat/KM peptide1) − RT ln(kcat/KM peptide2)
References
- [1].Khochbin S, Verdel A, Lemercier C, Seigneurin-Berny D. Functional significance of histone deacetylase diversity. Curr Opin Genet Dev. 2001;11:162–166. doi: 10.1016/s0959-437x(00)00174-x. [DOI] [PubMed] [Google Scholar]
- [2].Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep. 2011;1 doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- [5].Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- [6].Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- [8].Lemoine M, Younes A. Histone deacetylase inhibitors in the treatment of lymphoma. Discov Med. 2010;10:462–470. [PubMed] [Google Scholar]
- [9].Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- [10].Tong L, Denu JM. Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim Biophys Acta. 2010;1804:1617–1625. doi: 10.1016/j.bbapap.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Riester D, Hildmann C, Grunewald S, Beckers T, Schwienhorst A. Factors affecting the substrate specificity of histone deacetylases. Biochem Biophys Res Commun. 2007;357:439–445. doi: 10.1016/j.bbrc.2007.03.158. [DOI] [PubMed] [Google Scholar]
- [12].Wegener D, Wirsching F, Riester D, Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol. 2003;10:61–68. doi: 10.1016/s1074-5521(02)00305-8. [DOI] [PubMed] [Google Scholar]
- [13].Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dowling DP, Gattis SG, Fierke CA, Christianson DW. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry. 2010;49:5048–5056. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gantt SL, Joseph CG, Fierke CA. Activation and inhibition of histone deacetylase 8 by monovalent cations. J Biol Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Haider S, Joseph CG, Neidle S, Fierke CA, Fuchter MJ. On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg Med Chem Lett. 2011;21:2129–2132. doi: 10.1016/j.bmcl.2011.01.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schultz BE, Misialek S, Wu J, Tang J, Conn MT, Tahilramani R, Wong L. Kinetics and comparative reactivity of human class I and class IIb histone deacetylases. Biochemistry. 2004;43:11083–11091. doi: 10.1021/bi0494471. [DOI] [PubMed] [Google Scholar]
- [18].Smith BC, Denu JM. Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J Biol Chem. 2007;282:37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- [19].Buggy JJ, Sideris ML, Mak P, Lorimer DD, McIntosh B, Clark JM. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem J. 2000;350:199–205. Pt 1. [PMC free article] [PubMed] [Google Scholar]
- [20].Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, Kass SU. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000;478:77–83. doi: 10.1016/s0014-5793(00)01813-5. [DOI] [PubMed] [Google Scholar]
- [21].Gurard-Levin ZA, Kilian KA, Kim J, Bahr K, Mrksich M. Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem Biol. 2010;5:863–873. doi: 10.1021/cb100088g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gurard-Levin ZA, Mrksich M. The activity of HDAC8 depends on local and distal sequences of its peptide substrates. Biochemistry. 2008;47:6242–6250. doi: 10.1021/bi800053v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gurard-Levin ZA, Kim J, Mrksich M. Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. Chembiochem. 2009;10:2159–2161. doi: 10.1002/cbic.200900417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reger AS, Carney JM, Gulick AM. Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry. 2007;46:6536–6546. doi: 10.1021/bi6026506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gantt SL. Chemistry. The University of Michigan; Ann Arbor: 2006. Human Histone Deacetylase 8: Metal Dependence and Catalytic Mechanism. [Google Scholar]
- [26].Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- [27].Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- [28].Huang X, Hernick M. A fluorescence-based assay for measuring N-acetyl-1-D-myo-inosityl-2-amino-2-deoxy-alpha-D-glucopyranoside deacetylase activity. Anal Biochem. 2011;414:278–281. doi: 10.1016/j.ab.2011.04.001. [DOI] [PubMed] [Google Scholar]
- [29].Bergmeyer HU, Bergmeyer J, Grassl M. Methods of enzymatic analysis. 3rd Verlag Chemie, Weinheim; Deerfield Beach, Fla.: 1983. [Google Scholar]
- [30].Guynn RW, Gelberg HJ, Veech RL. Equilibrium constants of the malate dehydrogenase, citrate synthase, citrate lyase, and acetyl coenzyme A hydrolysis reactions under physiological conditions. J Biol Chem. 1973;248:6957–6965. [PubMed] [Google Scholar]
- [31].Udenfriend S, Stein S, Bohlen P, Dairman W, Leimgruber W, Weigele M. Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science. 1972;178:871–872. doi: 10.1126/science.178.4063.871. [DOI] [PubMed] [Google Scholar]
- [32].Verdin E. Histone deacetylases : transcriptional regulation and other cellular functions. Humana Press; Totowa, N.J.: 2006. [Google Scholar]
- [33].Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- [34].Fersht A. Structure and mechanism in protein science : a guide to enzyme catalysis and protein folding. W.H. Freeman; New York: 1999. [Google Scholar]
- [35].Riester D, Wegener D, Hildmann C, Schwienhorst A. Members of the histone deacetylase superfamily differ in substrate specificity towards small synthetic substrates. Biochem Biophys Res Commun. 2004;324:1116–1123. doi: 10.1016/j.bbrc.2004.09.155. [DOI] [PubMed] [Google Scholar]
- [36].Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K. Origin of attraction and directionality of the pi/pi interaction: model chemistry calculations of benzene dimer interaction. J Am Chem Soc. 2002;124:104–112. doi: 10.1021/ja0105212. [DOI] [PubMed] [Google Scholar]
- [37].Sinnokrot MO, Valeev EF, Sherrill CD. Estimates of the ab initio limit for pi-pi interactions: the benzene dimer. J Am Chem Soc. 2002;124:10887–10893. doi: 10.1021/ja025896h. [DOI] [PubMed] [Google Scholar]
- [38].Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fierke CA, Kuchta RD, Johnson KA, Benkovic SJ. Implications for enzymic catalysis from free-energy reaction coordinate profiles. Cold Spring Harb Symp Quant Biol. 1987;52:631–638. doi: 10.1101/sqb.1987.052.01.072. [DOI] [PubMed] [Google Scholar]
- [40].Schuetz A, Min J, Allali-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.