

Abstract
Aqueous reversible addition-fragmentation chain transfer (RAFT) polymerization was employed to prepare a series of linear copolymers of N,N-dimethylacrylamide (DMA) and 2-hydroxyethylacrylamide (HEAm) with narrow Đ values over a molecular weight range spanning three orders of magnitude (103 to 106 Da). Trithiocarbonate-based RAFT chain transfer agents (CTAs) were grafted onto these scaffolds using carbodiimide chemistry catalyzed with DMAP. The resultant graft chain transfer agent (gCTA) was subsequently employed to synthesize polymeric brushes with a number of important vinyl monomer classes including acrylamido, methacrylamido, and methacrylate. Brush polymerization kinetics were evaluated for the aqueous RAFT polymerization of DMA from a 10 arm gCTA. Polymeric brushes containing hydroxyl functionality were further functionalized in order to prepare 2nd generation gCTAs which were subsequently employed to prepare polymers with a brushed-brush architecture with molecular weights in excess of 106 Da. These resultant single particle nanoparticles (SNPs) were employed as drug delivery vehicles for the anthracycline-based drug doxorubicin via copolymerization of DMA with a protected carbazate monomer (bocSMA). Cell-specific targeting functionality was also introduced via copolymerization with a biotin-functional monomer (bioHEMA). Drug release of the hydrazone linked doxorubicin was evaluated as function of pH and serum and chemotherapeutic activity was evaluated in SKOV3 ovarian cancer cells.
Introduction
The American Cancer Society projects that in 2013 there were 1,660,290 new cases of cancer with approximately 580,350 Americans succumbing to the disease, making it the second most common cause of death in the United States.1 While 5-year survival rates for cancer as a whole have improved to 68% the 5-year survival rate for advanced ovarian cancer is only 30%.2 Ovarian cancer is disproportionately more deadly because of a lack of effective early detection methods and the absence of early warning symptoms. These factors generally result in a poor prognosis with 60 % of women presenting with stage III or stage IV cancer that has spread beyond the ovaries.3,4 Despite the pressing need for new treatments, there have been few new therapeutics and the most promising advance has been the demonstration of improved overall survival using intraperitoneal (IP) administration of cisplatin (with and without paclitaxel).5 However, widespread adoption of IP chemotherapy has been limited largely due to associated toxicities.
The use of nanoparticle-based therapies to deliver cytotoxic agents has the potential to significantly improve the prognosis and quality of life for women suffering from ovarian cancer. Chemotherapeutic nanoparticle formulations such as Doxil (liposomal encapsulated doxorubicin), exhibit higher circulation times than the unencapsulated drugs yet can exhibit substantially fewer deleterious side affects.6 In the case of Doxil the risk of cardiotoxicity is 7-fold lower than the free drug despite the large difference in circulation half-lives. Tumor specificity for untargeted nanoparticles is generally attributed to enhanced permeation and retention (EPR), where the leaky tumor vasculature and poor lymphatic drainage result in nanoparticle accumulation.7-10 Many tumors however do not display the EPR effect or have cores that are not well perfused. The use of antibody-targeting has been shown to provide significant enhancements in chemotherapeutic efficacy while substantially reducing side effects by directing these agents to tumor-associated antigens and thus limiting exposure to normal organs.11,12 Lipids and polymers have been employed extensively to build nanoparticles such as micelles, liposomes, and polymersomes for the controlled delivery of both hydrophilic and hydrophobic drugs.13,14 These systems can substantially improve the bioavailability and pharmacokinetic properties of the encapsulated drugs and are capable of integrating other important functional components such as cell-specific targeting and intracellular responsive segments.15-18 Despite the success of nanoparticle-based drug delivery systems the stability and morphological identity of these systems once introduced into the complex in vivo environment is difficult to ascertain.19-21 Direct polymer-drug conjugates that utilize a reversible covalent linkage to tether the therapeutic agent to the polymeric scaffold have been developed as a means of overcoming some of the limitations typically associated with physically encapsulating drug delivery systems. For example, Davie et al. have prepared polymer-drug conjugates with the chemotherapeutic drug camptothecin (CPT).22,23,24 CPT is a cytotoxic quinoline alkaloid that shows potent anticancer activity but low aqueous solubility and high adverse drug reactions. Conjugation of the CPT hydroxyl at the 20 position to form ester bonds with cyclodextrin-based polymers substantially improves the solubility of camptothecin while reducing side effects. In another study, Cameron et. al. conjugated doxorubicin to biodegradable dendrimers via hydrazone linkages.25 The resultant hydrazone linked polymeric prodrugs exhibit half-lives of approximately 16 hours under circulation conditions but rapidly degrade under acidic environments such as those found in intracellular compartments and many tumor microenvironments. Efficacy studies of the hydrazone linked doxorubicin, performed with BALB/c mice bearing s.c. C-26 tumors, at 20 mg/kg DOX equivalents resulted in complete tumor regression and 100% survival over course of the 60-day experiment.
Recent advances in controlled radical polymerization (CRP) methodologies have the potential to revolutionize the development of sophisticated multifunctional drug delivery systems.26-28 Reversible addition-fragmentation chain transfer (RAFT) polymerization in particular has allowed previously unattainable polymeric architectures to be prepared under industrially feasible conditions. A variety of advanced polymeric architectures have been prepared by RAFT polymerization methodology including stars, brushes, and bottlebrushes.29,30-41
These materials have the potential to reduce the cost and complexity of preparing multifunctional drug delivery systems while also overcoming stability and heterogeneity issues associated with physically assembled colloids. These single polymer nanoparticles (SPNs) can be synthesized with cleavable bonds linking polymeric segments to the central scaffold facilitating hydrolytic or enzymatic degradation allowing for renal elimination. Herein we detail the development of 1st and 2nd generation polymeric brushes for applications in targeted drug delivery.
Results and discussion
Evaluation of the aqueous RAFT polymerization of DMA in the presence of the ECT
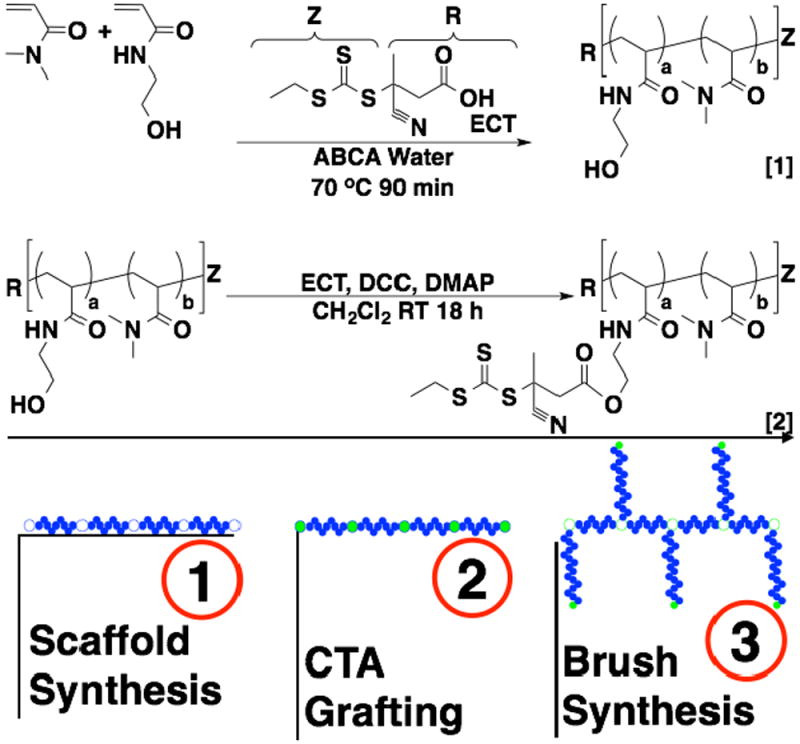
The preparation of polymeric brushes via RAFT may be accomplished by covalently conjugating multiple RAFT CTAs to a central core via either the R or Z group. The R group approach is advantageous in that the growing polymer chains propagate outwards away from the central core reducing steric hindrance and increasing accessibility of the thiocarbonyl thio groups to growing polymeric radicals. This strategy can, however, produce increased levels of radical-radical coupling because of the close spatial proximity of growing polymeric radicals. In these studies the trithiocarbonate-based RAFT agent, ECT, was covalently linked to hydroxyl functionalities, that were introduced into the polymeric backbone via copolymerization of HEAm with DMA, using carbodiimide chemistry catalyzed with DMAP (Scheme 1). The RAFT polymerization of DMA under both aqueous and organic conditions has been extensively evaluated by our group and others. 42-47 In the present studies the objective was to determine conditions that could be used to prepare well-defined polymers over a large molecular weight range with short polymerization times and higher than usual CTA to initiator ratios. The latter condition is important for limiting termination and deleterious chain transfer reactions when creating sophisticated polymer architectures such as brushes and block copolymers with more than two discrete segments. Aqueous polymerizations were investigated as acrylamido monomers, similar to other hydrophilic monomers, show exceptionally high propagation rates under these conditions facilitating the use of low initiator concentrations and short polymerization times.42,48-52 Rapid polymerizations are also particularly attractive for aqueous polymerizations as they minimize potential hydrolysis and/or aminolysis of the polymeric thiocarbonyl thio functionality.53,54 Shown in Fig. 1a are the molecular weight chromatograms for the aqueous RAFT polymerization of DMA targeting DPS of between 100 and 10 000 with a fixed initial ratio of ECT to ABCVA of 50 at 70 °C. As can be seen from Fig. 1a, the molecular weight distributions are narrow, symmetric and yield molecular weights that are easily controllable by simple manipulation of the initial monomer to CTA ratio. Significantly, all polymerizations reach moderate to high conversions even at large target DPs where the initiator concentration is quite low (e.g. [ABCVA]o = 675 μM at a target DP of 100 and 9 μM at a target DP of 7500). In all cases low Đ values are observed even for polymers where the resultant molecular weight is in excess of half a million Daltons. The high degree of control observed under these conditions over a wide molecular weight range is exemplified by these polymerizations where molecular weight and Đ values of 5100 Da/1.03 and 575 200 Da/1.19 are obtained for target DPs of 100 and 7500 respectively. The ability of these conditions to prepare well-defined block copolymers was also evaluated as shown in Fig. 1d. Here block copolymers with four discrete segments were prepared using a target DP of 200. These polymerizations show Đ values as low as 1.01 with clean evolution of the molecular weight distributions to lower elution volumes with each successive block copolymerization. Kinetic analysis of the aqueous RAFT polymerization of DMA targeting a DP of 5 000 provides additional evidence supporting the controlled nature of these synthetic conditions even at very high target molecular weights (Fig. 1c, d). As can be seen in Fig. 1c, the kinetics are linear following a brief induction period. Given the short reaction times (ie. 90 minutes) and low radical concentrations employed in these studies, this induction period is likely a result of the time needed for the aqueous solution to reach 70 °C and generate a sufficient primary radical concentration. Initial Đ values of around 1.3 are observed for conversions less than 15 % but quickly reach lower values around 1.15 at higher conversions. At polymerization times longer than 90 minutes, where conversion exceeds approximately 60 %, an increase in the Đ values is observed (data not shown). The pseudo first order plot (Fig. 1d) remains linear throughout the course of the polymerization suggesting that termination is negligible even at the low initial CTA concentrations employed. Taken together these results suggest that the aqueous RAFT polymerization of DMA can be controlled over a substantial range of target DPs.
Scheme 1.
(a) Synthetic strategy for the preparation of copolymers consisting of N,N-dimethylacrylamide (DMA) and Hydroxyethyl acrylamide (HEAm) at a 90:10 molar feed ratio (DMA:HEAm) with narrow Đ values over a range of molecular weights via RAFT. (b) Synthetic strategy for conjugation of ECT to the polymeric scaffolds using DCC and DMAP in methylene chloride.
Fig. 1.
Determination of polymerization conditions suitable for preparing well-defined polymeric scaffolds over a large range of molecular weights. Kinetic evaluation of the RAFT polymerization of DMA in water was conducted at an initial monomer concentration of 3 mol/L at 70 °C with ECT and ABCVA as the RAFT CTA and initiator respectively. (a) Mn- and Đ vs conversion (b) pseudo first order rate plot for the polymerization of DMA targeting a degree of polymerization (DP) of 4000 with an initial CTA to initiator ratio of 50 to 1. (c) RAFT polymerization of DMA targeting a range of DPs between 100 and 10 000. All polymerizations were conducted at [M]o and [CTA]o/[I]o values of 3 mol/L and 50:1 respectively. Mn (Da), target DP, and Đ values for the series were determined to be: 1 (100/5100/1.03), 2 (250/13,500/1.04), 3 (500/21,300/1.05), 4 (1000/59,400/1.04), 5 (2500/234,500/1.12), 6 (5000/402,400/1.16), 7 (7500/575,200/1.19). (d) Preparation of tetra-block copolymers via RAFT. The macroCTA and subsequent di-, tri-, and tetra- block copolymers (B1-B4) were prepare with [M]o:[CTA]o:[I]o = 200:1:0.02. Mn and Đ values for B1-B4 are as follows: (B1) 13 300/1.01 (B2) 32 500/1.01 (B3) 32500/1.03 (B4) 50500/1.06.
Evaluation of the carbodiimide mediated coupling of ECT to hydroxy-functional polymeric scaffolds
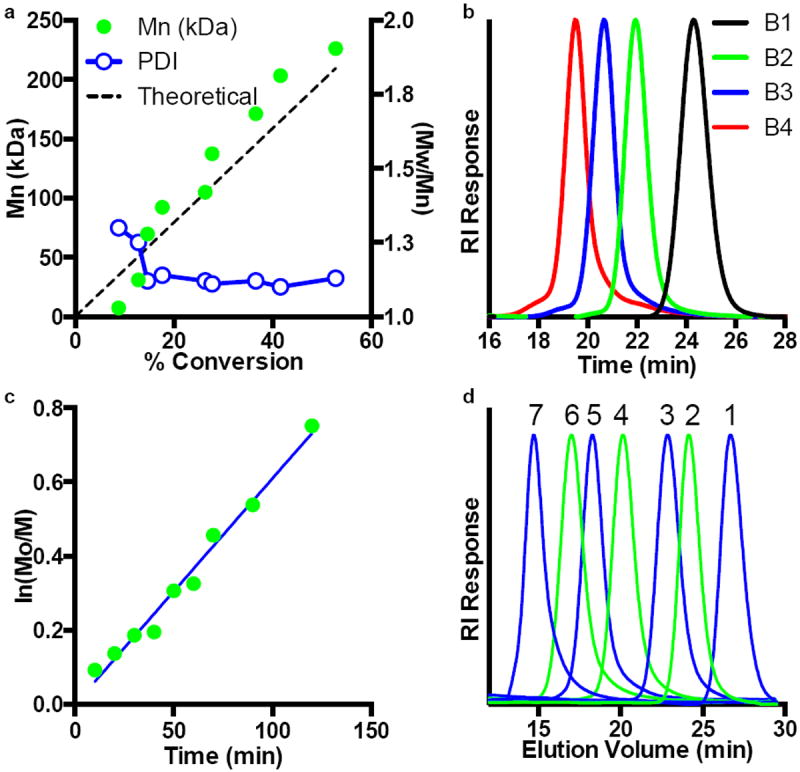
Based on the high degree of control observed for the aqueous homopolymerization of DMA, similar conditions were employed for copolymers of DMA and HEAm. In all cases the molar feed ratio of the two monomers (DMA:HEAm) was held constant at 90:10. This feed ratio was selected in order to limit the CTA graft density along the polymeric backbone, reduce steric hinderance of the growing polymer chains, and minimize deviations from the already established polymerization conditions. Reverse phase HPLC was used to confirm that these monomers are incorporated into the polymer backbone at ratios similar to the feed (not shown). The use of a high molar feed ratio of DMA also yields scaffolds and corresponding graft chain transfer agents (gCTAs) that can be used to prepare polymeric brushes under aqueous or organic conditions. Coupling of the polymeric hydroxyl groups to the carboxylic acid functional RAFT CTA was accomplished using carbodiimide chemistry catalyzed by DMAP in methylene chloride. Shown in Fig. 2, is the UV absorbance at 310 nm (corresponding to the CTA thiocarbonyl thio group) for molecular weight chromatograms before and after CTA conjugation. As can be seen in Fig. 2., the unfunctionalized mCTA (blue trace), which contains only a single thiocarbonyl thio group, shows low absorbance at 310 nm. Following CTA conjugation a substantial increase in the absorbance at 310 nm is observed for conjugation reactions conducted at an initial polymer concentration of between 0.5 and 10 wt. %. Also apparent from Fig. 2., is the appearance of a substantial high molecular weight coupling peak for conjugation reactions conducted at higher polymer concentrations (black traces). In strong contrast, coupling reactions conducted at a polymer concentration of 0.5 mg/mL (green trace) show a significant increase in the UV absorbance at 310 nm while remaining largely unimodal with only a small amount of polymer coupling. In all cases the injected polymer was held constant, which can be seen by the similar peak areas observed in the RI traces (vide infra). Based on these results all subsequent CTA coupling reactions were conducted at this concentration. The resultant CTA grafts are linked via ester bonds to the R group yielding polymeric gCTAs that are expected to polymerize from the central scaffold to form brush-like materials. Evidence supporting the formation of gCTAs containing multiple ester linked RAFT agents is provided by 1H NMR spectroscopy where the appearance of broad resonances at chemical shifts that are consistent with ECT (Fig. 2b). Particularly apparent is the ester resonance at 4 ppm, which is cleanly resolved from resonances associated with the acrylamido-based scaffold.
Fig. 2.
Overlay of UV absorbance at 310 nm for molecular weight distributions for poly(DMAcoHEAm) (90:10 mole fraction) before and after ECT conjugation at a constant mass of injected polymer. Before ECT grafting (blue trace) the absorbance of mCTA at 310 nm is low reflecting the presence of a single thiocarbonyl thio functionality per polymer chain. A substantial increase in the absorbance at 310 nm is observed for all of the molecular weight distributions following ECT conjugation. Conjugation reactions conducted at higher polymer concentrations (black traces) show substantial high molecular weight shoulders that are consistent with polymer crosslinking. In contrast conjugation reactions conducted at 0.5 wt % (green trace) show minimal high molecular weight shouldering while maintaining high levels of ECT grafting. CTA grafting reactions were conducted at between 0.5 and 10 wt % polymer in methylene chloride using dicyclohexyl carbodiimide (DCC) and dimethylaminopyridine (DMAP). The initial molar ratio of ECT to polymeric hydroxyl groups was 2 with equimolar quantities of DCC and DMAP relative to ECT. d. NMR spectrum for a generation1 gCTA in CDCl3 confirming successful conjugation of ECT to the poly(DMAcoHEAm) scaffold.
Preparation of gCTAs from scaffolds with different molecular weights
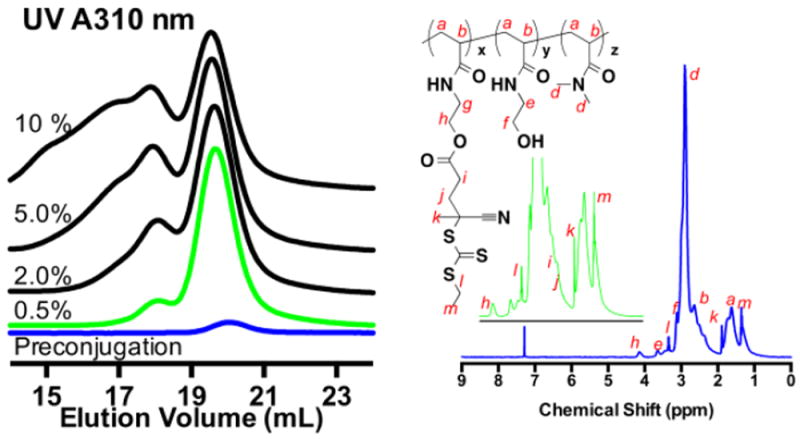
A series of gCTAs with between 10 and 60 ECT residues per polymer were prepared by first synthesizing scaffolds over a range of molecular weights. ECT was then coupled to the mCTAs using the conditions established above. Shown in Fig. 3., are the UV and RI traces for these materials before (green traces) and after CTA conjugation (blue traces). As can be seen in Fig. 3., the RI traces, which are proportional to the injected polymer mass, show similar peak areas at constant injection concentration/volumes. In contrast a substantial increase in UV absorbance is observed for all four polymers following CTA conjugation with only a slight increase in the Đ. The use of ester functionality to link the polymeric arms to a central scaffold is potentially advantageous for materials that are designed for use in vivo as it provides a mechanism by which high molecular weight polymer can be degraded to molecular weights suitable for clearance by hydrolysis and potentially via in vivo esterase activity. Based on the absolute molecular weight of the gCTAs (determined by GPC) as well as the extinction coefficient of ECT, which was determined to be 9422 mol L-1 cm-1 in DMF, the average numbers of trithiocarbonate groups per polymer as well as the average molar concentration of CTA per mass polymer was determined. Molecular weight, Đ, and average number of ECT functional groups per polymer for mCTAs 1 through 4 and the corresponding gCTAs were determined to be: (1) 15,500 Da/1.03/1 (1e, graft) 19,590 Da/1.05/10 (2) 35,600 Da/1.03/1 (2e, graft) 44,800 Da/1.09/20 (3) 69,800 Da/1.04/1 (3e, graft) 86,400 Da/1.11/36 (4) 116,500 Da/60/1.06 (4e, graft) 147 800 Da/1.14/60.
Fig. 3.
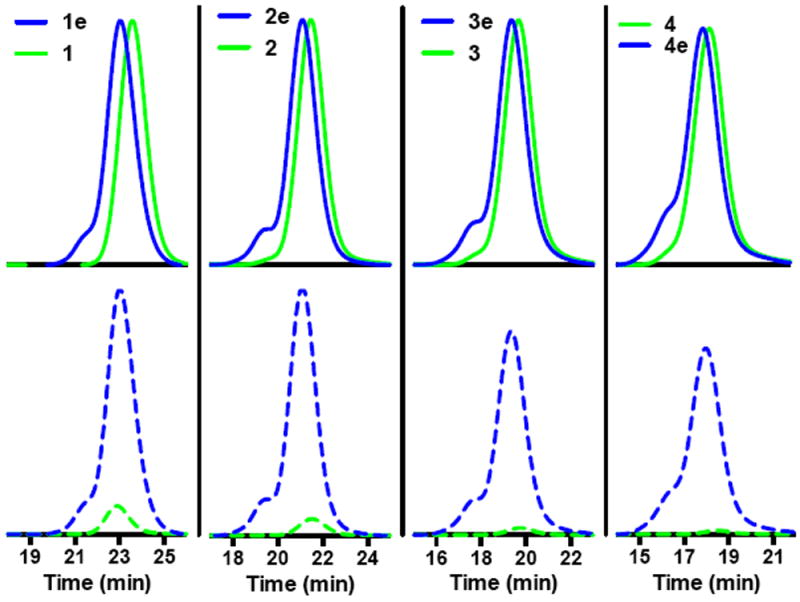
GPC chromatograms showing conjugation of ECT to polymeric scaffolds with different molecular weights. Poly(DMAcoHEAm) before (green traces) and after CTA (blues traces) conjugation was analyzed via GPC at a constant injection volume of 0.5 mg/mL. (top) Overlay of the RI response which is proportional to the injected mass. (bottom) Overlay of the UV response at 310 nm where thiocarbonyl thio groups show strong absorbance. Molecular weight, Đ, and number of ECT functional groups per polymer are as follows: (1) 15 500/1.03/1 (1e, graft) 19 590 Da/1.05/10 (2) 35 600 Da/1.03/1 (2e, graft) 44 800 Da/1.09/20 (3) 69 800 Da/1.04/1 (3e, graft) 86 400/1.11/36 (4) 116 500/60/1.06 (4e, graft) 147 800/1.14/60. Absolute molecular weights and Đ values where determined via SEC in DMF eluent. The extinction coefficient of ECT in DMF at 310 nm (9422 L/mol) was used to determine the concentration of polymer conjugated RAFT agent. The number of ECT functional groups per polymer was then determined by comparing these values to the respective absolute molecular weights determined by SEC.
Investigation of brush polymerization kinetics
Polymerization control as a function of monomer conversion was investigated for the aqueous RAFT polymerization of DMA from gCTA 1. The initial monomer ([M]o) to total polymer grafted CTAs ([gCTA]o) to initiator ([I]o) ratio was 200:1:0.02 respectively at an initial monomer concentration of 3M. The results of these studies are shown in Fig. 3a-c. Under these conditions a plot of the experimentally determined Mn versus conversion remains linear and are in good agreement with theoretical values (solid line) up to high monomer conversion with low Đ values (Fig. 4a). Analysis of the resultant copolymers show that an overall monomer conversion of ~86 % is reached at 90 min with a molecular weight and Đ of 159 000 Da (theoretical Mn = 189 000 Da) and 1.20, respectively. Given the close agreement between the theoretical and experimental molecular weights under these conditions, it is likely that brush-brush coupling and degradative chain transfer reactions are not occurring to a significant extent. Similarly, the pseudo first order rate plot versus time (Fig. 4b) is also linear suggesting that termination reactions remain low throughout the course of the polymerization. Moreover, analysis of the molecular weight distributions (Fig. 4c) shows that the peaks remain unimodal and symmetric throughout the course of the polymerization and shift to lower elution volumes with time. The lack of significant low molecular weight tailing in the molecular weight distributions coupled with the absence of high molecular weight shouldering suggest that the RAFT polymerization of DMA from the 10 arm gCTA is indeed living under these conditions. Evidence supporting the proposed brush architecture is shown Fig. 4d. Shown here are the molecular weight distributions for mCTA 3 (blue trace), as well as the corresponding DMA brush (green trace), and the resultant brush fragments (red trace) following transesterification with methanol catalyzed by DMAP. Following reflux in methanol containing DMAP for 96 hours complete disappearance of the brush peak is observed with the appearance of two peaks (red traces) at 19.2 and 23.1 mL with molecular weights that are consistent with the anticipated values for the polymeric scaffold cleaved arm product respectively.
Fig. 4.
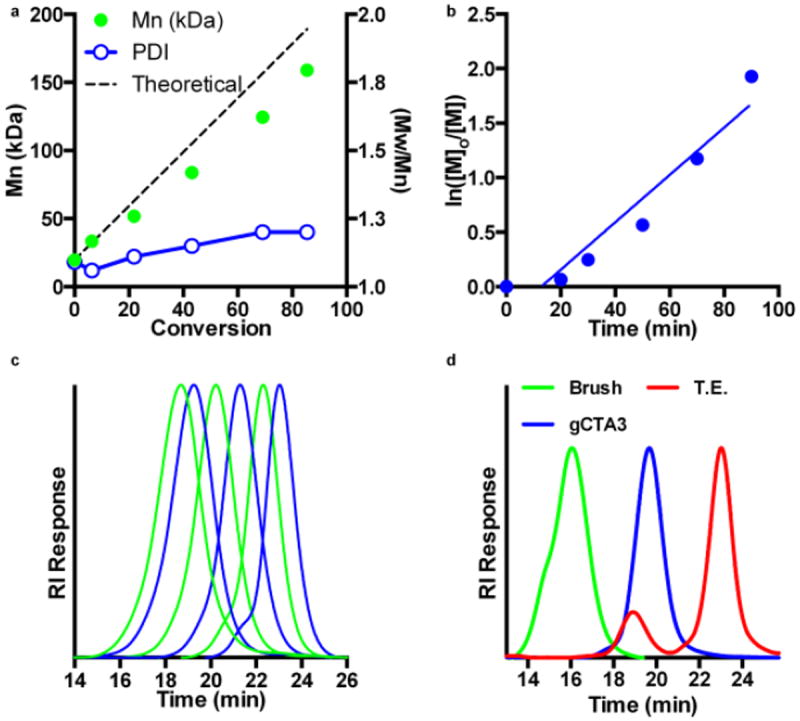
Kinetic data for the RAFT polymerization of DMA from a polymeric graft chain transfer agent (gCTA) containing an average of 10 CTAs/chain at 70 °C in molecular grade water with ABCVA as the initiator. The initial monomer ([M]o) to total polymer grafted CTAs ([gCTA]o) to initiator ([I]o) ratio was 200:1:0.02 respectively at an initial monomer concentration of 3M. a. Mn versus conversion b. pseudo first order rate plot. c. Evolution of the molecular weight distributions with time. d. Transesterification of a poly(DMA) brush in refluxing methanol for 96 h catalyzed by DMAP yields cleavage fragments corresponding to the polymeric scaffold and arms at elution times of 19 and 23 minutes respectively.
Synthesis of polymeric brushes
The gCTAs detailed in Fig. 3 were subsequently employed to prepared a series of polyDMA brushes targeting DPs of 100, 200, and 400, which are represented by the green, blue, and red traces respectively. The parent gCTAs (traces shown in black) have molecular weights and CTA grafting values (ECT groups/polymer chain) of 19 600 Da/10, 35 600 Da/20, 86 000 Da/36, and 116 500 Da/60 for gCTAs 1 through 4 respectively (Fig. 5). All polymeric brushes were preparing using conditions similar to those used to prepare the parent scaffolds. In all cases a clear shift in the molecular weight chromatograms (refractive index traces shown) are observed. Exceptionally good polymerization control is observed for the synthesis of brushes with 10 and 20 arms. These polymerizations yield materials with narrow symmetric molecular weight distributions that cleanly shift to lower elution volumes. Moreover the resultant chromatograms show only small amounts of brush-brush coupling as evidenced by the lack of significant high molecular weight shouldering. Also apparent is the lack of peaks at longer elusions volumes, which confirms the absence of observable low molecular weight unimeric polymer species. Under these conditions brushes with molecular weights and Đ values of 193 000 Da/1.19 and 352 000 Da/1.16 are observed for the 10 and 20 arm gCTAs targeting a DP of 400. This corresponds to an average molecular weight per arm of 17 800 and 15 400 Da respectively. Relatively good control is also observed for the 36 arm and 60 arm brushes although the appearance of high molecular weight shoulder is apparent especially at the higher target DPs. The appearance of a high molecular shoulder is most prevalent in the 60 arm brush where all target DPs show moderate coupling. Similar to the lower arm gCTAs however these materials all cleanly shift to lower elution volumes without appreciable low molecular weight populations. Molecular weight and Đ values for the polymeric brushes shown in Fig. 5 are detailed in Table 1. The ability to prepare polymeric brush for different monomer classes was also investigated. In these studies gCTA 2, which contains an average number of ECT residues per chain of 20, was employed to probe the polymerization behavior of several important monomer classes including acrylamido, methacrylamido, and methacrylate (Fig. 6). Polymers of HPMA have been shown to be hydrophilic and biocompatible making them an important material in a number of biotechnology applications.55-57 For this reasons it was of interest to develop polymerization conditions facilitating the synthesis of well-defined brushes of HPMA with controllable molecular weights and low Đ values. Previously it has been shown that HPMA polymerizes rapidly with a high degree of control under aqueous conditions but requires significantly higher levels of initiator when polymerizations are conducted in organic media. 55,58 Aqueous RAFT polymerizations of HPMA with an initial stoichiometric ratio of monomer, chain transfer agent, and initiator ([M]o:[CTA]o:[ABCC]o) equal to 150:1:0.05 were selected with[M]o equal to 15 wt %. SEC analysis of the resultant HPMA brush following polymerization at 70 °C in molecular grade water yielded an overall molecular weight, Đ, and molecular weight per arm of 271 200 Da, 1.13, and 11 500 Da respectively. Previously we also outlined conditions allowing NIPAM to be polymerized with a high degree of control via RAFT at room temperature. 42,59In the present studies, however, the LCST of the resultant polyNIPAM brush was below room temperature (data not shown) necessitating the development of organic polymerization conditions. Polymerizations conducted in DMSO at [M]o = 20 wt % and [M]o:[CTA]o:[I]o = 400:1:0.025 for 3 hours at 70 °C yielded a polymeric brush with an overall molecular weight, molecular weight per arm, and Đ of 472 000 Da, 21 500 Da, and 1.15 respectively. Similarly the preparation of a polyDMAEMA brush was also conducted in DMSO with [M]o = 30 wt % and [M]o:[CTA]o:[I]o = 200:1:0.02 for 4 hours at 90 °C. Characterization of the resultant polyDMAEMA brush yielded corresponding values of 472 000, 1.15, and 21 500 Da respectively. Brushes composed of the hydrophilic monomer DMA with glycine acrylamide (GlyAM) which has hydroxyl-reactive carboxylate were also prepared. These materials could potentially be employed to, for example, conjugate hydroxyl containing drugs (e.g. taxanes, etoposide, camptothecin) to polymeric scaffolds via degradable ester bonds with precisely tuned polymer and drug compositions.
Fig. 5.
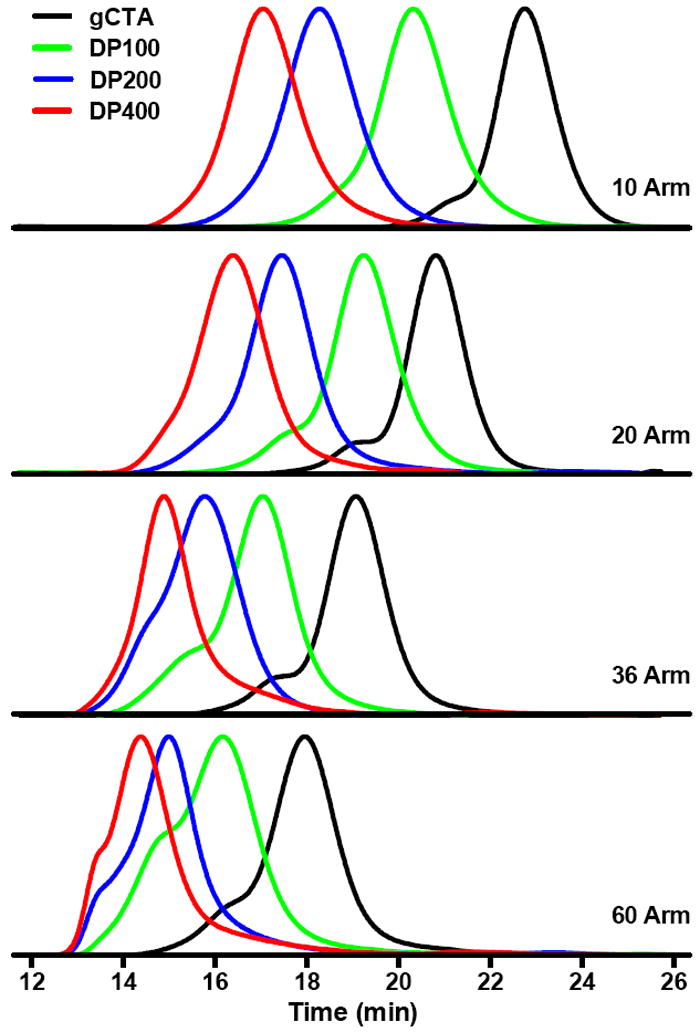
Normalized refractive index response versus time for the RAFT polymerization of DMA from graft chain transfer agents containing 10, 20, 36, and 60 thiocarbonyl thio groups per polymer. Polymerizations were conducted in molecular grade water at an initial monomer concentration of 3M for 90 minutes at 70 °C with ABCVA as the initiator. Target degrees of polymerization evaluated were 100, 200, and 400 with an initial thiocarbonyl thio group to initiator ratio of 0.02. Molecular weight, Đ values, and molecular weight per arm for the macromolecular brushes were determined to be: 10 arm DP 100 (63,000/1.15/4800) DP 200 (135,000/1.17/12,000) DP 400 (193,000/1.23/17800) (20 arm) DP 100 (101,700/1.15/2900) DP 200 (230,100/1.17/9300) DP 400 (352,200/1.16/15,400) (36 arm) DP 100 (276,500/1.25/5300) DP 200 (551,200/1.22/12,900) DP 400 (749,000/1.18/18,400) (60 arm) DP 100 (549,600/1.32/6700) DP 200 (1,198,000/1.24/17,500) DP 400 (1,700,000/1.24*/25,900)-(*at the exclusion limit of the columns).
Table 1.
Summary of theoretical and experimentally determined molecular weights and Đ values for polymeric brushes synthesized from 10, 20, 36, and 60 arm gCTAs as a function of target DP.
| Polymer # | Target DP | Arms Per Polymer | Monomer Conv. (%) | Mn Theory (Da) | Mn Exp. (Da) | Đ |
|---|---|---|---|---|---|---|
| 1 | 400 | 10 | 48 | 208400 | 193000 | 1.23 |
| 2 | 400 | 20 | 53 | 465500 | 352200 | 1.16 |
| 3 | 400 | 36 | 48 | 772700 | 749000 | 1.18 |
| 4 | 400 | 60 | 48 | 1291600 | 1700000 | 1.24 |
| 5 | 200 | 10 | 66 | 150200 | 135000 | 1.17 |
| 6 | 200 | 20 | 62 | 292300 | 230100 | 1.17 |
| 7 | 200 | 36 | 51 | 453700 | 551000 | 1.32 |
| 8 | 200 | 60 | 63 | 902600 | 1198000 | 1.24 |
| 9 | 100 | 10 | 60 | 79500 | 63000 | 1.15 |
| 10 | 100 | 20 | 59 | 162600 | 101700 | 1.15 |
| 11 | 100 | 36 | 48 | 258000 | 276500 | 1.25 |
| 12 | 100 | 60 | 38 | 373100 | 549600 | 1.32 |
As determined by size exclusion chromatography using Tosoh SEC TSK-GEL α-3000 and α-4000 columns (Tosoh Bioscience, Montgomeryville, PA) connected in series to an Agilent 1200 Series Liquid Chromatography System (Santa Clara, CA) and Wyatt Technology miniDAWN TREOS, 3 angle MALS light scattering instrument and Optilab TrEX, refractive index detector (Santa Barbara, CA). HPLC-grade DMF containing 0.1 wt.% LiBr at 60 °C was used as the mobile phase at a flow rate of 1 mL/min.
As determiend by 1H NMR in CDCl3 by comparison of the DMA vinyl resonances between 5.60 and 6.78 to the backbone resonances between 1 and 1.8 ppm.
As determined by the equation Mn theory = [M]o*CM*FWM/[ECT]o where [ECT]o is the total concentration of ECT groups linked to the gCTA
Fig. 6.
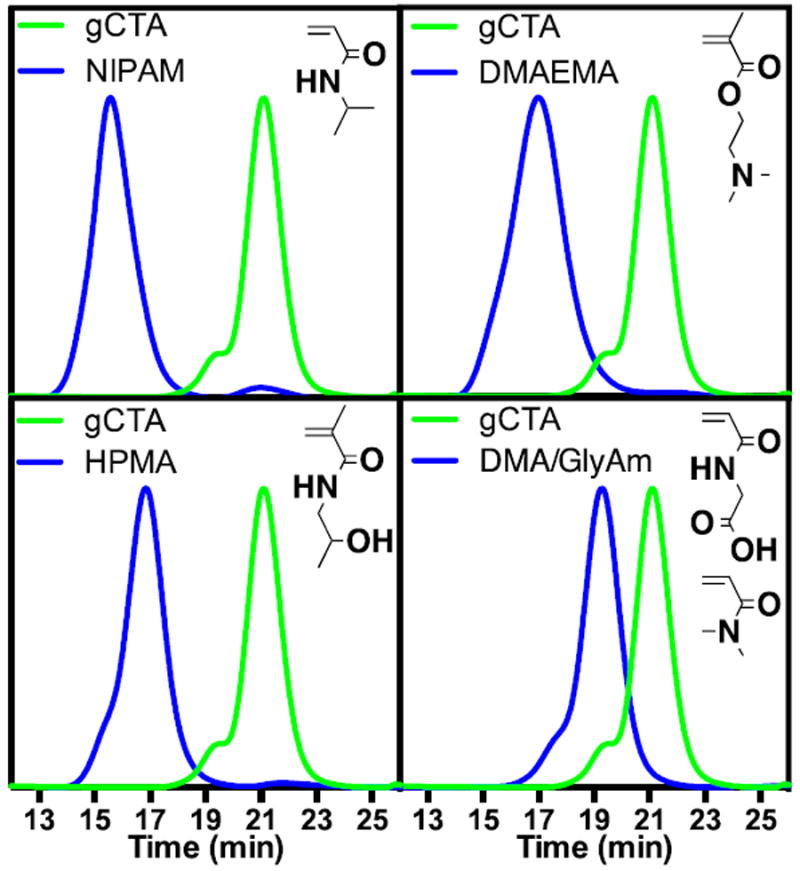
Mn, Đ, and molecular weight per arm values for polymeric brushes prepared from a 20-arm gCTA (Mn 44,800; Đ 1.09) (a) NIPAM 472,000/1.15/21,500 (b) DMAEMA 352,500/1.19/15,500 (c) HPMA 271 200/1.13/11,500 (d) DMAcoGlyAM 123,000/1.19/4000. Polymerization Conditions: (NIPAM) [M]o:[CTA]o:[ABCVA]o and [M]o = 400:1:0.025 and 20 wt % in DMSO for 3h at 70 °C. (DMAEMA) [M]o:[CTA]o:[ABCC]o and [M]o = 200:1:0.02 and 30 wt % in DMSO for 4h at 90 °C. (HPMA) [M]o:[CTA]o: [ABCC]o and [M]o = 150:1:0.05 and 15 wt % in molecular grade water for 4h at 70 °C.
Second generation brushed-brushes for application in controlled drug delivery
High molecular weight polymers typically show enhanced in vivo circulation times but can present clearance problems. In an attempt to circumvent these issues a new polymer architecture was developed based on the 2nd generation brushed-brushes. This approach, which is outlined Scheme 2, consists of a central scaffold from which a 1st generation brush is grown via RAFT polymerization from conjugated thiocarbonyl thio groups. Following the initial brush synthesis a second CTA grafting reaction is conducted. Subsequent RAFT polymerization from the 2nd generation gCTA is then performed to yield the final 2nd generation SPN, which consists of a polymeric brush where the individual polymeric arms are also brushes. This architecture, which has some similarities with dendrimers, is composed of relatively small polymer segments (e.g. 5-10 kDa) of polyDMA linked together via hydrolytically cleavable ester bonds. Conjugation of doxorubicin to the hydrophilic megamolecular scaffolds via degradable hydrazones bonds is accomplished by adding a boc-protected carbazide monomer (bocSMA) at 5 mol % to the feed. Following removal of the boc groups via treatment with TFA for 1 hour the resultant carabazide groups can then be reacted with doxorubicin to yield the desired hydrazone linked polyvalent drug conjugates. The brushed-brushes were also designed to integrate streptavidin (SA) linked cancer-specific antibodies (AB) by incorporating biotin groups into the scaffold. Biotin spontaneously forms an exceptionally strong non-covalent bond with SA providing a facile route by which SA-ABs can be linked to the polyvalent drug carrier spontaneously simply by mixing buffered solutions of the reagents. This approach provides a modular route for preparing polymeric drug conjugates with a range of antibodies specifically tailored to the target tissue. As can be seen in Fig. 7a, synthesis of the 1st generation scaffold and subsequent ECT grafting yields a strong increase in the UV absorbance at 310 nm. The resultant 1st generation gCTA has a molecular weight and Đ of 20 100 Da and 1.12 respectively with an average number of ECT groups per polymer chain of 9.3. Polymerization of DMA and HEAm (90:10 molar feed ratio) from this gCTA yields a polymeric brush with a molecular weight, Đ, and molecular weight per segment of 140 000 Da, 1.19, and 13 000 Da. The latter values were calculated by subtracting the molecular weight of the 1st generation gCTA and then dividing by the average number of CTA groups (including the thiocarbonyl thio group at the chain terminus). As can be seen from Fig. 7b, upon brush formation a clear shift in the molecular weight distribution (refractive index channel) to shorter elution volumes is observed while remaining narrow and symmetric. Successful formation of the 2nd generation gCTA is supported, as before, by the substantial increase in the UV absorbance at 310 nm (Fig. 7c). The resultant 2nd generation gCTA has a molecular weight of 171 000 Da and dispersity of 1.27 with an average number of ECT groups per polymer chain of 75.5. The final drug delivery system was then prepared by polymerizing DMA, bocSMA, and BioHEMA from the 2nd generation gCTA to yield a brushed-brush with a molecular weight and Đ of 1 152 000 Da and 1.25 respectively. The average number of carbazide and biotin residues per brushed-brush was determined to be 385 and 65 via TNBSA and HABA assays, respectively. An interesting aspect of this approach is that it allows for more sterically bulky monomers such as the methacrylate monomer DMAEMA to be polymerized from scaffolds prepared from less bulky acrylamido (DMA) scaffolds. Typically RAFT polymerizations require the more sterically bulky monomer to be synthesized first in order to avoid slow fragmentation from the intermediate radical species. Since a large fraction of the CTA residues present in the generation 1 and 2 gCTAs contain a cyanovalerate R the polymerizations remain unimodal with slow fragmentation from the thiocarboyl thio group at the chain terminus not showing significant deleterious effects on the polymerization control. This behavior is exemplified by the controlled RAFT polymerization of DMAEMA from a 2nd generation DMA gCTA (not shown).
Scheme 2.
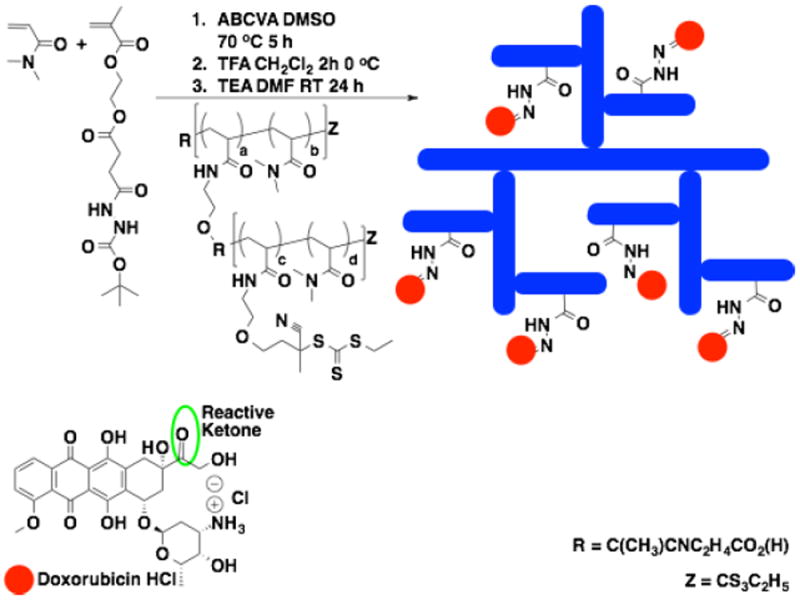
Synthetic scheme for the preparation of 2nd generation brushed-brushes from a generation 2 gCTA containing polyvalent hydrolytically cleavable doxorubicin moieties.
Fig. 7.
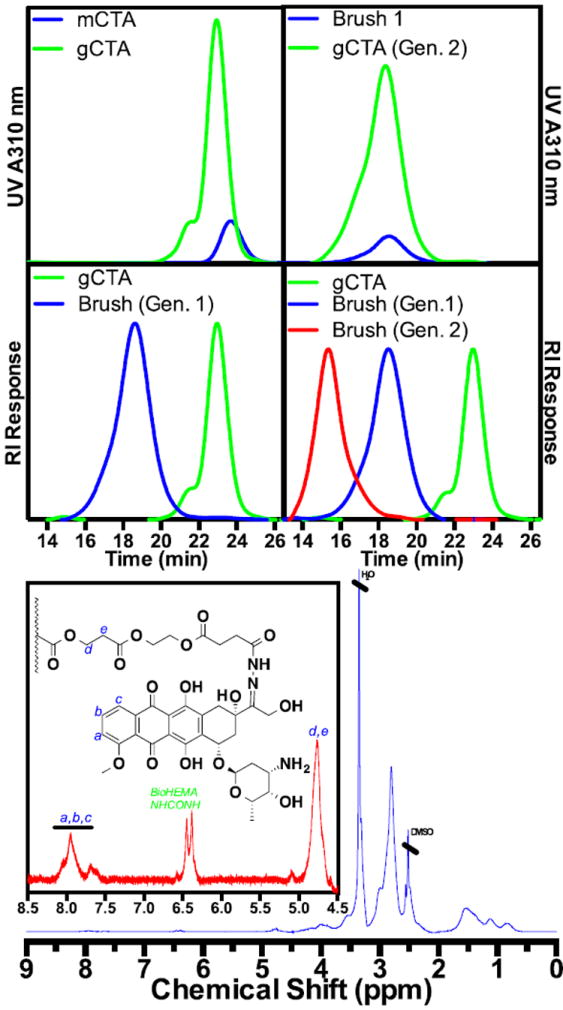
SEC data supporting the formation of brushed-brushes incorporating chemical functionality suitable for binding SA-AB conjugates and bocprotected carbazate residues capable of binding doxorubcin (following deprotection with TFA) via hydrolytically cleavable hydrazone bonds. Molecular weight, Đ, and thiocarbonyl thio groups per polymer: (mCTA) 15,700/1.01/1, (gCTA Gen. 1) 20,100/1.12/9.30, (Brush Gen. 1) 140,000/1.19/n.d., (gCTA Gen. 2) 171,000/1.27/75.5, (Brush Gen. 2) 1,152,000/1.25/n.d. The average molecular weight per arm for the 1st and 2nd generation brush are 16,200 and 13,000 respectively. The average number of carbazate (following deptrotection) and biotin residues was determined to be 385 and 65 respectively.
Conjugation of Doxorubicin to 2nd generation brushed-brushes via hydrolytically cleavable hydrazone bonds
Conjugation of doxorubicn to the polymeric scaffold was subsequently accomplished through reaction of the deprotected carbazide residues with doxorubicin in DMF in the presence of TEA. Following purification 1H NMR (Fig. 8c) and UV spectroscopy (Fig. 8d) were employed to confirm successful hydrazone formation. The appearance of aromatic resonances associated with doxorubicin can be seen in Fig. 8c. along with residues associated with the biotin NHCONH residues. Comparison of the doxorubicin concentration, determined by UV spectroscopy at 480 nm, to that of the polymer determined by absolute SEC-MALLS yielded an average number of drug molecules per brushed-brush of 195. Even with the large number of drug molecules per polymer the carriers remain soluble in PBS with an average hydrodynamic volume of 31 nm as determined by DLS. With the high cost of antibody-targeted therapies such as Trastuzumab, which currently costs approximately $70,000 for a full course of treatment, the capability of the brushed-brushes to carry high numbers of covalently linked drug molecules could potentially provide substantial reductions in the cost of AB-targeted therapies by minimizing the amount expensive antibodies needed. Release of the doxorubcin as a function of pH and in the presence of serum was evaluated using HPLC as shown in Fig. 8d. The hydrazone linkage tethering doxorubicin to the polymeric scaffold was found to be quite stable at pH 7.4 with only 30 % cleavage observed following a 24 h incubation period. In contrast, conjugates incubated under acidic conditions show high levels of hydrolysis even at short incubation times. This cleavage was found to be most significant at pH 5.8 where nearly 90 % of the hydrazone linked doxorubicin was hydrolyzed from the polymeric scaffold at 1 h incubation. Conjugates incubated at pH 6.6 showed intermediate levels of hydrolysis between these two extremes with 44 % drug release at 24 hours. As can be seen in Fig. 8, both the free drug and polymer drug conjugates show similar chemotherapeutic activity against SKOV3 ovarian cancer cells which are somewhat resistant to doxorubicin. For example, cell viability was found to be 34 % at a doxorubicin conjugate concentration of 10 M as compared to 39 % for the free drug. Significant enhancements in chemotherapeutic activity for the SNP linked Dox could however be observed in vivo where differences in pharmacokinetic properties, short resonances times, and off-target affects limit the maximum achievable dose for free Dox. Shown in Fig. 9, are live cell deconvoluted fluorescent images for SKOV3 cells treated with free Dox, Dox conjugated to the 2nd generation brushed-brushes, Dox conjugated to the 2nd generation brushed-brushes with SA-CD19 linked antibody (negative control antibody), and 2nd generation brushed-brushes with SA-Trastuzumab linked antibody. The latter antibody is specific for HER 2 receptors, which are overexpressed on many breast and ovarian cancer cell lines. As can be seen in Fig. 9, the 1 hour treatment with free Dox (left column) shows low fluorescence (green) relative to all of the polymer-Dox treatments when analyzed at comparable imaging settings. At 1 hour both the polymer-Dox conjugate and CD19 linked polymer-Dox conjugate show modest green fluorescence while the Trastuzumab linked-polymer conjugate shows intense green fluorescence. This result is consistent with enhance antibody-mediated internalization of the polymeric Dox conjugates via the HER 2-specific Trastuzumab. At 4 hours the differences between the antibody-targeted and untargeted conjugates are less pronounced suggesting that given sufficient time the SPNs are taken up via nonspecific endocytosis.
Fig. 8.
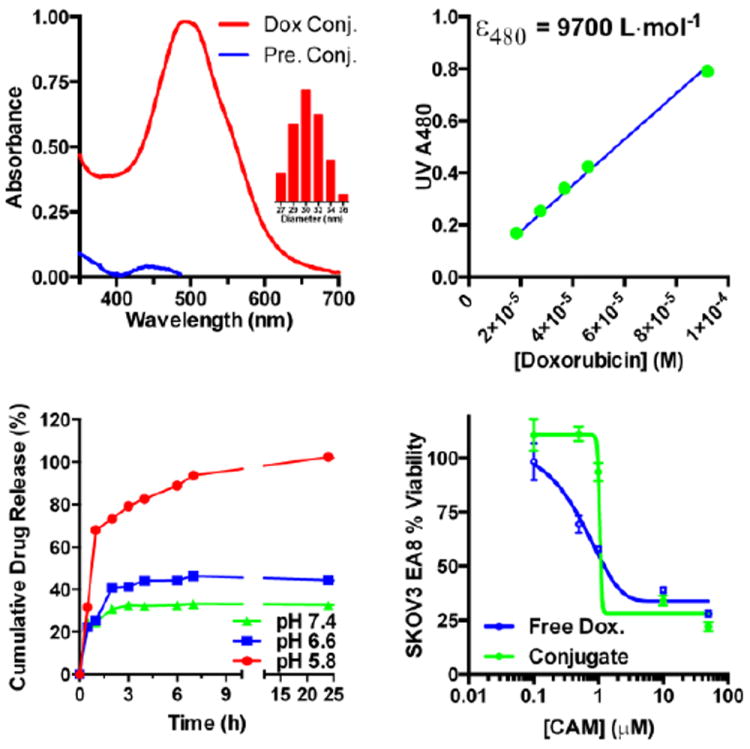
(a) UV absorbance spectrum for the 2nd generation brushed brushes before (blue) and after (red) Dox conjugation in PBS as pH 7.4. (b) Beers law plot for free Dox in PBS. Drug release kinetics for Dox linked to the brushed brushes via degradable hydrazone bonds. At pH 7.4 the hydrazone linkages are relatively stable with only 30 % cleavage at 24 h. In contrast, conjugates incubated under acidic conditions show high levels of hydrolysis even at short incubation times. This cleavage was found to be most significant at pH 5.8 where nearly 90 % of the hydrazone linked doxorubicin was hydrolyzed from the polymeric scaffold at 1 h incubation. Conjugates incubated at pH 6.6 showed intermediate levels of hydrolysis between these two extremes with 44 % drug release at 24 hours. (d) In vitro toxicity of the doxorubicin conjugates in SKOV3 cells, as determined by MTS, following a 72 h incubation period.
Fig 9.
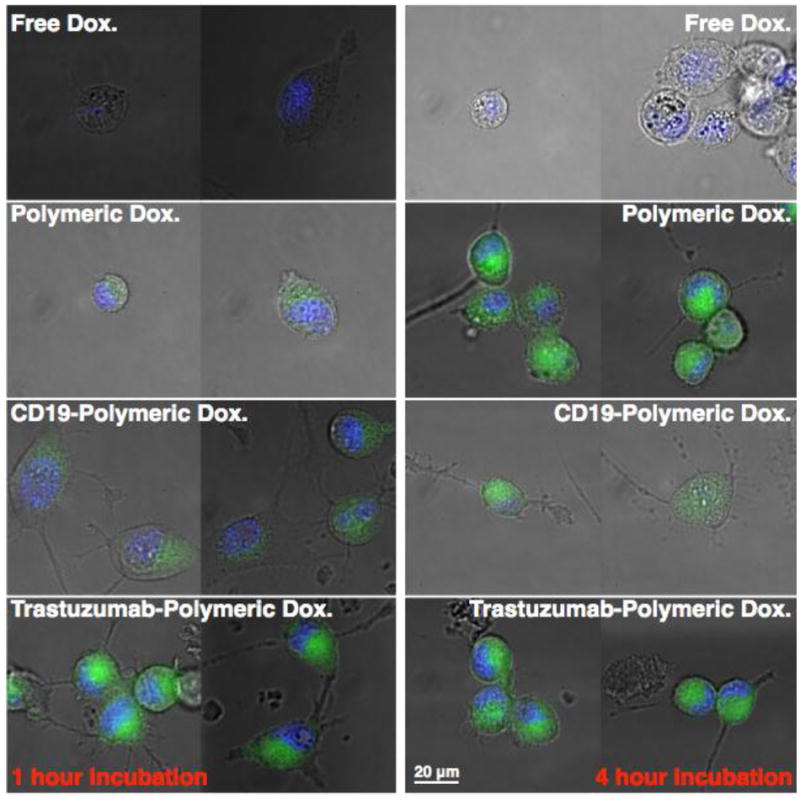
Live cell deconvoluted fluorescence microscopy images showing the time-dependent dynamics of antibody-mediated enhanced uptake in SKOV3 cells. Cells were treated with free doxorubcin (Dox), 2nd generation brushed brushes conjugated to Dox via degradable hydrazone linked Dox. 2nd generation brushed brushes conjugated to Dox with SA-CD19 (negative control antibody) and 2nd generation brushed brushes conjugated to Dox with SA-Trastuzumab (HER 2 specitic antibody). SKOV3 cells were treated with Dox, which is fluorescent (green fluorescence channel), or the indicated Dox conjugates for 1 and 4 hours. Thirty minutes prior to imaging Hoechst nuclear stain (blue fluorescence channel) was added to the cells after which time the media was replaced with fresh PBS. All images were taken at identical imaging settings. At both 1 and 4 hours free doxorubicin shows relatively low green fluorescence compared to the polymer and polymer-antibody conjugates at comparable imaging settings. At 1 hour incubation the CD19 conjugates show fluorescence intensities comparable to the untargeted polymer conjugates while the Trastuzumab-polymer conjugates show a substantial increase in green fluorescence. At 4 hours the differences between targeted and untargeted polymer conjugates is smallar suggesting accumulation of the polymer conjugates in intracellular compartments.
Conclusions
Aqueous RAFT copolymerizations of DMA and HEAm in the presence of the trithiocarbonate-based RAFT agent ECT yield polymers with narrow and symmetric molecular weight distributions over a range of molecular weights between 1000 and 1 000 000 Da. The resultant hydroxyl functional copolymers can be employed as scaffolds to which RAFT CTAs can be grafted using DCC DMAP chemistry. These gCTAs retain the narrow and symmetric molecular weight distributions of the parent scaffolds while showing substantial increases in UV absorbance at 310 nm which is attributed to the addition of multiple CTAs. Polymerization kinetics, conducted from a 10 arm gCTA, show that polymeric brushes prepared via this approach remain living throughout the course of the reaction as evidenced by the linear Mn vs. conversion and pseudo first order rate plots. Polymeric brushes containing hydroxyl functionality can be used to prepared 2nd generation gCTAs allowing for the controlled synthesis of 2nd generation brushed-brushes. These SPNs can have molecular weights in excess of 1 million Da with low Đ values and cleavable ester functionality linking the polymeric segments. Introduction of carbazate and biotin functionality for drug and antibody binding respectively was confirmed by a combination of 1H NMR, UV spectroscopy, HABA and TNBSA. The chemotherapeutic activity of these materials was evaluated in SKOV3 ovarian cancer cells with the anthracycline-based drug doxorubicin via copolymerization of DMA with a protected carbazate monomer (bocSMA). Cell specific targeting functionality was also introduced via copolymerization with a biotin-functional monomer (bioHEMA). Enhanced release of the doxorubicin from hydrazone linkages was confirmed via drug release studies at neutral and acidic pH values.
Experimental
Materials
Chemicals and all materials were supplied by Sigma-Aldrich unless otherwise specified. Dimethylaminoethyl methacrylate (DMAEMA), dimethylacrylamide (DMA), and, hydroxyethylacrylamide (HEAm) were distilled under reduced pressure. NIPAM was recrystazllized from hexanes. 2-hydroxypropyl methacryalmide (HPMA) was purchased from polysciences. Spectra/Por regenerated cellulose dialysis membranes (6-8 kDa cutoff) where obtained from Fisher Scientific. 4-Cyano-4-(ethylsulfanylthiocarbonyl) sulfanylvpentanoic acid (ECT) was synthesized as described previously35. 1,1’-Azobis(cyclohexane-1-carbonitrile) (ACC) was obtained from Wako Pure Chemicals. Sephedex G-25 prepacked PD10 columns were obtained from GE Life Sciences. It is particularly important to completely remove all of the inhibitor from the DMA and HEAm monomers as well as uncoupled RAFT agents following CTA conjugation as degradative or degenerate chain transfer to these impurities can cause the formation of unimeric polymer species during brush synthesis. Distillation should be used over flash chromatography for these polar monomers. TNBSA and HABA assays were conducted as described previously.
Gel permeation chromatography (GPC)
Absolute molecular weights and polydisperity indices were determined using using Tosoh SEC TSK-GEL α-3000 and α-e4000 columns (Tosoh Bioscience, Montgomeryville, PA) connected in series to an Agilent 1200 Series Liquid Chromatography System (Santa Clara, CA) and Wyatt Technology miniDAWN TREOS, 3 angle MALS light scattering instrument and Optilab TrEX, refractive index detector (Santa Barbara, CA). HPLC-grade DMF containing 0.1 wt.% LiBr at 60 °C was used as the mobile phase at a flow rate of 1 ml/min.
Dynamic Light Scattering
Particle sizes of polymeric brushes was measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS. Lyophilized polymer was dissolved in acetone at 50 mg/mL, then diluted 50-fold into the appropriate buffer solution.
Determination of comonomer conversion via HPLC
Comonomer conversion was determined using reverse-phase high-performance liquid chromatography (RP-HPLC). RP-HPLC was performed using a Finnigan Surveyor LC Pump Plus with UV-Vis Detector and a WAVELENGTH mm Vydac 218TP C18 5 column (Part 218TP52). Aliquots taken prior to and after copolymerization (100 μL) were diluted into 900 μL of eluent. A 20 μL aliquot of the solution was then diluted with 20 μL of hexafuoro-2-propanol and then injected with a 20 μL sample loop. A gradient was set from 5% to 95% acetonitrile over 15 minutes and UV absorbance was measured to observe the monomer concentration.The percent of each monomer converted was measured by integrating absorbance signal of the respective monomers before and after polymerization.
Mono-2-(Methacryloyloxy)ethyl succinate tertiarybutylcarbazate (bocSMA)
To 2.76 g (12 mmol) of mono-2-(methacryloyloxy)ethyl succinate in 75 mL of CH2Cl2 was added 1.45 g (11 mmol) of tert-butyl carbazate followed by 2.49 g (13 mmol) of N-(3-dimethylaminopropyl)-N’-ethylcarbodimide hydrochloride. After stirring at RT for 5 h, CH2Cl2 was evaporated and the residue was dissolved in 100 mL of ethyl acetate and washed with brine (3 × 50 mL). The organic layer was dried over Na2SO4, and then evaporated under reduced pressure. Flash column chromatography with ethyl acetate/hexane (1:1) yielded 3.33 g of SMA carbazate (89%). 1H NMR (300 MHz, CDCl3) δ 1.45 (s, 9H, C(CH3)3), 1.94 (s, 3H, CH3), 2.52 (t, J = 6.9 Hz, 2H, HNCOCH2), 2.72 (t, J = 6.9 Hz, 2H, OCOCH2), 4.34 (s, 4H, -OCH2CH2O-), 5.59 (s, 1H, CH2=CCH3 trans), 6.12 (s, 1H, CH2=CCH3 cis), 6.57 (s, 1H, HNCOCH2), 7.76 (s, 1H, (CH3)3COONH). MS (ESI) m/z = 367.1 (M +Na).
Synthesis of bioHEMA monomer
Biotin (5 g) was dissolved in dimethylacetamide (30 mL). A 2-fold molar excess of hydroxyethylmethacrylate (HEMA), N,N-diisopropylcarbodiimide (DIC), and 4-dimethylaminopyridine (DMAP) were dissolved in the biotin solution and reacted overnight at 4 °C. Reaction solution was filtered to remove dicyclohexyl urea by-product, then precipitated in cold diethylether (4X) to remove DMAP. Finally the product was dissolved in dimethylsulfoxide and precipitated in 1M HEPES buffer pH 8.4 to remove free biotin. The pellet was washed with ddH2O and the final product was lyophilized into a dry powder. The purity of the bioHEMA monomer was confirmed by 1H-NMR and mass spectrometry (See Supporting Information).
RAFT polymerizations of DMA
RAFT polymerizations of DMA targeting a range of target DPs between 100 and 10 000 were conducted with ECT and ABCVA as the CTA and initiator respectively in molecular grade water at 70 °C. Polymerizations targeting an initial monomer to CTA ratio ([M]o/[CTA]o) between 100 and 5 000 where conducted at a fixed initial CTA to initiator ratio ([CTA]o/[I]o) of 50 and an initial monomer concentration of 3M. Polymerizations targeting higher [M]o/[CTA]o ratios were conducted at [CTA]o/[I]o of 100 at an initial monomer concentration of 4M. Polymerizations were purged with nitrogen for 1 hour and then allowed to react at 70 °C for 90 minutes. A representative procedure for the RAFT polymerization of DMA targeting a DP of 1500 is as follows. To a 10 mL round bottom flask was added DMA (1.00 g, 10.88 mmol), ECT (1.77 mg, 6.72 μmol as 100 μL of a 17.71 mg/mL ECT stock in ethanol), and ABCVA (0.037 mg, 0.134 μmol as 100 μL of a 3.7 mg/mL ABCVA stock in ethanol). The solution was then diluted with 2.37 mL of molecular graded water sealed with a rubber septum and purged with nitrogen for 60 minutes. After this time, the polymerization solution was transferred to a preheated oil bath at 70 °C and allowed to polymerize for 90 minutes. The copolymer was subsequently isolated by dialysis against deionized water at 5 °C and lyophilisation. The final monomer conversion was determined by comparison of the total vinyl resonances (3H) between 5.6 and 6.7 ppm to the backbone resonances between 1.0 and 1.72 ppm. Chain extension of poly(DMA) macroCTAs were conducted up to a maximum of six blocks using conditions similar to those outlined for the ECT mediated homopolymerization of DMA except that subsequent block copolymerizations employed additional molecular grade water equal to one half the mass of added macroCTA (m/v).
RAFT copolymerization of N,N-dimethylacrylamide (DMA) and 2-hydroxyethylacrylamide (HEAm)
Synthesis of Poly[(DMA)-co-HEAm)] was prepared with ECT and ABCVA as the RAFT chain transfer agent (CTA) and radical initiator respectively. The molar feed composition for all copolymerization was 90% DMA and 10 % HEAm with total initial monomer to CTA ratios ([M]o/[CTA]o) between 500 and 4000 at a fixed initial CTA to initiator ratio ([CTA]o/[I]o) of 50 and an initial monomer concentration of 3M in molecular grade water. Copolymerizations were purged with nitrogen for 1 hour and then allowed to react at 70 °C for 90 minutes. A representative procedure for the synthesis of poly(DMA-co-HEAm) targeting a degree of polymerization (DP) of 500 is as follows. To a 50 mL round bottom flask was added DMA0 (4.65 g, 46.9 mmol), HEAm (0.600 g, 5.21 mmol), ECT (27.5 mg, 0.104 mmol), ABCVA (0.58 mg, 2.08 μmol as 100 μL of a 5.8 mg/mL ABCVA stock in ethanol). After the ECT had completely dissolved in the liquid monomer the solution was diluted with 17.4 mL of molecular grade water. The polymerization solution was then sealed with a rubber septum and purged with nitrogen for 60 minutes. After this time, the polymerization solution was transferred to a preheated oil bath at 70 °C and allowed to polymerize for 90 minutes. Following polymerization, the total monomer conversion was determined via HPLC by comparison of the UV absorbance peak areas for the polymerization solution relative to samples taken at time 0. The copolymer was subsequently isolated by dialysis against deionized water at 5 °C and lyophilisation.
ECT grafting to Poly(DMA-co-HEAm)
CTA grafting reactions were conducted in methylene chloride at 0.5 wt % copolymer using dicyclohexyl carbodiimide (DCC) and dimethylaminopyridine (DMAP). The initial molar ratio of ECT to polymeric hydroxyl groups was 2 with equimolar quantities of DCC and DMAP relative to ECT. The polymer, DMAP, and ECT were first dissolved in methylene chloride and then allowed to cool to -20 °C after which the DCC was dissolved. The solution was then allowed to react overnight at 5 °C after which time the polymerization solution was spiked with methanol, filtered to remove the DCU. The filtrate was then reduced to approximately 20 % of the original volume and then isolated by repeated precipitation from acetone into diethyl ether. A representative procedure is as follows: To a 500 mL round bottom flask was added poly(DMAcoHEAm) (Mn = x.xx, PDI = x.xx) (90:10 mol/mol DMA/HEAm) 1.25 g (1.24 mmol HEAm OH), ECT (0.654 g, 2.48 mmol), DMAP (0.303 g, 2.48 mmol), and dichloromethane (249 g, 187 mL). The solution was then cooled to – 20 °C at which time DCC (0.512 g, 2.48 mmol) was added. Following complete dissolution of the DCC the solution was placed in the refrigerator and allowed to react at 5 °C overnight. After this time the cold solution was spiked with 50 mL of methanol and then filtered through a plug of cotton to remove the precipitated dicyclohexyl urea. The volume of the filtrate was then reduced to approximately 20 % of the original volume via rotary evaporation under reduced pressure at 25 °C. The final gCTA was then isolated by adding 5 mL of the reaction solution to 45 mL of diethyl ether in 50 ml conical tubes. After vortexing the solutions were centrifuged at 4200 rpm for 5 minutes. After this time the clear ether solution was decanted and the yellow/orange polymer oil was diluted 1 to 1 with acetone and reprecipitated into ether as described above (x8). The final graft copolymer was then dried under high vacuum for 24 hours. Grafting studies as a function of polymer concentration were conducted as described above with the solvent concentration systematically varied between 0.5 and 10 wt %.
Kinetic analysis of the RAFT polymerization of DMA from a 10-arm gCTA
Kinetic evaluation of the RAFT polymerization of DMA from a 10-arm gCTA were conducted with ABCVA as initiator at [M]o:[CTA]o, [CTA]o:[I]o, and [M]o equal to 100:1, 50:1, and 3M respectively at 70 °C. Individual polymerization solutions were transferred to a septa-sealed vial and purged with nitrogen for 30 minutes. After this time, the polymerization vials were transferred to a preheated oil bath at 70 °C and allowed to polymerize for the prescribed time period. To a 22 mL scintillation vial was added gCTA (0.3 g, 0.147 mmol ECT equivalents) (Mn = 19 600, PDI = 1.05), ABCVA (0.824 mg, 2.94 μmol as 100 μL of a 8.24 mg/mL ABCVA stock in ethanol), and DMA (2.91 g, 29.4 mmol). After the gCTA had completely dissolved the solution was diluted with 9.8 mL of molecular grade water and transferred to individual septa-sealed round bottom flasks. The polymerization vials were then purged with nitrogen for 30 minutes and then transferred to a preheated oil bath at 70 °C and allowed to polymerize for the prescribed time period.
RAFT polymerization of NIPAM from a 20-arm gCTA
RAFT polymerization of NIPAM from a 20-arm gCTA was conducted in DMSO with ABCVA as initiator at [M]o:[CTA]o, [CTA]o:[I]o, and [M]o equal to 400:1, 40:1, and 20 wt % (weight fraction monomer disregards the mass of the added gCTA). To a 20 mL scintillation vial was added gCTA (0.10 g, 44.25 μmol ECT equivalents) (Mn = 44.800, Đ = 1.12), ABCVA (0.31 mg, 1.10 μmol as 100 μL of a 3.1 mg/mL ABCVA stock in ethanol), NIPAM (2.03 g, 17.9 mmol), and DMSO (8.03 mL). After the gCTA and monomer were completely dissolved the solution was then septa sealed and purged with nitrogen for 60 minutes. The round bottom flask was then transferred to a preheated oil bath at 70 °C and allowed to polymerize for 3 hours. Mn = 472,000, Đ =1.15.
RAFT polymerization of HPMA from 20-arm gCTA
The RAFT polymerization of HPMA from a xx-arm gCTA was conducted in molecular grade water with ABCVA as initiator at [M]o:[CTA]o, [CTA]o:[I]o, and [M]o equal to 150:1, 20:1, and 15 wt % (weight fraction monomer disregards the mass of the added gCTA). To a 20 mL scintillation vial was added gCTA (0.10 g, 44.25 μmol ECT equivalents) (Mn = 44,800, Đ = 1.12), ABCVA (0.62 mg, 2.21 μmol as 100 μL of a 6.2 mg/mL ABCVA stock in ethanol), HPMA (0.95 g, 29.4 mmol), and molecular grade water (5.39 mL). After the gCTA and monomer were completely dissolved the solution was then septa sealed and purged with nitrogen for 30 minutes. The round bottom flask was then transferred to a preheated oil bath at 70 oC and allowed to polymerize for 4 hours. Mn = 271,200 g/mol, PDI =1.13.
RAFT polymerization of DMAEMA from 20-arm gCTA
The RAFT polymerization of DMAEMA from a 20-arm gCTA was conducted in molecular grade water with ABCC as initiator at [M]o:[CTA]o, [CTA]o:[I]o, and [M]o equal to 200:1, 50:1, and 30 wt % (weight fraction monomer disregards the mass of the added gCTA). To a 20 mL scintillation vial was added gCTA (0.10 g, 44.25 μmol ECT equivalents) (Mn = 44,800, Đ = 1.12), ABCC (0.216 mg, 0.88 μmol as 100 μL of a 2.16 mg/mL ABCC stock in ethanol), DMAEMA (1.39 g, 8.84 mmol), and DMSO (3.25 mL). After the gCTA and monomer were completely dissolved the solution was then septa sealed and purged with nitrogen for 30 minutes. The round bottom flask was then transferred to a preheated oil bath at 70 °C and allowed to polymerize for 4 hours. Mn = 352,500, Đ = 1.19.
Conjugation of Doxorubicin to 2nd generation brushed-brushes
Generation 2 brushed-brushes containing boc-protected carbazate groups were dissolved in a solution of methylene chloride and TFA (2:1 by volume) for 2 h and then precipitated into a large excess of cold diethyl ether. The precipitate was then dried under vacuum and then further purified by dialysis against water at 5 °C followed by lyophilization. The resultant carbazate functional polymer (0.10 g, 33.4 μM hydrazine equivalents) was then reacted with doxorubicin (19.4 mg, 33.4 μM) in 5 mL of DMF in the presence of 40 μL TFA. The conjugates were then purified by dialysis against deionized water at 5 °C followed by two PD10 columns conducted in series.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (Grant R01EB002991 and Grant 1R21EB014572- 01A1), the Defense Threat Reduction Agency (DTRA) (HDTRA1-13-1-0047), the Life Science Discovery Fund (Grant 2496490), and the Oregon Community Fund.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Siegel R, Naishadham D, Jemal A. CA: A Cancer Journal for Clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Su Z, Graybill WS, Zhu Y. Clinica Chimica Acta. 2013;415:341–345. doi: 10.1016/j.cca.2012.10.058. [DOI] [PubMed] [Google Scholar]
- 3.Stirling D, Evans DGR, Pichert G, Shenton A, Kirk EN, Rimmer S, Steel CM, Lawson S, Busby-Earle RMC, Walker J, Lalloo FI, Eccles DM, Lucassen AM, Porteous ME. JCO. 2005;23:5588–5596. doi: 10.1200/JCO.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 4.Robert J, Morgan J, Alvarez RD, Armstrong DK, Burger RA, Chen L-M, Copeland L, Crispens MA, Gershenson DM, Gray HJ, Hakam A, Havrilesky LJ, Johnston C, Lele S, Martin L, Matulonis UA, Penson RT, Powell MA, Remmenga SW, Sabbatini P, Santoso JT, Schink JC, Teng N, Werner TL, Dwyer MA, Hughes M. J Natl Compr Canc Netw. 2013;11:1199–1209. doi: 10.6004/jnccn.2013.0142. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 6.Oerlemans C, Bult W, Bos M, Storm G, Nijsen JFW, Hennink WE. Pharm Res. 2010;27:2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barenholz YC. Journal of Controlled Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt CJ, Dietrich S, Ho AD, Witzens-Harig M. Ann Hematol. 2012;91:391–397. doi: 10.1007/s00277-011-1308-y. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien MER, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C CAELYX Breast Cancer Study Group. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 10.Sparano JA, Makhson AN, Semiglazov VF, Tjulandin SA, Balashova OI, Bondarenko IN, Bogdanova NV, Manikhas GM, Oliynychenko GP, Chatikhine VA, Zhuang SH, Xiu L, Yuan Z, Rackoff WR. J Clin Oncol. 2009;27:4522–4529. doi: 10.1200/JCO.2008.20.5013. [DOI] [PubMed] [Google Scholar]
- 11.Senter PD. Curr Opin Chem Biol. 2009;13:235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 12.Gray JC, Kohler JA. Pediatric Blood & Cancer. 2009;53:931–940. doi: 10.1002/pbc.22153. [DOI] [PubMed] [Google Scholar]
- 13.Allen TM, Cullis PR. Advanced Drug Delivery Reviews. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 14.Pegoraro C, Cecchin D, Gracia LS, Warren N, Madsen J, Armes SP, Lewis A, Macneil S, Battaglia G. Cancer Lett. 2013;334:328–337. doi: 10.1016/j.canlet.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Roy D, Berguig GY, Ghosn B, Lane DD, Braswell S, Stayton PS, Convertine AJ. Polymer Chemistry. 2014;5:1791–1799. doi: 10.1039/C3PY01404E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palanca-Wessels MC, Convertine AJ, Cutler-Strom R, Booth GC, Lee F, Berguig GY, Stayton PS, Press OW. Mol Ther. 2011;19:1529–1537. doi: 10.1038/mt.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, Stayton PS. Biomacromolecules. 2010;11:2904–2911. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. J Control Release. 2009;133:221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller T, Breyer S, van Colen G, Mier W, Haberkorn U, Geissler S, Voss S, Weigandt M, Goepferich A. Int J Pharm. 2013;445:117–124. doi: 10.1016/j.ijpharm.2013.01.059. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Shi Y, Kim JY, Park K, Cheng J-X. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 21.Lu J, Owen SC, Shoichet MS. Macromolecules. 2011;44:6002–6008. doi: 10.1021/ma200675w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eliasof S, Lazarus D, Peters CG, Case RI, Cole RO, Hwang J, Schluep T, Chao J, Lin J, Yen Y, Han H, Wiley DT, Zuckerman JE, Davis ME. PNAS. 2013;110:15127–15132. doi: 10.1073/pnas.1309566110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han H, Davis ME. Mol Pharm. 2013;10:2558–2567. doi: 10.1021/mp300702x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng J, Khin KT, Jensen GS, Liu A, Davis ME. Bioconjugate chemistry. 2003;14:1007–1017. doi: 10.1021/bc0340924. [DOI] [PubMed] [Google Scholar]
- 25.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Fréchet JMJ, Dy EE, Szoka FC. Proc Natl Acad Sci U S A. 2006;103:16649–16654. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moad G, Rizzardo E, Thang SH. Australian Journal of Chemistry. 2012;65:985–1076. [Google Scholar]
- 27.Chu DSH, Schellinger JG, Shi J, Convertine AJ, Stayton PS, Pun SH. Acc Chem Res. 2012;45:1089–1099. doi: 10.1021/ar200242z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roth PJ, Boyer C, Lowe AB, Davis TP. Macromolecular Rapid Communications. 2011;32:1123–1143. doi: 10.1002/marc.201100127. [DOI] [PubMed] [Google Scholar]
- 29.Stenzel MH, Davis TP. Journal of Polymer Science Part A: Polymer Chemistry. 2002;40:4498–4512. [Google Scholar]
- 30.Stenzel Rosenbaum MH, Davis TP, Fane AG, Chen V. Angewandte Chemie. 2001;113:3536–3540. doi: 10.1002/1521-3773(20010917)40:18<3428::aid-anie3428>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Stenzel Rosenbaum M, Davis TP, Chen V, Fane AG. Journal of Polymer Science Part A: Polymer Chemistry. 2001;39:2777–2783. [Google Scholar]
- 32.Li A, Ma J, Sun G, Li Z, Cho S, Clark C, Wooley KL. Journal of Polymer Science Part A: Polymer Chemistry. 2012;50:1681–1688. [Google Scholar]
- 33.Hong Chun-Yan, You A Ye-Zi, Pan C-Y. Chemistry of materials, 2005 [Google Scholar]
- 34.Roy Debashish, Guthrie A James T, Perrier S. Macromolecules. 2005;38:10363–10372. [Google Scholar]
- 35.Hao X, Nilsson C, Jesberger M, Stenzel MH, Malmström E, Davis TP, Östmark E, BarnerKowollik C. Journal of Polymer Science Part A: Polymer Chemistry. 2004;42:5877–5890. [Google Scholar]
- 36.Zhang L, Bernard J, Davis TP, Barner Kowollik C, Stenzel MH. Macromolecular Rapid Communications. 2008;29:123–129. [Google Scholar]
- 37.Liu J, Tao L, Xu J, Jia Z, Boyer C, Davis TP. Polymer. 2009;50:4455–4463. [Google Scholar]
- 38.Liu J, Liu H, Jia Z, Bulmus V, Davis TP. Chem Commun (Camb) 2008:6582–6584. doi: 10.1039/b817037a. [DOI] [PubMed] [Google Scholar]
- 39.Nese A, Kwak Y, Nicolaÿ R, Barrett M, Sheiko SS. Macromolecules. 2010;43:4016–4019. [Google Scholar]
- 40.Zheng G, Pan C. Polymer. 2005;46:2802–2810. [Google Scholar]
- 41.Boyer C, Stenzel MH, Davis TP. Journal of Polymer Science Part A: Polymer Chemistry. 2011;49:551–595. [Google Scholar]
- 42.Convertine AJ, Lokitz BS, Vasileva Y, Myrick LJ, Scales CW, McCormick Charles L. Macromolecules. 2006;39:1724–1730. [Google Scholar]
- 43.Lokitz BS, Convertine AJ, Ezell RG, Heidenreich A, Li Y, McCormick CL. Macromolecules. 2006;39:8594–8602. [Google Scholar]
- 44.Quek JY, Zhu Y, Roth PJ, Davis TP, Lowe AB. Macromolecules. 2013 [Google Scholar]
- 45.Crownover EF, Convertine AJ, Stayton PS. Polymer Chemistry. 2011;2:1499–1504. [Google Scholar]
- 46.Keddie DJ, Guerrero-Sanchez C, Moad G, Rizzardo E, Thang SH. Macromolecules. 2011;44:6738–6745. [Google Scholar]
- 47.DE P, Sumerlin BS. Macromolecular Chemistry and Physics. 2013;214:272–279. [Google Scholar]
- 48.Seabrook SA, Gilbert RG. Polymer. 2007;48:4733–4741. [Google Scholar]
- 49.Seabrook SA, Tonge MP, Gilbert RG. Journal of Polymer Science Part A: Polymer Chemistry. 2005;43:1357–1368. [Google Scholar]
- 50.Seabrook SA, Pascal P, Tonge MP, Gilbert RG. Polymer. 2005;46:9562–9573. [Google Scholar]
- 51.Kuchta Frank-Dieter, Alexander A, van Herk M, German AL. Macromolecules. 2000;33:3641–3649. [Google Scholar]
- 52.Lacík I, Učňová L, Kukučková S, Buback M, Hesse P, Beuermann S. Macromolecules. 2009;42:7753–7761. [Google Scholar]
- 53.Henry SM, Convertine AJ, Benoit DSW, Hoffman AS, Stayton PS. Bioconjugate chemistry. 2009;20:1122–1128. doi: 10.1021/bc800426d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas David B, Convertine Anthony J, Hester Roger D, Lowe A Andrew B, McCormick Charles L. Macromolecules. 2004;37:1735–1741. [Google Scholar]
- 55.Scales CW, Vasilieva YA, Convertine AJ, Lowe AB, McCormick CL. Biomacromolecules. 2005;6:1846–1850. doi: 10.1021/bm0503017. [DOI] [PubMed] [Google Scholar]
- 56.Ulbrich K, Etrych T, Chytil P, Jelınková M, Ríhová B. Journal of Controlled Release. 2003;87:33–47. doi: 10.1016/s0168-3659(02)00348-6. [DOI] [PubMed] [Google Scholar]
- 57.Rıhová B, Etrych T, Pechar M, Jelınková M, Štastnýa M, Hovorkaa O, Kovářa M, Ulbrich K. Journal of Controlled Release. 2001;74:225–232. doi: 10.1016/s0168-3659(01)00320-0. [DOI] [PubMed] [Google Scholar]
- 58.Xu J, Boyer C, Bulmus V, Davis TP. Journal of Polymer Science Part A: Polymer Chemistry. 2009;47:4302–4313. [Google Scholar]
- 59.Convertine AJ, Lokitz BS, Lowe AB, Scales CW, Myrick LJ, McCormick CL. 2005;26:791–795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.