

Abstract
Up to 20% of patients with pilocytic astrocytoma (PA) experience a poor outcome. BRAF alterations and Fibroblast growth factor receptor 1 (FGFR1) point mutations are key molecular alterations in Pas, but their clinical implications are not established. We aimed to determine the frequency and prognostic role of these alterations in a cohort of 69 patients with PAs. We assessed KIAA1549:BRAF fusion by fluorescence in situ hybridization and BRAF (exon 15) mutations by capillary sequencing. In addition, FGFR1 expression was analyzed using immunohistochemistry, and this was compared with gene amplification and hotspot mutations (exons 12 and 14) assessed by fluorescence in situ hybridization and capillary sequencing. KIAA1549:BRAF fusion was identified in almost 60% of cases. Two tumors harbored mutated BRAF. Despite high FGFR1 expression overall, no cases had FGFR1 amplifications. Three cases harbored a FGFR1 p.K656E point mutation. No correlation was observed between BRAF and FGFR1 alterations. The cases were predominantly pediatric (87%), and no statistical differences were observed in molecular alterations–related patient ages. In summary, we confirmed the high frequency of KIAA1549:BRAF fusion in PAs and its association with a better outcome. Oncogenic mutations of FGFR1, although rare, occurred in a subset of patients with worse outcome. These molecular alterations may constitute alternative targets for novel clinical approaches, when radical surgical resection is unachievable.
Key Words: BRAF, Brain tumor, FGFR1, Glioma, Molecular diagnosis, Pilocytic astrocytoma, Prognosis
INTRODUCTION
Pilocytic astrocytomas (PAs) are the major solid neoplasms in children and teenagers (1, 2). According to data from the Central Brain Tumor Registry of the United States, it is the main neoplasm in the 5- to 14-year-old range in the United States (3). Similarly in Brazil, PAs are the second most common neoplasm in pediatric patients after leukemias (4), accounting for almost 20% of primary brain tumors in children (5). Pilocytic astrocytomas are less frequent in adults in whom they are associated with more aggressive clinical courses (1, 6). According to the World Health Organization (WHO), PAs are grade I tumors because of their well-limited and usually indolent nature (1). The 5-year survival rate is >90% in children (1, 7), and 52% in adults (8). Despite the overall good prognosis of PAs, up to 20% of patients will have a poor outcome, with recurrence, growth of incompletely resected lesions, or dissemination through the cerebrospinal fluid, and ultimately death due to disease (1, 7).
Pilocytic astrocytomas can occur throughout the neuraxis, but the most common location of sporadic tumors is the cerebellum (1). Extracerebellar tumors, particularly those located in the cerebral hemispheres and in the optic pathways, have a known association with neurofibromatosis 1 (NF1), a familial tumor predisposition syndrome with autosomal dominant inheritance (1, 9). Approximately 10% of all PAs are related to NF1 (NF1-PAs), and conversely, PAs are the most frequent brain tumor related to NF1 (49% of cases) (10, 11). When these PAs arise in locations where gross total resection is difficult to achieve, they usually follow a more benign course than sporadic PAs (11).
Molecular studies based on the relationship between PAs and NF1 allowed the discovery of germline and somatic mutations with silencing of the tumor suppressor gene NF1 in NF1-PAs. These were recently defined as point mutations, splice mutations or nonsense mutations (germline mutations) and loss of heterozygosity and epigenetic changes, such as methylation of the gene (somatic mutations) (9). These mutations result in loss of expression of neurofibromin, which is a negative signaling regulator of RAS proteins; this results in an increase of activated RAS levels and further activation of the mitogen-activated protein kinase (MAPK) pathway (12). The constitutive activation of the MAPK pathway increases survival and proliferation of cancer cells in various neoplasms (13, 14).
MAPK is a key signaling pathway in the development of PAs; it is altered in up to 90% of cases (7, 15, 16). The major alterations leading to constitutive activation of MAPK in PAs are gene fusions and point mutations involving the oncogene BRAF (7, 17–23). Gene fusions between KIAA1549 and BRAF (KIAA1549:BRAF fusion) leading to the overexpression of the fusion protein affects up to 80% of PAs. There are decreasing rates with age, varying from 79% in children younger than 10 years to 7% in patients older than 40 years (16, 20); this is associated with a better prognosis in low-grade gliomas, including PAs (21). Less frequent fusions, such as SRGAP3-RAF1 (24) and FAM131B-BRAF (25, 26), have also been described. Another mechanism of sustained BRAF activation in PAs is the point mutation V600E, which results in an amino acid substitution at codon 600 in BRAF, from a valine (V) to a glutamic acid (E) in the majority of cases, leading to the activation of the kinase domain of this oncogene (7, 18, 22, 27). Nevertheless, this finding is infrequent in PAs (approximately 6%) and may be detected more frequently in other brain tumor types, such as glioblastomas (22, 28), gangliogliomas, and particularly, pleomorphic xanthoastrocytomas (>60%) (16, 22).
Recent studies have identified upstream alterations in the MAPK pathway, mainly in the tyrosine-kinase receptor Fibroblast growth factor receptor 1 (FGFR1), leading to constitutive activation of the growth cascade in PAs (15, 29). In contrast to the FGFR1 amplification frequently observed in breast, ovary, and lung cancer (30, 31), gene fusions and duplications are described at low frequencies in brain tumors such as glioblastomas (32) and pediatric diffuse astrocytomas (16), respectively. In PAs, the newly described alterations of FGFR1 are point mutations in the hotspot tyrosine kinase region, affecting mainly the codons 546 (p.N546K –asparagine-to-lysine substitution) and 656 (p.K656E –lysine-to-glutamate substitution) of the gene in extracerebellar PAs (15).
Despite the great improvement in the knowledge on the molecular oncogenesis of PAs in the last years, the main established prognostic factors for PAs remain in the clinical features, such as patient’s age, feasibility of radical resection of lesion (33–35), exposure to radiation therapy (1), and the sporadic or hereditary nature of the tumor (12). The prognostic implications of BRAF and FGFR1 alterations have not been fully explored, and advances in this field might identify potential targets for clinical treatment of PA, particularly for the tumors located in eloquent areas where radical resection is rarely achieved.
In this study, we aimed to determine the frequency of the molecular alterations in BRAF and FGFR1 and to evaluate the prognostic role of these oncogenes in a series of Brazilian patients with PAs.
MATERIALS AND METHODS
Patients
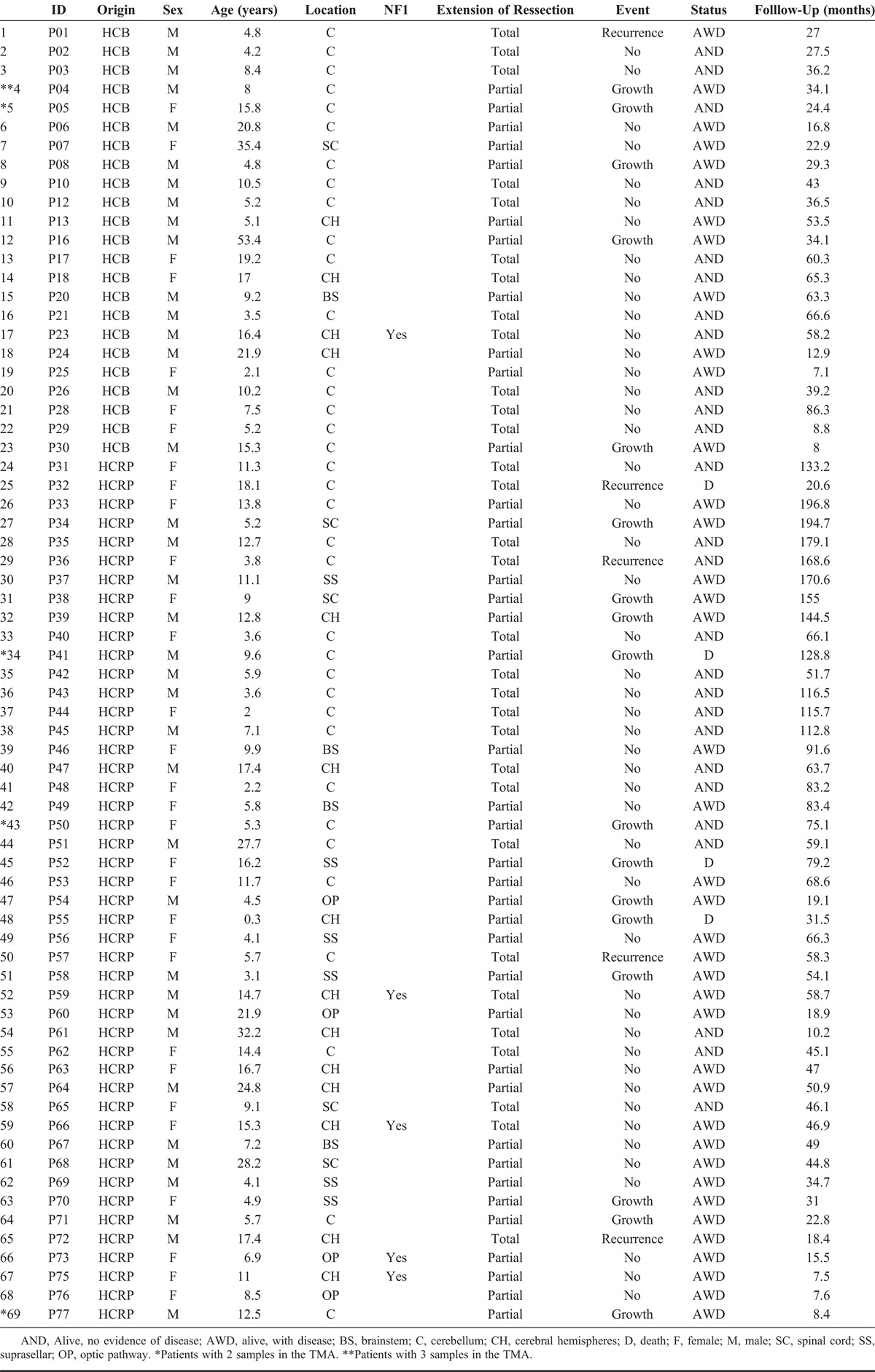
Sixty-nine patients from the Barretos Cancer Hospital (HCB) and the Hospital of Clinics of Faculty of Medicine of Ribeirao Preto (HCRP), from 1993 to 2013, were included in this study. The patients were clustered according to sex, age group (≤19 years old vs ≥20 years old), clinical diagnosis of NF1 (confirmed by standardized clinical criteria), and lesion location (cerebellar vs extracerebellar). The outcome of patients was classified as “favorable” (i.e. patients without any events and/or with Karnofsky index ≥80 at follow-up) and “unfavorable” (i.e. occurrence of some event and/or Karnofsky index ≤70 at follow-up). We defined “event” as death, growth of a partially resected lesion, or the recurrence of a completely resected lesion confirmed by immediate postsurgical computed tomography (36), detected either clinically and/or through neuroradiologic examinations. The study was approved by both local Ethics Committee (protocols HCB 87362 and HCRP 212.313).
The series included 38 male and 31 female patients (ratio, 1.2:1), with ages ranging from 0.3 to 53.4 years old (median, 9.1 years old). The 5-year and 10-year survival of the series were > 95% and 80%, respectively. Overall, 35 cases (50.7%) had unfavorable outcomes according to our criteria: 8 patients (11.5%) had relapsing or growing residual tumors; 23 patients (33.3%) developed moderate to severe clinical deficits (Karnofsky index, 50–70); and 4 patients (5.8%) died of disease. The deaths occurred after 1.7, 2.6, 6.5, and 10.7 years of the diagnosis, respectively. Two deceased patients had cerebellar lesions with subsequent medullary dissemination of the tumor, 1 patient had a suprasellar lesion, and the other had an insular tumor. Table 1 summarizes clinical data of the patients.
TABLE 1.
Clinicoepidemiologic Data of the PA Series
Of the 69 patients included in this study, 5 had relapsed lesions analyzed, and 1 of these had yet a second relapsed lesion analyzed, totaling 75 samples. All cases were reviewed by 2 neuropathologists, according to the 2007 WHO diagnostic criteria (1); negative immunohistochemical reaction to mutated IDH1 was found in all cases (37, 38).
We constructed 2 blocks of tissue microarray (TMA) from the formalin-fixed, paraffin-embedded (FFPE) samples, using the Beecher Instruments TMA platform, with tissue cores at 1 mm diameter for the HCB cases and 1.5 mm for the HCRP cases. Because of the histologic heterogeneity of the PAs, we obtained up to 8 cores of each case (average, 3.6 cores/case), representing the different histologic patterns of the tumors. In 9 cases, adjacent nonneoplastic cerebellar tissue was available and included in the TMA.
Immunohistochemistry
Automated immunohistochemistry using Ventana BenchMark Ultra equipment (Ventana-Roche, Tucson, AZ) was performed in the TMA slides with 4-μm-thick tissue sections, according to the manufacturer’s protocols. First, the sections were deparaffinized and dehydrated, then the antigen retrieval process was done with a mixed citrate/EDTA buffer (pH 6.0, at 125 °C for 4 minutes and 95 °C for 25 minutes in pressure cooker). The monoclonal antibody used was anti-FGFR1 (Cell Signaling, Danvers, MA, clone D8E4, dilution 1:50). As external controls for the immunohistochemical reaction, we used prostate epithelium and liver; internal control was the endothelial cytoplasmic reaction.
The cytoplasmic expression of FGFR1 was evaluated in a double-blind fashion following semiquantitative criteria based on the intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong) and extension of the reaction (0, 0% of positive cells; 1, <25% of positive cells; 2, 25%–50% of positive cells; and 3, >50% of positive cells) (39). With the sum of these analyses, we achieved scores ranging from 0 to 6. Samples with scores 0 to 2 were considered negative; those with scores 3 to 6 were considered positive (39). Tissues sections were also evaluated for nuclear expression; ≥25% nuclear staining was considered positive, and cases with <25% of nuclear staining were considered negative. In the cases with more than 1 tissue core, we calculated the average score.
KIAA1549-BRAF and FGFR1 Fluorescence In Situ Hybridization Assay
For analysis of KIAA1549-BRAF fusion, fluorescence in situ hybridization (FISH) probes were created from BAC clones containing human DNA from regions homologous to the KIAA1549 and BRAF genes on chromosome 7, as identified through the Ensembl Genome Browser (GRCh37). The BRAF DNA was validated by polymerase chain reaction (PCR) using the following sequences as primers: 5′-CAGAGTTTGTCAGATGGTCCCTTT-3′ (forward) and 5′-ACCATATAATAGAAGCGCCTCCCA-3′ (reverse). For the KIAA1549 DNA, the validation sequences were 3′-AGGTATTGTTGGAACATTGAAGGCT-3′ (forward) and 5′-CAGTCAAATGCTCGCAATGAATGAA-3′ (reverse). DNA inserts were extracted from clone mini-cultures, purified and subjected to whole genome amplification using the REPLI-g Midi Kit from Qiagen (Cat# 150045, Qiagen, Düsseldorf, Germany).
An aliquot of 1 μg of each purified BRAF and KIAA1549 DNA were labeled, respectively, with SpectrumRed and SpectrumGreen conjugated dUTPs using the Vysis Nick Translation Kit (Cat# 32–801300, Abbott Molecular, Des Plaines, IL), as previously reported (40). Labeled DNA was coprecipitated with herring sperm DNA as carrier (1:50) and human Cot-1 DNA (1:10) for blocking of repetitive sequences then diluted 1:10 in t-DenHyb hybridization buffer (Insitus Biotechnologies, Albuquerque, NM). The labeled FISH probe mix was validated for chromosome mapping and quality of hybridization in interphase and metaphase cells prior to this study.
The FFPE slides were deparaffinized and dehydrated according to previously established protocols (41). The probe was applied to the selected areas, and hybridization was allowed to occur at 37 °C for 40 to 67 hours and, finally, the chromatin was counterstained with DAPI/anti-fade (0.3 μg/mL in Vectashield mounting medium, Vector Laboratories, Burlingame, CA).
Analysis was performed on an epifluorescence microscope using single interference filter sets for green (fluorescein isothiocyanate), red (Texas red), and blue (DAPI), as well as dual (red/green) and triple (blue, red, and green) band pass filters. For each interference filter, monochromatic images were acquired and merged using CytoVision (Leica Microsystems Inc., Wetzlar, Germany). A minimum of 50 tumor nuclei was evaluated.
The specimen was considered positive for the KIAA1549:BRAF fusion when there were doublets of red and green signals very close or partially overlapping observed, as opposed to signals separated by >2 signal diameters, which characterize alleles with native status.
FGFR1 Amplification
The FGFR1/CEP8 enumeration assay measured 2 genomic targets using 2 commercial FISH probes provided as Analyte Specific Reagents (ASR) by Abbott Molecular (Ref. 08 N21-020 and Ref. 06 J37-018, respectively): Vysis LSI FGFR1 SpectrumRed FISH probe, which contains the entire FGFR1 gene, labeled with SpectrumRed fluorophore, and the CEP 8 (D8Z2) FISH probe, labeled with SpectrumGreen fluorophore.
The FFPE slides were processed and evaluated as previously described for the KIAA1549-BRAF fusion. The determination of low and high level of FGFR1 gene amplification followed the criteria proposed by Schultheis et al (42) based on the ratio FGFR1/CEP 8 ≥ 2.0, or the average number of FGFR1 signals per nucleus ≥6 copies.
BRAF and FGFR1 Point Mutation Analyses
We first obtained serial 10-μm-unstained sections of FFPE blocks. One adjacent hematoxylin and eosin–stained section was used for identification and selection of tumor area by the pathologist. DNA was isolated from 1 or 2 unstained section from each specimen, depending on the size of the tissue fragment, as previously described (43). Briefly, tissues were deparaffinized and dehydrated. Selected areas of tumor were macrodissected using a sterile needle (18G × 1½) (Becton Dickinson, Curitiba, Brazil), and carefully collected into a microtube. DNA was isolated using QIAamp DNA Micro Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions, followed by evaluation of DNA quantity and quality by Nanodrop 2000 (Thermo Scientific, Wilmington, DE). DNA samples were then diluted to a final concentration of 50 ng/μL and stored at −20 °C for further molecular analysis.
The whole exons of BRAF (exon 15; codon 600) and FGFR1 (exons 12 and 14; codons 546 and 656) were analyzed by PCR, followed by direct sequencing, with emphasis in the hotspot loci, as previously described (15, 28). Briefly, the PCR reaction was performed in a final volume of 15 μL, under the following conditions: 1× PCR buffer (Invitrogen, Carlsbad, CA); 2 mmol/L MgCl2 (Invitrogen); 10 mmol/L dNTPs (Invitrogen); 0.3 mmol/L of both sense and antisense primers (Sigma Aldrich, St. Louis, MO); 1 unit of Platinum Taq DNA polymerase (Invitrogen); and 50 ng of DNA. The BRAF primers used were TCATAATGCTTGCTCTGATAGGA (sense) and GGCCAAAAATTTAATCAGTGGA (antisense) (28), for FGFR1 exon 12 TCAAGTCCCAGGGAAAAGCAG (sense) and AGGCCTTGGGACTGATACCC (antisense), and for FGFR1 exon 14 GACAAGTCGGCTAGTTGCAT (sense) and CCCACTCCTTGCTTCTCAGAT (antisense). The PCR was performed in Veriti 96-Wll Thermal Cycler (Applied Biosystems, Austin, TX ). The PCR products were evaluated by agarose gel electrophoresis prior to capillary sequencing.
The PCR products of each analyzed exon were firstly purified with EXOSAP-IT (GE Technology, Cleveland, OH), then, PCR products were submitted to a sequencing reaction using 1 μL of BigDye (Applied Biosystems), 1.5 μL of sequencing buffer (Applied Biosystems) and 3.2 μmol/L of primer. The sequencing reaction was followed by post-sequencing purification with EDTA, alcohol and sodium citrate. The purified products were eluted in HiDi (formamide) and incubated at 90 °C for 5 minutes and at 4 °C for at least 5 minutes. Direct sequencing was carried out on a Genetic Analyzer ABI PRISM® 3500 (Applied Biosystems). The analysis of each sample was done by comparison of eletropherogram with Ensembl GeneBank sequence (BRAF: ENSG00000157764 and FGFR1: ENSG00000077782).
All cases with mutations were confirmed twice with a new PCR and direct sequencing starting from extracted DNA. In addition, for quality controls, a new DNA isolation and further mutation analyses were performed in 10% of cases.
Statistical Analysis
Statistical analyses were performed with SPSS version 20 for Windows™ (IBM, Chicago, IL) with statistically significant values of p < 0.05. Differences in molecular alterations of BRAF and FGFR1 between groups were verified by the Fisher exact and the Pearson chi-square tests. Overall survival (OS) and event-free survival (EFS) curves were determined by the Kaplan-Meier method.
RESULTS
Molecular Characterization of BRAF
The FISH assay for KIAA1549:BRAF fusion detection was successful in 64 (92.8%) of 69 primary lesions and in 4 of 5 relapsing lesions, which maintained the expression pattern of their primary counterparts (Fig. 1A, B). Thirty-seven primary lesions (57.8 %) displayed KIAA1549:BRAF fusion, with strong positive association with a cerebellar location (p < 0.001), and negative association with clinical diagnosis of NF1 (p = 0.011) (Table 2). There was a tendency for this alteration to be detected in the younger group, with 55.0% and 44.4% of patients positive for KIAA1549:BRAF fusion in the pediatric and adult group, respectively (Table 2). Nevertheless, this difference was not statistically significant. In addition, there were no differences between groups for sex or outcome (Table 2).
FIGURE 1.
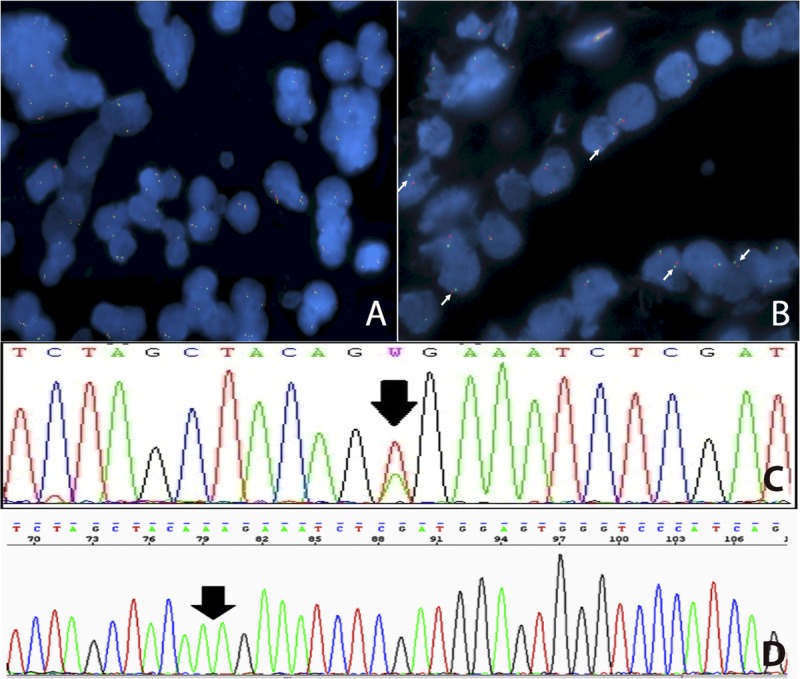
Molecular alterations in BRAF. (A, B) FISH assay for detection of KIAA1549:BRAF fusion showing a positive (A) and a negative (B) case (white arrows). (C, D) Point mutations detected by Sanger sequencing for V600E (C) and V600K (D).
TABLE 2.
Clinical Features of Patients and Their Association with BRAF Changes

Because of compromised DNA quality in some specimens, we were able to obtain conclusive results of BRAF point mutations in 48 (69.6%) of 69 primary lesions and in 5 of 5 recurrences. Two cases (4.2%) showed BRAF point mutations (Fig. 1C, D). One recurrent suprasellar (hypothalamic) tumor of an 11-year-old male patient (P37, Table 1) had the p.V600E mutation, but the patient had only 1 sample available for molecular analysis, and this tumor was also positive for the KIAA1549:BRAF fusion in the FISH assay. In addition, a point mutation p.V600K, with a valine-to-lysine substitution at the codon 600 (Fig. 1D), was detected in a cerebellar PA of an 11-year-old female patient (P31, Table 1), who remains alive without evidence of disease after a long follow-up (11 years). None of these patients with tumors harboring mutated BRAF had the clinical diagnosis of NF1. Despite the small number of BRAF-mutated cases, we performed statistical analysis but did not identify significant associations between BRAF status and patients clinical features (Table 2).
Molecular Characterization of FGFR1
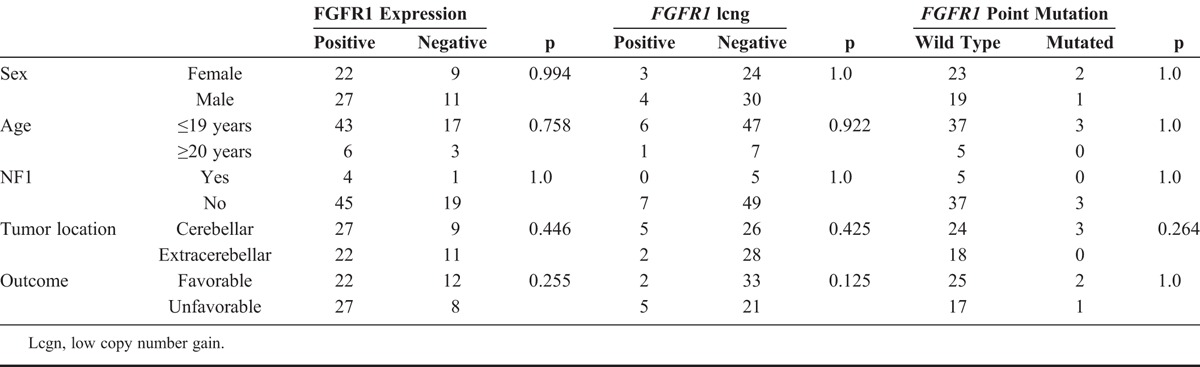
The immunohistochemical expression of FGFR1 was tested in 74 of 75 samples. Nonneoplastic cerebellum showed cytoplasmic staining only in Purkinje cells and was absent or faintly expressed in the nonneoplastic astrocytes (Fig. 2A, B). In tumor areas, cytoplasmic staining was detected in 51 (73.9%) of 69 primary tumors, regardless of the histologic pattern of the PA (Fig. 2C, D) and no nuclear staining was observed. Forty-nine cases (71%) had scores ≥3, and 19 cases (27.5%) were completely negative. No association was found between FGFR1 immunohistochemical staining with the presence of the KIAA1549:BRAF fusion (p = 0.272), or with BRAF point mutations (p = 0.456) (data not shown). No significant associations were seen between expression and clinical features of the patients (Table 3).
FIGURE 2.
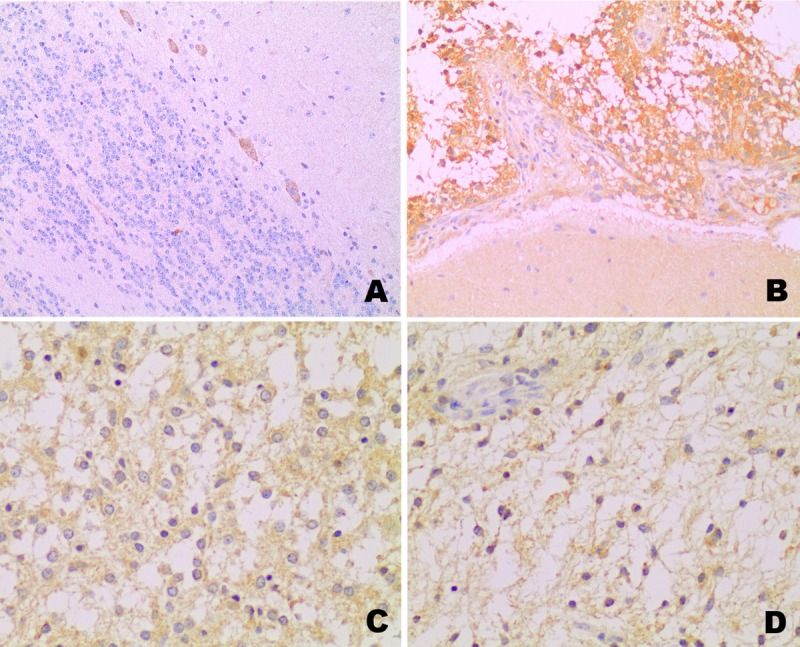
Immunohistochemical expression of FGFR1. (A) In normal cerebellum, expression is limited to Purkinje cells. (B) Neoplastic cells show overexpression when compared with nonneoplastic astrocytes (bottom). (C) Oligodendroglial pattern of PA, showing moderate FGFR1 expression. (D) Piloid pattern of PA with similar FGFR1 expression.
TABLE 3.
Clinical Features of Patients and Their Association with FGFR1 Changes
Sanger sequencing for FGFR1 was performed in 45 (65.2%) of 69 primary lesions and in 5 of 5 relapsed lesions. Among primary lesions, 3 (6.7%) of 45 carried the p.K656E point mutation (Fig. 3A): an 18-year-old female (P32), a 3-year-old male (P21), and a 2-year-old female (P44) patient. All of these patients had cerebellar lesions (Tables 1 and 3). The older patient had recurrence of a completely resected lesion 8 months after the first surgery with cerebrospinal fluid dissemination despite adjuvant chemotherapy, and she died 21 months after the original surgery. On the other hand, the youngest patient had the best outcome of the subgroup (i.e. no evidence of disease after 9 years of follow-up), had a tumor that was also positive for the KIAA1549:BRAF fusion by FISH assay. No association was observed between FGFR1 mutation and the patients’ clinicopathologic characteristics (Table 3) or FGFR protein expression (p = 0.086, data not shown), as 2 of the 3 mutated cases had positive scores and 1 had a negative immunohistochemical score.
FIGURE 3.
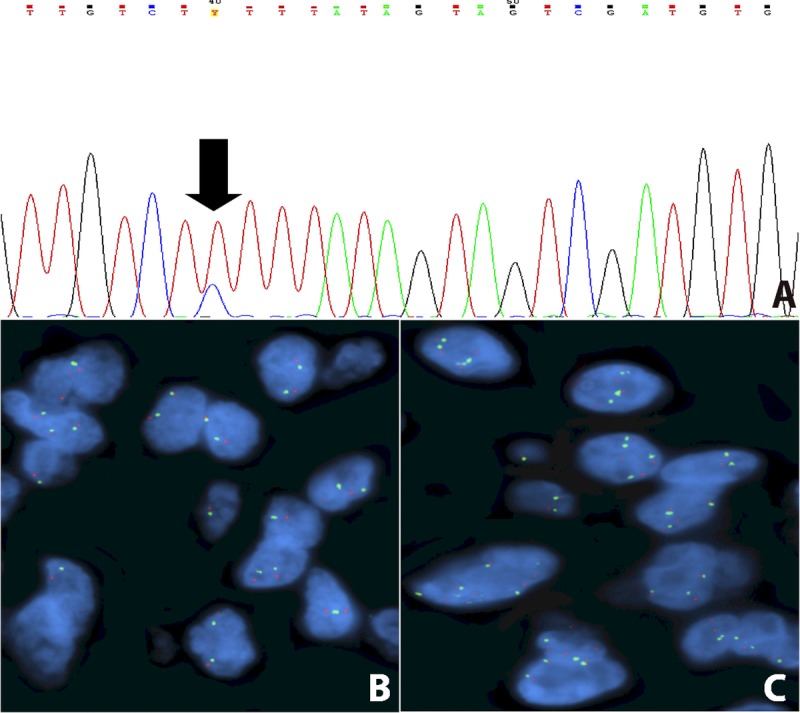
Molecular alterations of FGFR1. (A) Electropherogram showing the point mutation K656E. (B, C) FISH assay displaying a normal pattern (B), and a case with low-copy number gain of the FGFR1 signal (C). The amount of FGFR1 signals (green) did not reach the cutoff value needed for the diagnosis of gene amplification.
The FISH assay for FGFR1 was successful in 61 of the 69 primary lesions and in all 5 of the relapsed lesions; none showed gene amplification (Fig. 3B), but 7 (10.6 %) of 66 cases had a low level of copy number gain (Fig. 3C), which was not statistically related to any clinical feature (Table 3). The patient who had a poor outcome (P32) had a concomitant FGFR1 point mutation and low copy number gain of FGFR1 by FISH. The association between low copy number gain of FGFR1 and the immunohistochemical expression of FGFR1 was not significant (p = 0.091, data not shown).
Prognostic Role of BRAF and FGFR1
The Kaplan-Meier survival curves showed that the presence of KIAA1549:BRAF fusion was significantly associated with patients’ longer OS (p = 0.009) and EFS (p = 0.018) (Fig. 4A. B). BRAF point mutations were not associated with differences in the OS (p = 0.527), nor in the EFS (p = 0.317).
FIGURE 4.
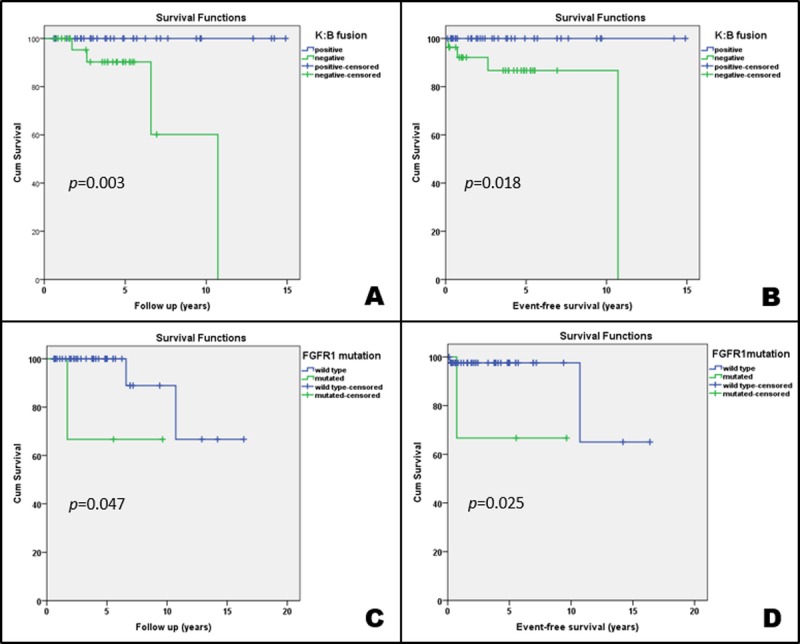
(A–D) Kaplan-Meier curves showing the impact of KIAA1549:BRAF (K:B) fusion (A, B) and FGFR1 p.K656E point mutation (C, D) in the overall survival and event-free survival of the patients.
FGFR1 immunohistochemical expression and low copy number gain were not correlated with OS (p = 0.103) or EFS (p = 0.923). On the other hand, patients with the FGFR1 p.K656E point mutation had significantly shorter OS (p = 0.047) and EFS (p = 0.025) when compared with patients with wild-type tumors (Fig. 4C, D).
Finally, we assessed the combined impact of BRAF and FGFR1 alterations in patients OS and EFS. We found that patients with tumors positive for KIAA1549:BRAF fusion showed longer survival regardless of FGFR1 status and FGFR1 immunohistochemical expression (Fig. 5A, B). Distinctively, among the tumors negative for KIAA1549:BRAF fusion, the ones with the FGFR1 pK656E point mutation had significantly worse prognosis (p = 0.002), whereas the overexpression of FGFR1 was related to a better prognosis (p = 0.03) (Fig. 5A, B).
FIGURE 5.
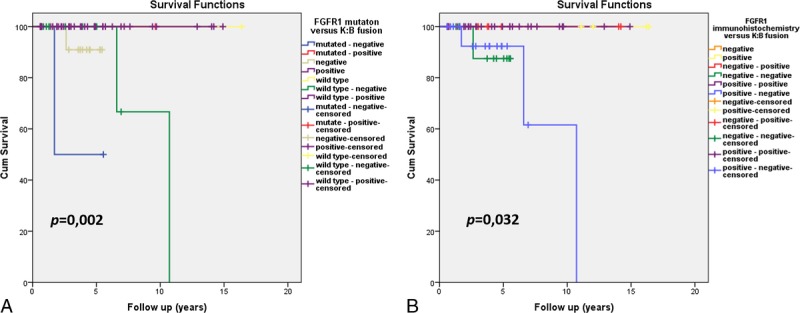
Kaplan-Meier curves comparing the simultaneous impact of KIAA1549:BRAF (K:B) fusion and FGFR1 alterations on the overall survival of patients. (A) Impact of FGFR1 p.K656E point mutation. (B) Impact of FGFR1 expression assessed by immunohistochemistry (positive score ≥3; negative score ≤2).
DISCUSSION
We have shown in a series of 69 WHO grade I PAs that KIAA1549:BRAF fusions are present in most of the cases and that they are associated with better prognosis. In addition, we found that FGFR1 is altered by oncogenic mutations in a small subset (∼7%) of cases that were associated with an adverse outcome.
Despite the emergent interest in children’s brain tumors, many studies have clustered PAs, diffuse astrocytomas (WHO grade II), and other neoplasms in a set of “low-grade gliomas” (16, 21, 23, 44–47). This has probably occurred because of the rarity of these brain tumors compared with the much more frequent adult tumors, such as glioblastoma (3). In addition, the studies tend to isolate adults from children (6, 8). As far as we are aware, this is the first study with a cohort composed exclusively of PAs, excluding the pilomyxoid astrocytoma variant of PA (WHO grade II), and comparing different age groups.
Herein, we were able to evaluate gene fusions and point mutations in 67 of 69 cases and observed that nearly 60% of them had alterations in BRAF and/or FGFR1, which are triggers of the MAPK pathway, the dominant oncogenic pathway of PAs (15, 19). The high incidence of KIAA1549:BRAF fusion and its predominance in cerebellar lesions are in line with previous studies (7, 15, 16, 24, 26), confirming it as the most frequent molecular change of PAs (7). In addition, 2 of 5 tumors that harbored BRAF or FGFR1 mutations had a coexisting KIAA1549:BRAF gene fusion. The occurrence of a concomitant KIAA1549:BRAF fusion and other changes in the same pathway is a rare occurrence, but it has been previously reported (7, 21, 26). Finally, the negative relationship between KIAA1549:BRAF fusions and clinical diagnosis of NF1 (thus with alterations in the NF1 gene) has been previously reported (7). Further studies on NF1 are necessary for a better understanding of this relationship.
The incidence of BRAF point mutations in our study (4.2%) is also similar to published data (22). The presence of this alteration did not show a clinical impact on prognosis, thereby confirming the previous findings of Bannykh et al, who showed that the BRAF V600E mutation did not imply higher aggressiveness to PAs (48). The usual p.V600E point mutation was detected only in an 11-year-old male patient who had an unstable hypothalamic lesion, which reinforces the occurrence of this mutation in extracerebellar lesions (7). Moreover, that patient also harbored the KIAA1549:BRAF fusion. We also observed 1 unusual BRAF point mutation in codon 600 of a cerebellar PA. The point mutation V600K was found in an 11-year-old female patient who had an excellent outcome after long follow-up (11.1 years). This mutation was previously described in 5% to 15% of melanomas and was related to metastatic disease and worse outcome (49–51); however, it has also been associated with a response to first-generation BRAF inhibitors (PLX4032, vemurafenib) (51). Patients with PAs have experienced adverse results after treatment with vemurafenib (52), as opposed to the good response observed in patients with high-grade tumors (53, 54), probably because of the overall low frequency of p.V600E. Nevertheless, the subset of patients with KIAA1549:BRAF positive tumors could potentially benefit from treatment with the second-generation BRAF inhibitors such as PLX-PB3, which specifically target the fusion protein (52).
Most tumors in our series showed strong immunohistochemical FGFR1 expression. These findings are in line with a previous study in gliomas, in which FGFR1 overexpression was detected, although the underlying molecular mechanism was not explained at the time (55). Our FISH assays largely eliminated amplification as the underlying mechanism in the PAs, contrary to what is seen in a subset of breast and lung cancers (30, 31). FGFR1 low copy number gain was rare and showed a nonstatistically significant trend toward immunohistochemical overexpression (p = 0.094, data not shown). The mutated form of FGFR1 was also not associated with protein overexpression (p = 1.0).
With respect to the FGFR1 mutation, we observed p.K656E point mutations at the tyrosine kinase domain in 6.7% of the PAs in this series, all of which were located in the cerebellum. The oncogenic FGFR1 mutations, p.K656E and p.N456K, were recently described by Jones et al (15) as recurrent events in extracerebellar PAs. Those mutations were further described in rosette-forming glioneuronal tumor of the fourth ventricle, but 1 of the patients in that series had an earlier extracerebellar (diencephalic) PA with pilomyxoid features, which also harbored the p.K656E mutation (29).
FGFR1 is currently an attractive therapeutic target, and the immunohistochemical assessment of FGFR1 may represent a good indicator of the management of PAs. Recent studies have related the efficacy of novel specific FGFR1 inhibitors, such as ponatinib (AP 24534), in cases of lung cancer with FGFR1 overexpression that were assessed by immunoblotting and mRNA quantification (56). Besides this drug, other FGFR1 inhibitors, such as lucitanib (57) and CH5183284/Debio 1347 (58), may constitute future alternatives for the treatment of inoperable PAs. Nevertheless, further preclinical and clinical studies are needed to determine whether FGFR1 expression and hotspot mutations will modulate and predict patient response to these FGFR1-specific tyrosine inhibitors.
Concomitant KIAA1549:BRAF fusion and FGFR1 mutations were not referred events in the study of Jones et al (15), but this was detected in 1 of the patients of our series. We further evaluated the impact of both the aforementioned alterations in the prognosis of the patients. The KIAA1549:BRAF fusion had a positive impact on patients’ OS and EFS and was confirmed as a prognostic factor, corroborating the tendency to better outcome of PAs, similar to what happens in the complex group of low-grade gliomas described by Hawkins et al (21).
On the other hand, FGFR1 mutations were significantly related to PA patients’ shorter OS and EFS when compared with the wild-type group; however, the significance of this finding needs to be confirmed in larger series. To our knowledge, this the first study to indicate the prognostic role of FGFR1 mutation in PAs and their occurrence in cerebellar lesions.
In conclusion, we confirmed the pivotal role of KIAA1549:BRAF fusion and, to a lesser extent, of FGFR1 in MAPK activation in PAs. More exactly, we showed the usefulness of evaluating the KIAA1549:BRAF fusion as a prognostic biomarker, while FGFR1 mutation may be a relevant prognostic marker in PAs. With further investigation, the molecular changes of BRAF and FGFR1 may constitute potential therapeutic targets for inoperable or recurrent PAs.
ACKNOWLEDGMENT
The authors thank Nathalia Campanella for the help in picture editing.
Footnotes
This study was partially supported by CNPq/Universal (475358/2011-2), and FAPESP (2012/19590-0) grants to RMR and to the NIH- P30CA046934 (CCSG Molecular Pathology/Cytogenetics) to MVG and DLA.
REFERENCES
- 1. Scheithauer BW, Hawkins C, Tihan T, et al. Pilocytic astrocytoma. In: David N, Louis MD, Hiroko Ohgaki PD, Otmar D, Wiestler MD, Webster K, Cavenee PD, editors. WHO Classification of Tumours of the Central Nervous System. World Health Organization Classification of Tumours. Lyon, France: IARC Press, 2007: 13– 21 [Google Scholar]
- 2. Ward E, DeSantis C, Robbins A, et al. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014; 64: 83– 103 [DOI] [PubMed] [Google Scholar]
- 3. Dolecek TA, Propp JM, Stroup NE, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol 2012; 14 (Suppl 5): v1– 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camargo Bd, Felipe CFP, Noronha CP, et al. Câncer na criança e no adolescente no Brasil dados dos registros de base populacional e de mortalidade. Rio de Janeiro: Instituto Nacional do Câncer, 2008 [Google Scholar]
- 5. Rosemberg S, Fujiwara D. Epidemiology of pediatric tumors of the nervous system according to the WHO 2000 classification: a report of 1,195 cases from a single institution. Child Nerv Sys 2005; 21: 940– 4 [DOI] [PubMed] [Google Scholar]
- 6. Theeler BJ, Ellezam B, Sadighi ZS, et al. Adult pilocytic astrocytomas: clinical features and molecular analysis. Neuro Oncol 2014 2014; 16: 841– 7. doi: 10.1093/neuonc/not246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones DT, Gronych J, Lichter P, et al. MAPK pathway activation in pilocytic astrocytoma. Cell Molec Life Sci 2012; 69: 1799– 811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson DR, Brown PD, Galanis E, et al. Pilocytic astrocytoma survival in adults: analysis of the Surveillance, Epidemiology, and End Results Program of the National Cancer Institute. J Neuro-oncol 2012; 108: 187– 93 [DOI] [PubMed] [Google Scholar]
- 9. Gutmann DH, McLellan MD, Hussain I, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res 2013; 23: 431– 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol 2008; 67: 240– 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Listernick R, Charrow J, Gutmann DH. Intracranial gliomas in neurofibromatosis type 1. Am J Med Gen 1999; 89: 38– 44 [DOI] [PubMed] [Google Scholar]
- 12. Tada K, Kochi M, Saya H, et al. Preliminary observations on genetic alterations in pilocytic astrocytomas associated with neurofibromatosis 1. Neuro Oncol 2003; 5: 228– 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dhanasekaran DN, Johnson GL. MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene 2007; 26: 3097– 9 [DOI] [PubMed] [Google Scholar]
- 14. Murphy T, Hori S, Sewell J, et al. Expression and functional role of negative signalling regulators in tumour development and progression. Int J Cancer 2010; 127: 2491– 9 [DOI] [PubMed] [Google Scholar]
- 15. Jones DT, Hutter B, Jager N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nature Gen 2013; 45: 927– 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nature Gen 2013; 45: 602– 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bar EE, Lin A, Tihan T, et al. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol 2008; 67: 878– 87 [DOI] [PubMed] [Google Scholar]
- 18. Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol 2011; 122: 11– 9 [DOI] [PubMed] [Google Scholar]
- 19. Forshew T, Tatevossian RG, Lawson AR, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 2009; 218: 172– 81 [DOI] [PubMed] [Google Scholar]
- 20. Hasselblatt M, Riesmeier B, Lechtape B, et al. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol 2011; 37: 803– 6 [DOI] [PubMed] [Google Scholar]
- 21. Hawkins C, Walker E, Mohamed N, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 2011; 17: 4790– 8 [DOI] [PubMed] [Google Scholar]
- 22. Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 2011; 121: 397– 405 [DOI] [PubMed] [Google Scholar]
- 23. Tian Y, Rich BE, Vena N, et al. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J Molec Diag 2011; 13: 669– 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones DT, Kocialkowski S, Liu L, et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 2009; 28: 2119– 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roth JJ, Santi M, Pollock AN, et al. Chromosome band 7q34 deletions resulting in KIAA1549-BRAF and FAM131B-BRAF fusions in pediatric low-grade gliomas. Brain Pathol 2015; 25: 182– 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 2011; 121: 763– 74 [DOI] [PubMed] [Google Scholar]
- 27. Chen YH, Gutmann DH. The molecular and cell biology of pediatric low-grade gliomas. Oncogene 2014; 33: 2019– 26 [DOI] [PubMed] [Google Scholar]
- 28. Basto D, Trovisco V, Lopes JM, et al. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol 2005; 109: 207– 10 [DOI] [PubMed] [Google Scholar]
- 29. Gessi M, Moneim YA, Hammes J, et al. FGFR1 mutations in rosette-forming glioneuronal tumors of the fourth ventricle. J Neuropathol Exp Neurol 2014; 73: 580– 4 [DOI] [PubMed] [Google Scholar]
- 30. Dutt A, Ramos AH, Hammerman PS, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PloS One 2011; 6: e20351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Theillet C, Adelaide J, Louason G, et al. FGFRI and PLAT genes and DNA amplification at 8p12 in breast and ovarian cancers. Genes Chromosome Cancer 1993; 7: 219– 26 [DOI] [PubMed] [Google Scholar]
- 32. Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012; 337: 1231– 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Colin C, Padovani L, Chappe C, et al. Outcome analysis of childhood pilocytic astrocytomas: a retrospective study of 148 cases at a single institution. Neuropathol Appl Neurobiol 2013; 39: 693– 705 [DOI] [PubMed] [Google Scholar]
- 34. Fernandez C, Figarella-Branger D, Girard N, et al. Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery 2003; 53: 544– 53; discussion 54–5 [DOI] [PubMed] [Google Scholar]
- 35. Paixao Becker A, de Oliveira RS, Saggioro FP, et al. In pursuit of prognostic factors in children with pilocytic astrocytomas. Childs Nerv Syst 2010; 26: 19– 28 [DOI] [PubMed] [Google Scholar]
- 36. Schneider JH, Jr, Raffel C, McComb JG. Benign cerebellar astrocytomas of childhood. Neurosurgery 1992; 30: 58– 62; discussion 3 [DOI] [PubMed] [Google Scholar]
- 37. Capper D, Weissert S, Balss J, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 2010; 20: 245– 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mellai M, Piazzi A, Caldera V, et al. IDH1 and IDH2 mutations, immunohistochemistry and associations in a series of brain tumors. J Neuro-oncol 2011; 105: 345– 57 [DOI] [PubMed] [Google Scholar]
- 39. Pinto F, Pertega-Gomes N, Pereira MS, et al. T-box transcription factor brachyury is associated with prostate cancer progression and aggressiveness. Clinical Cancer Res 2014; 20: 4949– 61 [DOI] [PubMed] [Google Scholar]
- 40. Toschi L, Finocchiaro G, Nguyen TT, et al. Increased SOX2 gene copy number is associated with FGFR1 and PIK3CA gene gain in non–small cell lung cancer and predicts improved survival in early stage disease. PloS One 2014; 9: e95303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aisner DL, Nguyen TT, Paskulin DD, et al. ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Molec Cancer Res 2014; 12: 111– 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schultheis AM, Bos M, Schmitz K, et al. Fibroblast growth factor receptor 1 (FGFR1) amplification is a potential therapeutic target in small-cell lung cancer. Mod Pathol 2014; 27: 214– 21 [DOI] [PubMed] [Google Scholar]
- 43. Yamane LS, Scapulatempo-Neto C, Alvarenga L, et al. KRAS and BRAF mutations and MSI status in precursor lesions of colorectal cancer detected by colonoscopy. Oncology Rep 2014; 32: 1419– 26 [DOI] [PubMed] [Google Scholar]
- 44. Bhattacharjee MB, Armstrong DD, Vogel H, et al. Cytogenetic analysis of 120 primary pediatric brain tumors and literature review. Cancer Gen Cytogen 1997; 97: 39– 53 [DOI] [PubMed] [Google Scholar]
- 45. Bigner SH, McLendon RE, Fuchs H, et al. Chromosomal characteristics of childhood brain tumors. Cancer Gen Cytogen 1997; 97: 125– 34 [DOI] [PubMed] [Google Scholar]
- 46. Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 2008; 118: 1739– 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sievert AJ, Jackson EM, Gai X, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol 2009; 19: 449– 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bannykh SI, Mirocha J, Nuno M, et al. V600E BRAF mutation in pilocytic astrocytoma is associated with a more diffuse growth pattern but does not confer a more aggressive clinical behavior. Cli Neuropathol 2014; 33: 388– 98 [DOI] [PubMed] [Google Scholar]
- 49. El-Osta H, Falchook G, Tsimberidou A, et al. BRAF mutations in advanced cancers: clinical characteristics and outcomes. PloS One 2011; 6: e25806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Menzies AM, Haydu LE, Visintin L, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clinical Cancer Res 2012; 18: 3242– 9 [DOI] [PubMed] [Google Scholar]
- 51. Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010; 8: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sievert AJ, Lang SS, Boucher KL, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA 2013; 110: 5957– 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bautista F, Paci A, Minard-Colin V, et al. Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Ped Blood Cancer 2014; 61: 1101– 3 [DOI] [PubMed] [Google Scholar]
- 54. Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014; 14: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ueba T, Takahashi JA, Fukumoto M, et al. Expression of fibroblast growth factor receptor-1 in human glioma and meningioma tissues. Neurosurgery 1994; 34: 221– 5; discussion 5–6 [DOI] [PubMed] [Google Scholar]
- 56. Wynes MW, Hinz TK, Gao D, et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI ensitivity across all lung cancer histologies. Clin Cancer Res 2014; 20: 3299– 309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Soria JC, DeBraud F, Bahleda R, et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann Oncol 2014; 25: 2244– 51 [DOI] [PubMed] [Google Scholar]
- 58. Nakanishi Y, Akiyama N, Tsukaguchi T, et al. The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Molec Cancer Ther 2014; 13: 2547– 58 [DOI] [PubMed] [Google Scholar]